
A Hyphenated Technique based on High-Performance Thin Layer Chromatography for Determining Neutral Sphingolipids: A Proof of Concept


Abstract
:1. Introduction
2. Experimental Section
2.1. Fluorophore
2.2. SL Standards and Plasma Samples
2.3. Sample Treatment
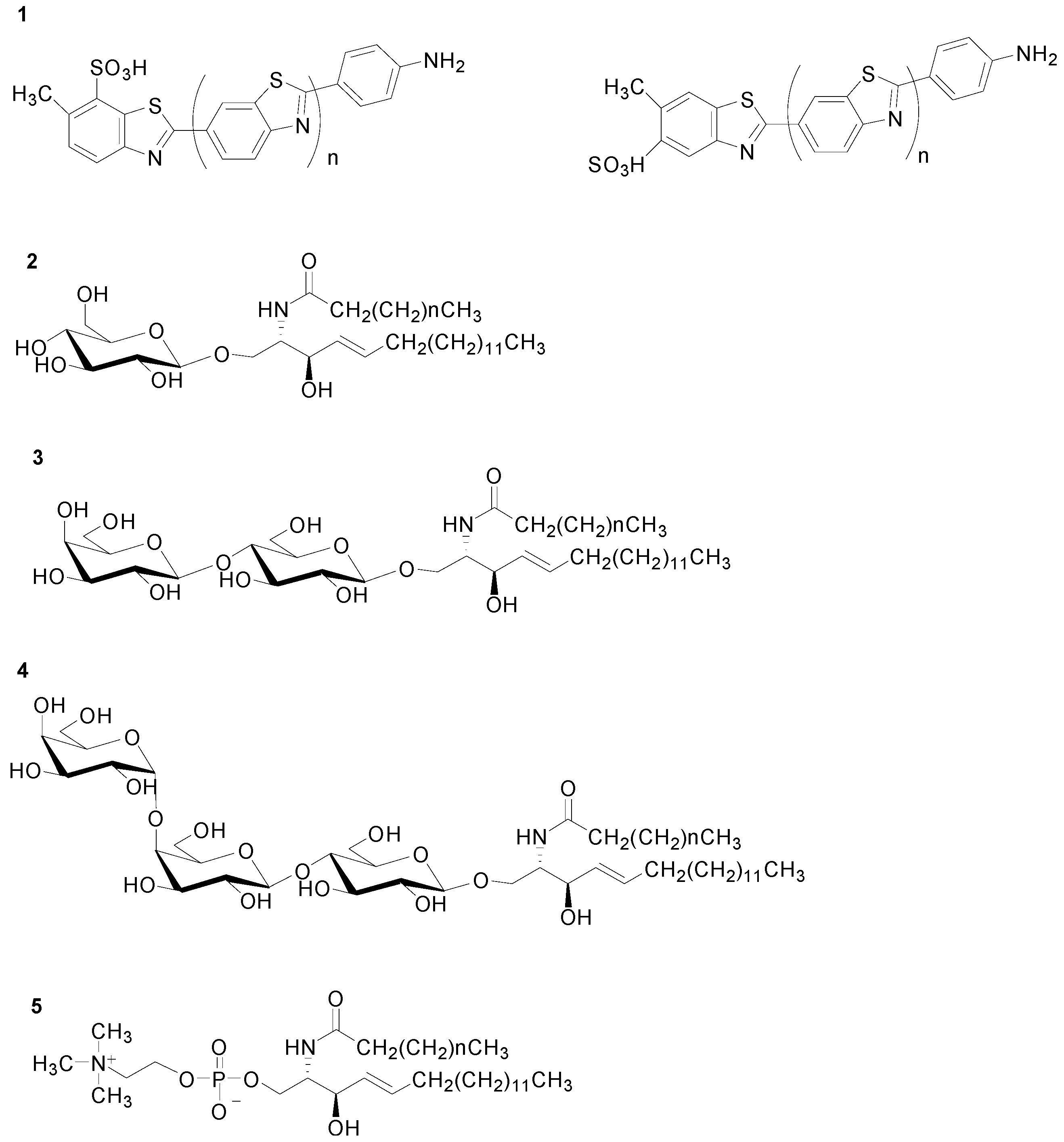
2.4. Plates for HPTLC Separation
2.5. AMD-FDIC-MS Hyphenated System: Description and Conditions for SL Analysis
2.5.1. Sample Application
2.5.2. HPTLC Using AMD

{kind=link}
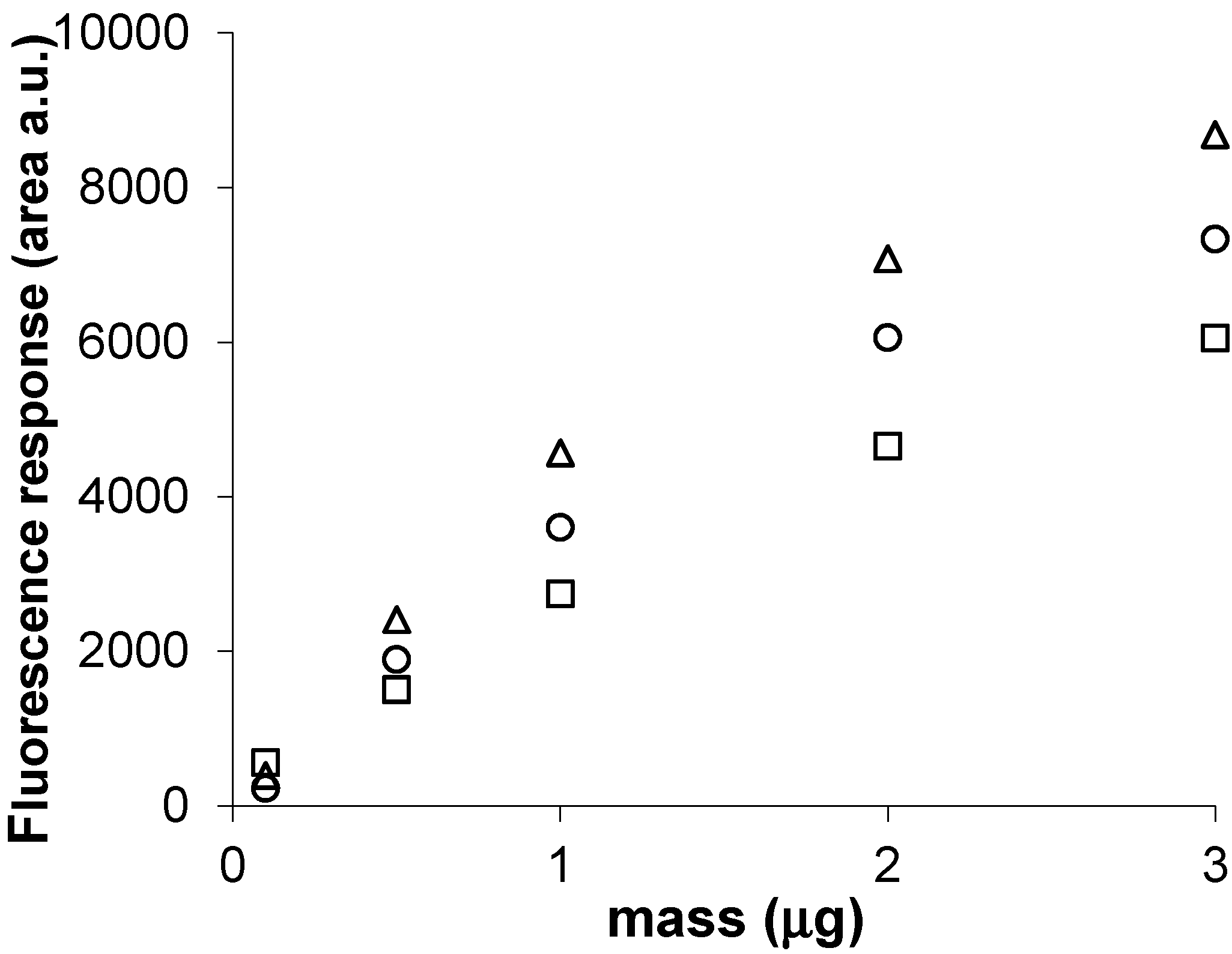{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MeOH (vol %) | DCM (vol %) | Migration (mm) | |
|---|---|---|---|
| Step 1 | 50 | 50 | 30 |
| Step 2 | 50 | 50 | 30 |
| Step 3 | 50 | 50 | 30 |
| Step 4 | 50 | 50 | 30 |
| Step 5 | 50 | 50 | 30 |
| Step 6 | 30 | 70 | 60 |
| Step 7 | 10 | 90 | 90 |
| MeOH (vol %) | DCM (vol %) | Migration (mm) | |
|---|---|---|---|
| Step 1 | 80 | 20 | 20 |
| Step 2 | 50 | 50 | 50 |
2.5.3. FDIC Using Fluorescence Scanning Densitometry
2.5.4. Coupling AMD-FDIC to API-MS
2.6. Repeatability of Sample Treatment
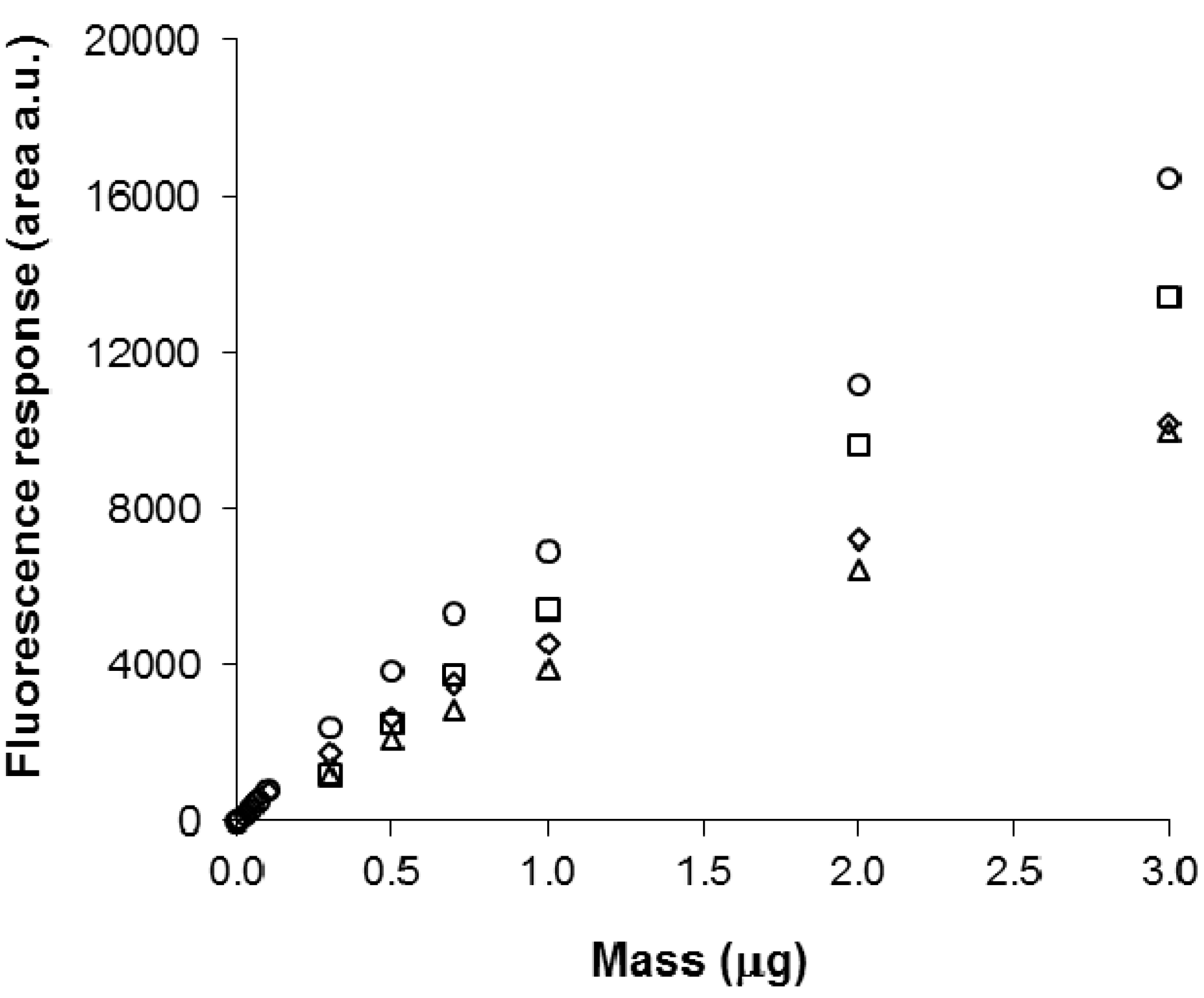
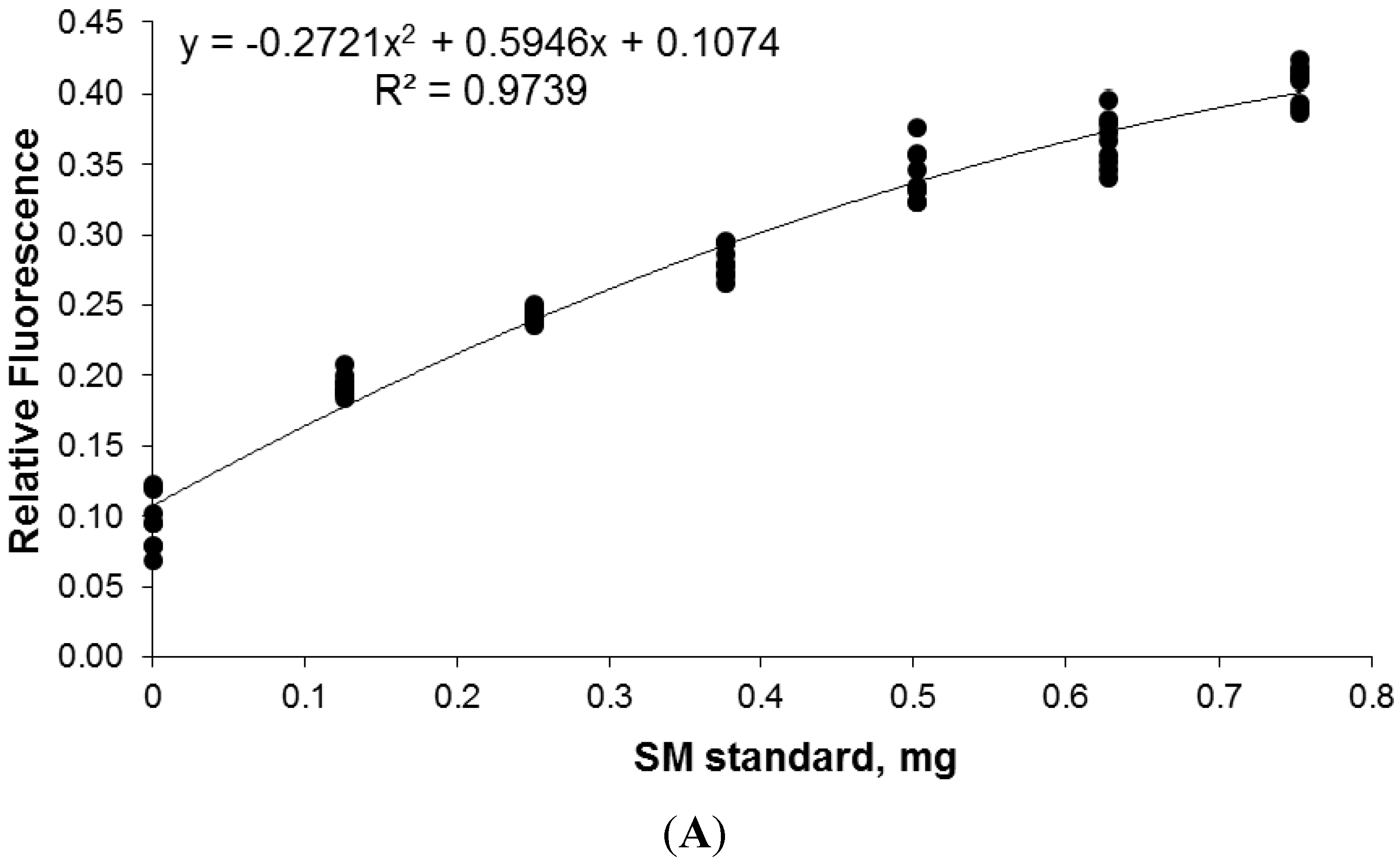
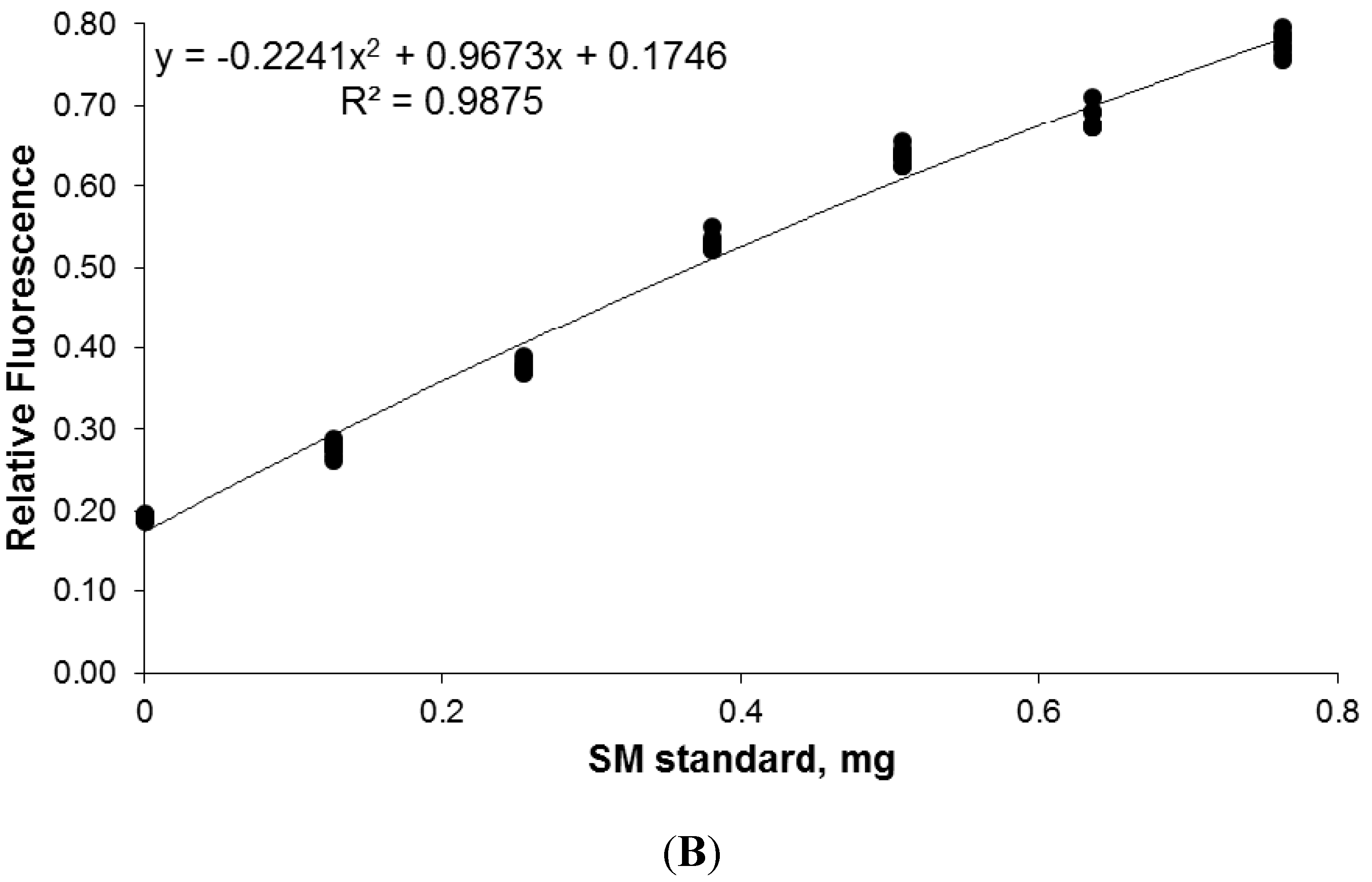
2.7. Calibration and Quantification of SM in Plasma Using FDIC
3. Results and Discussion
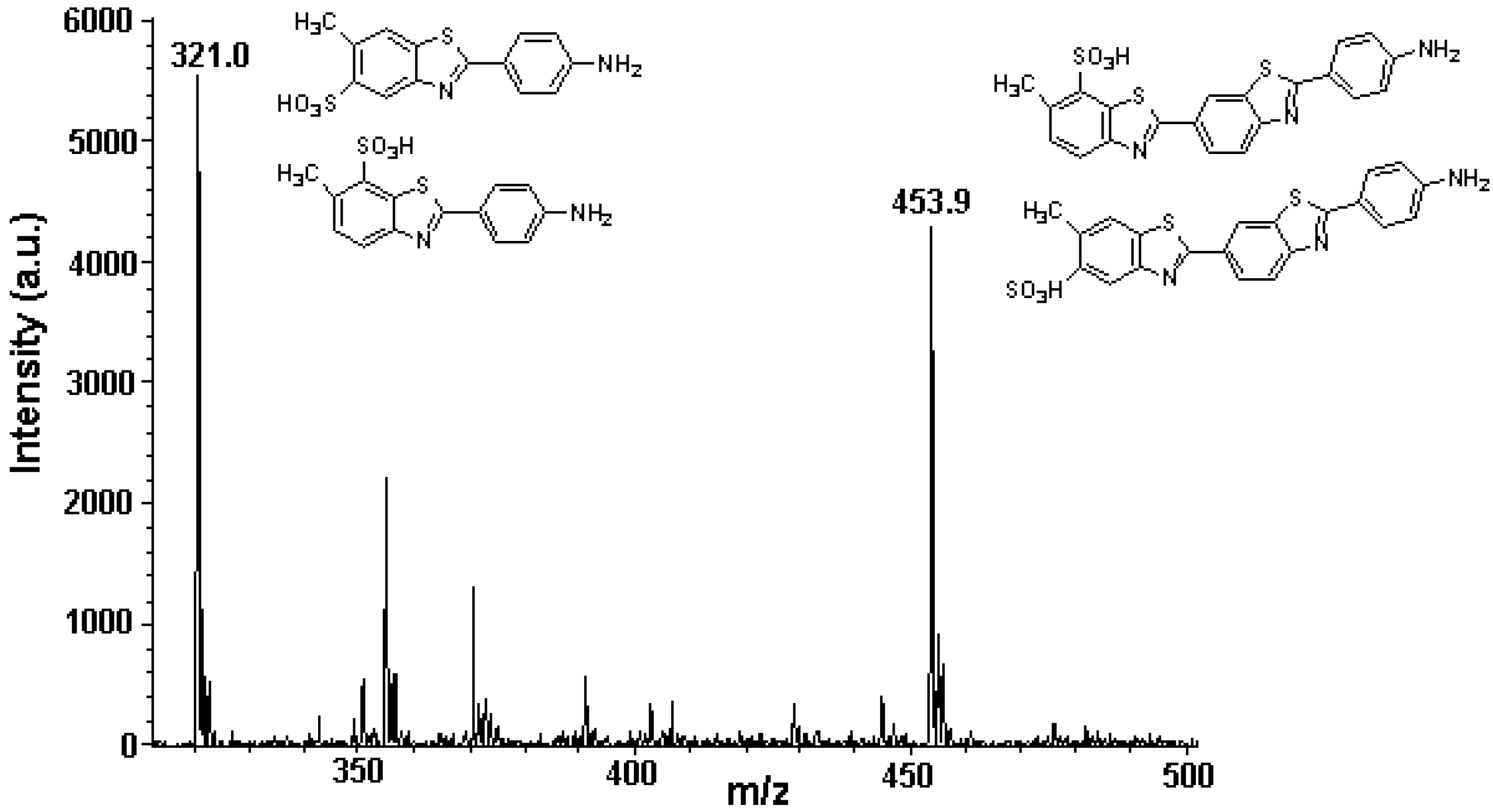
3.1. Primuline as an FDIC Fluorophore
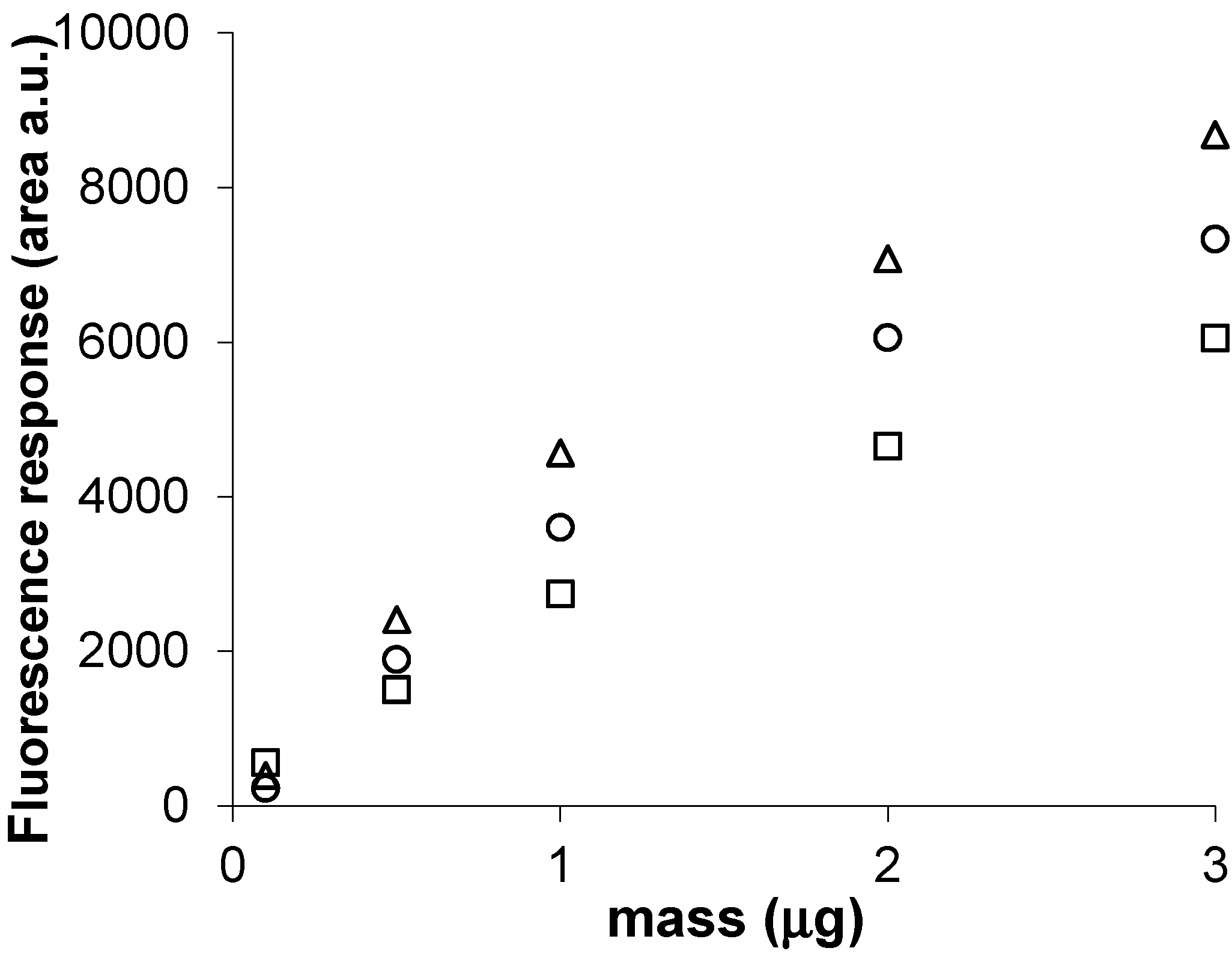
3.2. AMD-Gradient Separation of SLs

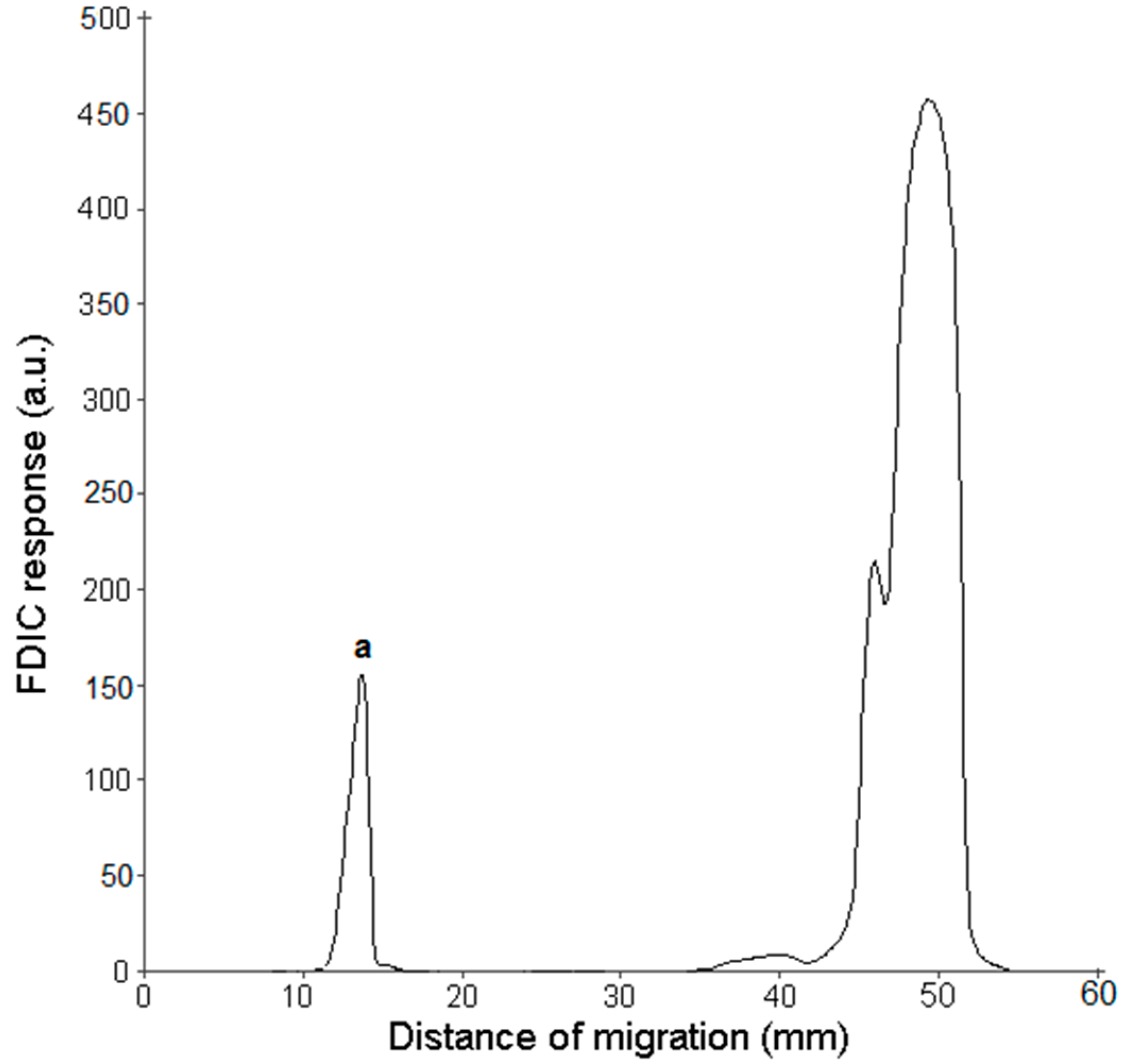
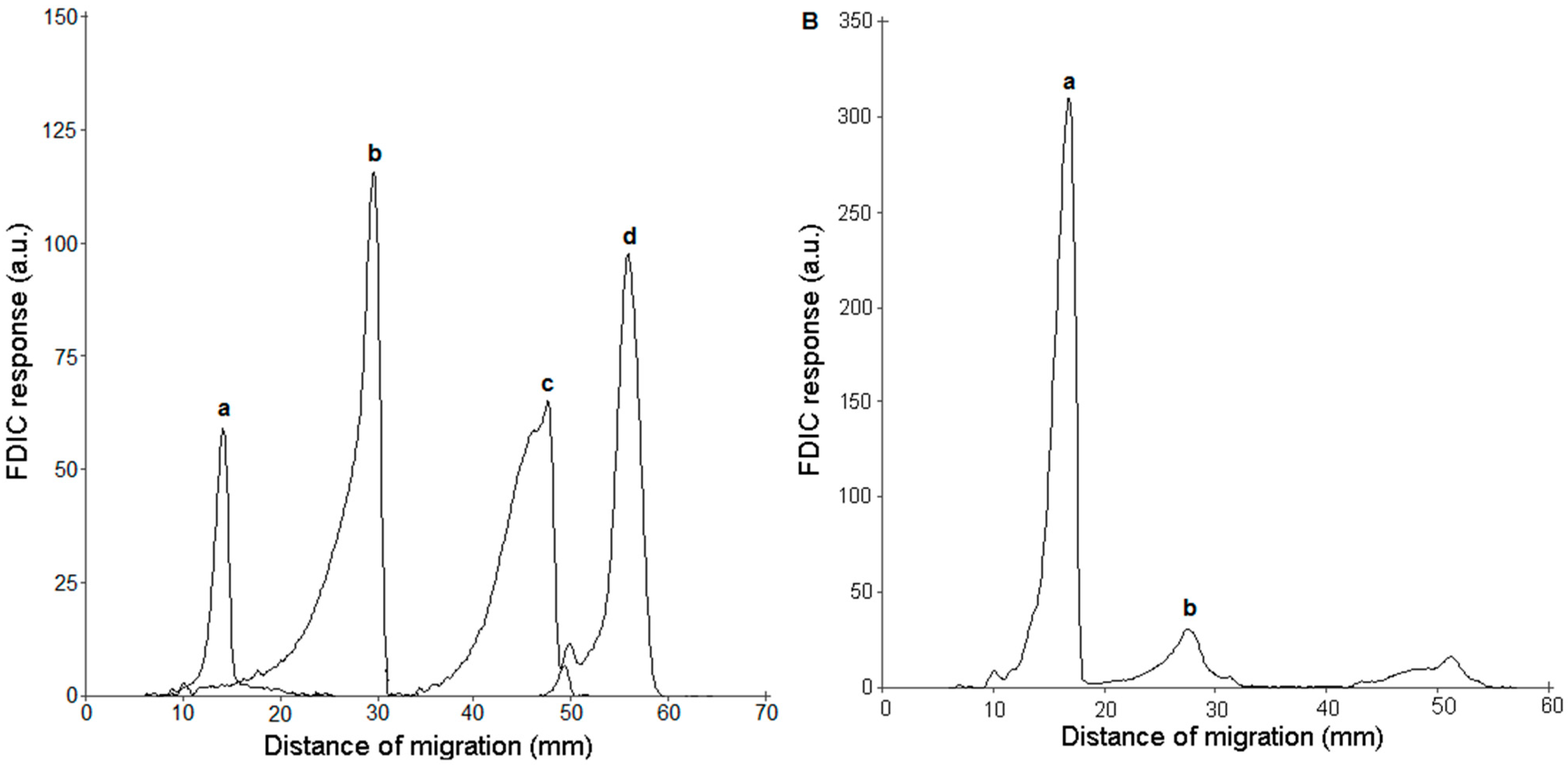
3.3. FDIC of Separated SLs Using Primuline
3.4. Online AMD-FDIC-MS Coupling
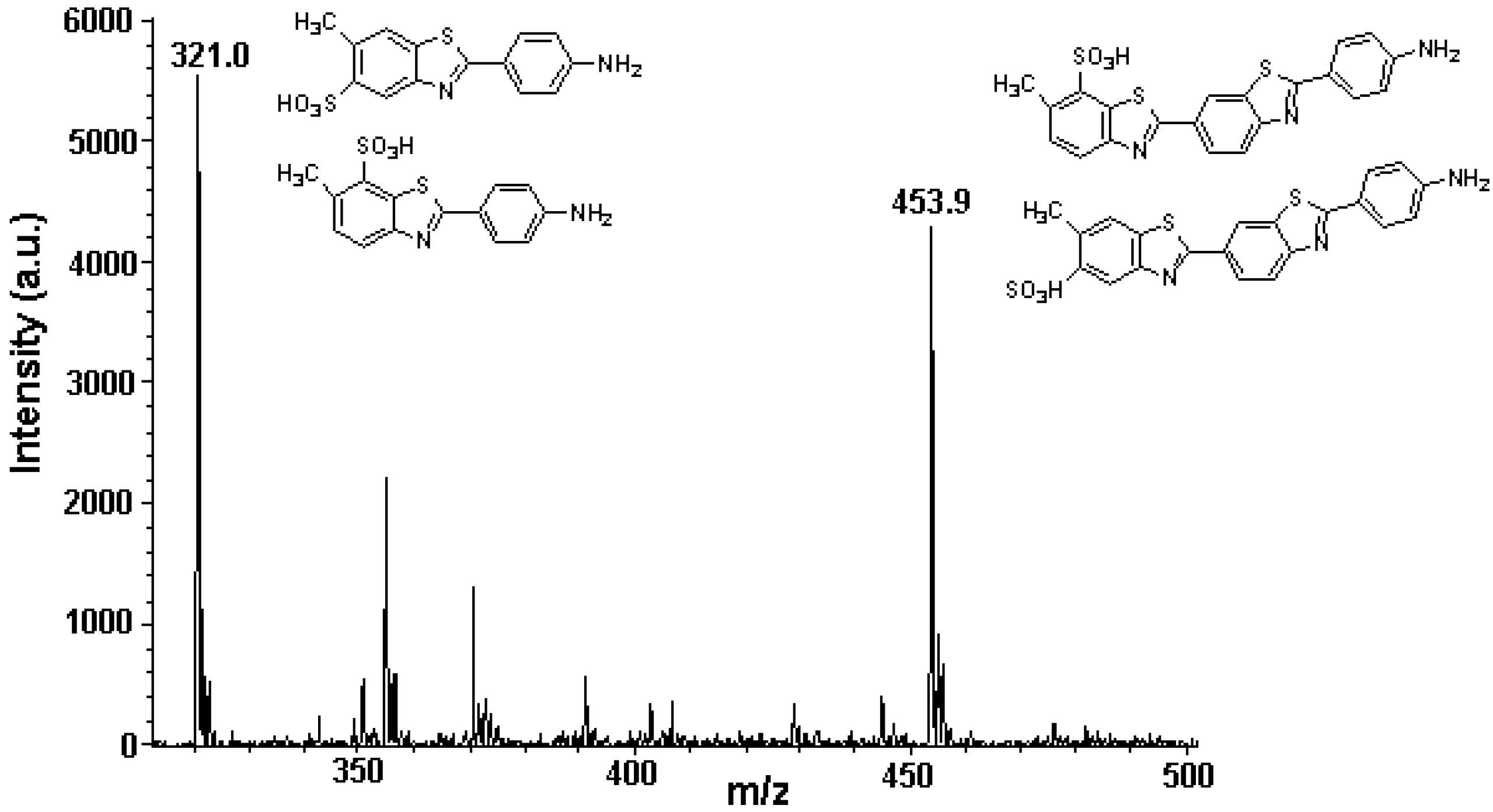
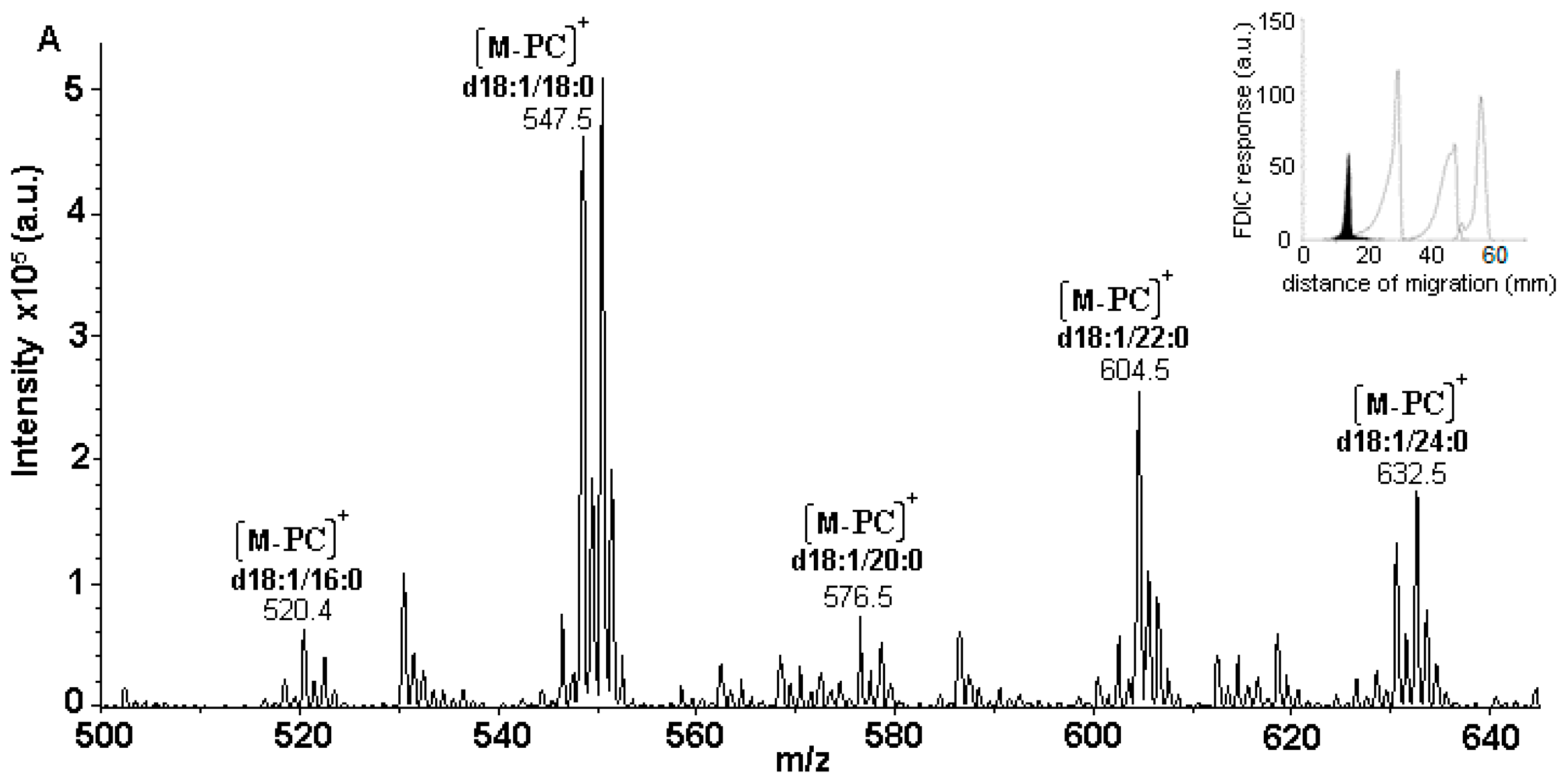
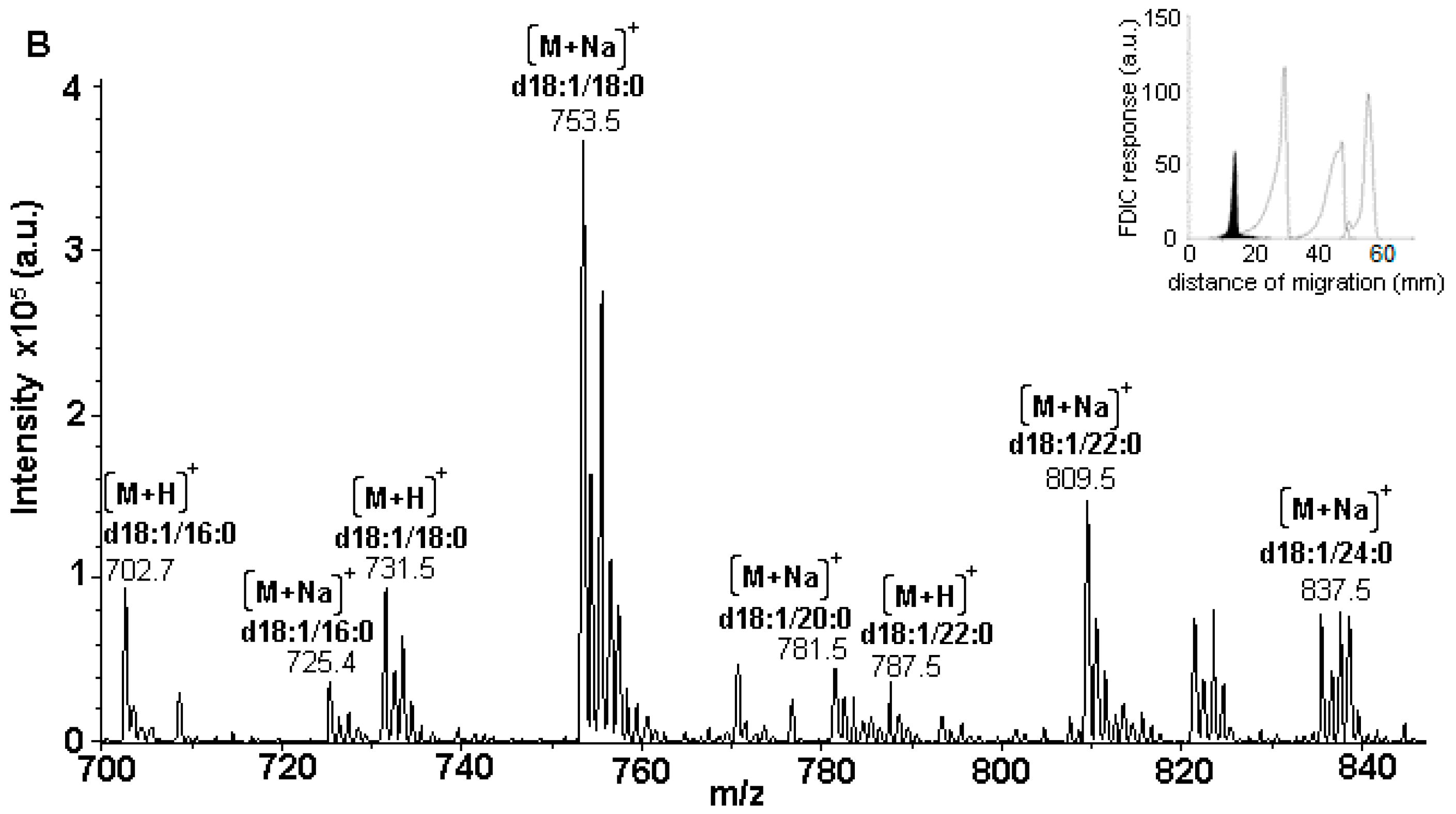
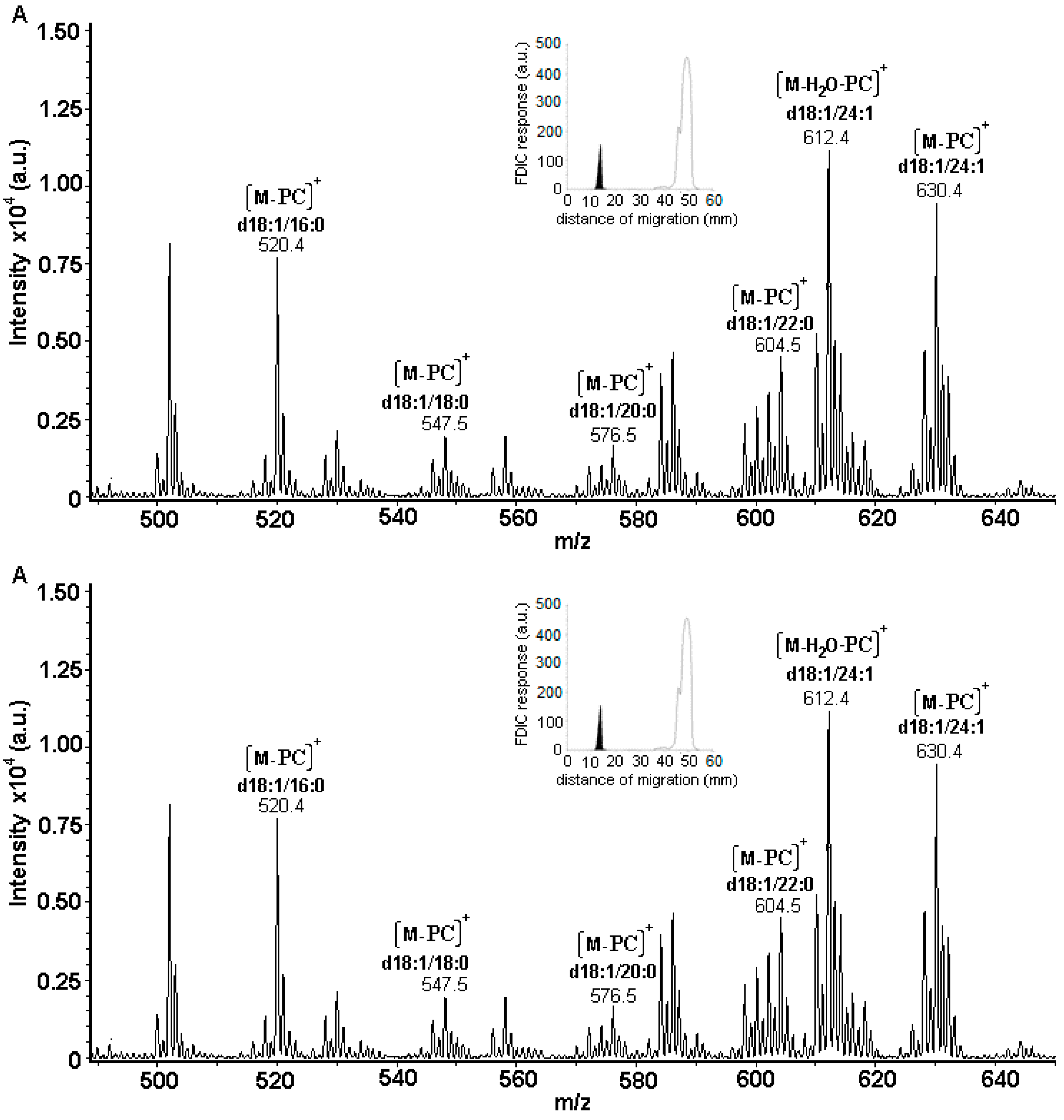
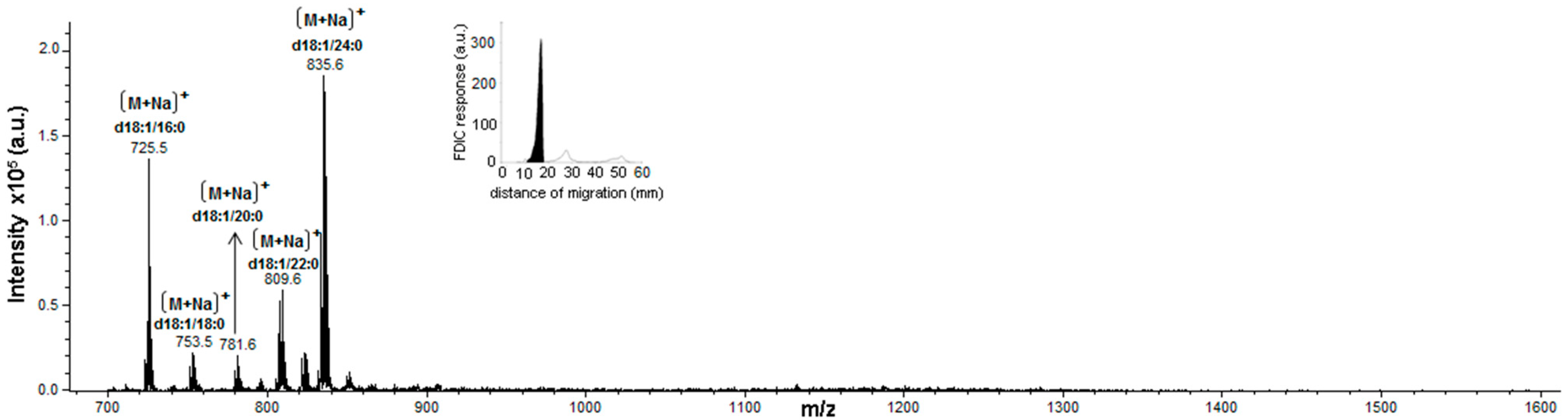
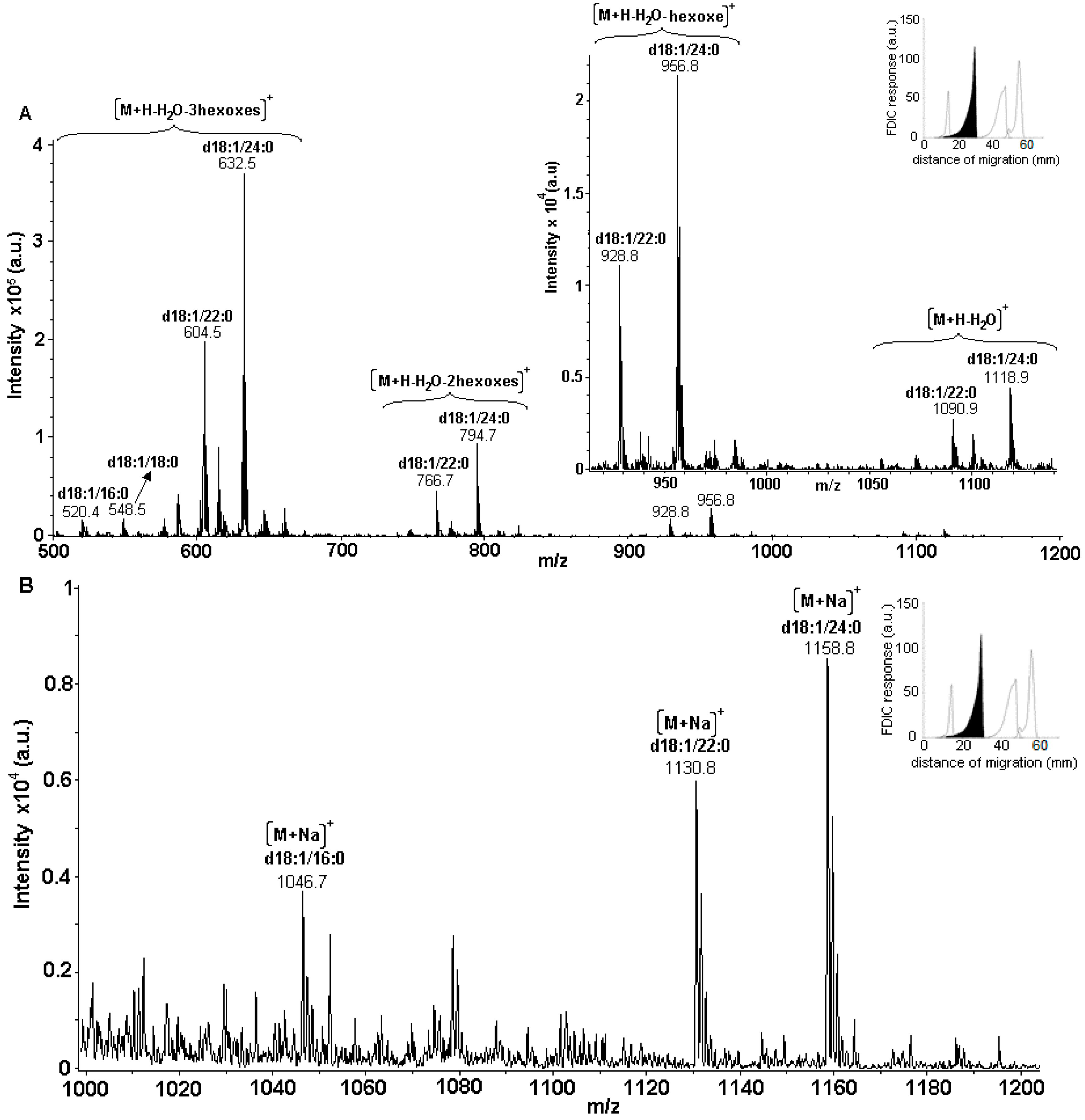
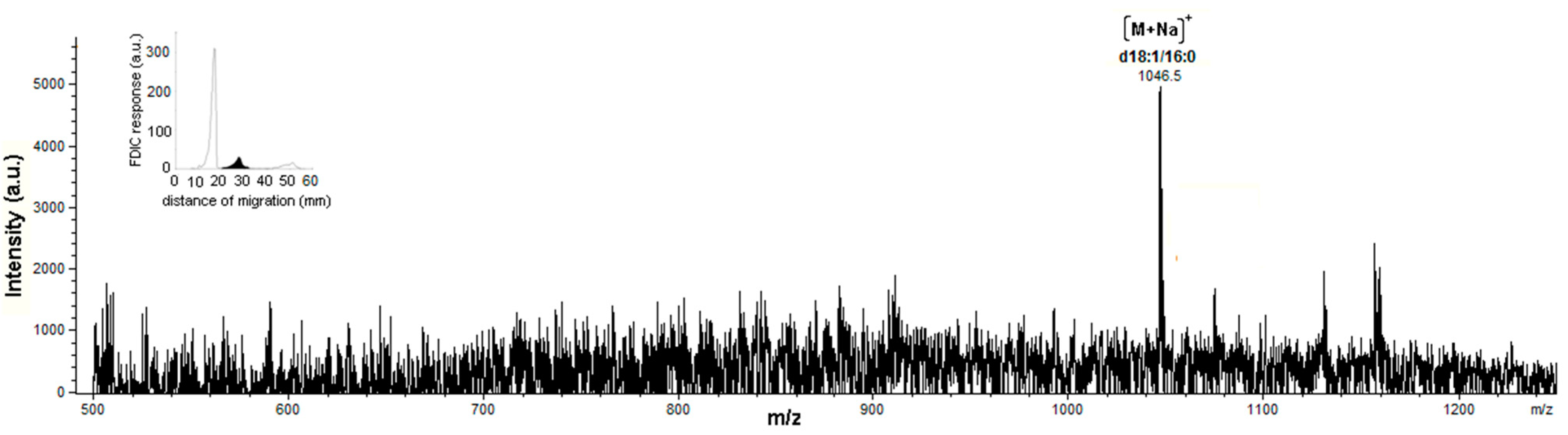
3.5. Identification of SLs Using AMD-FDIC-(API)-MS
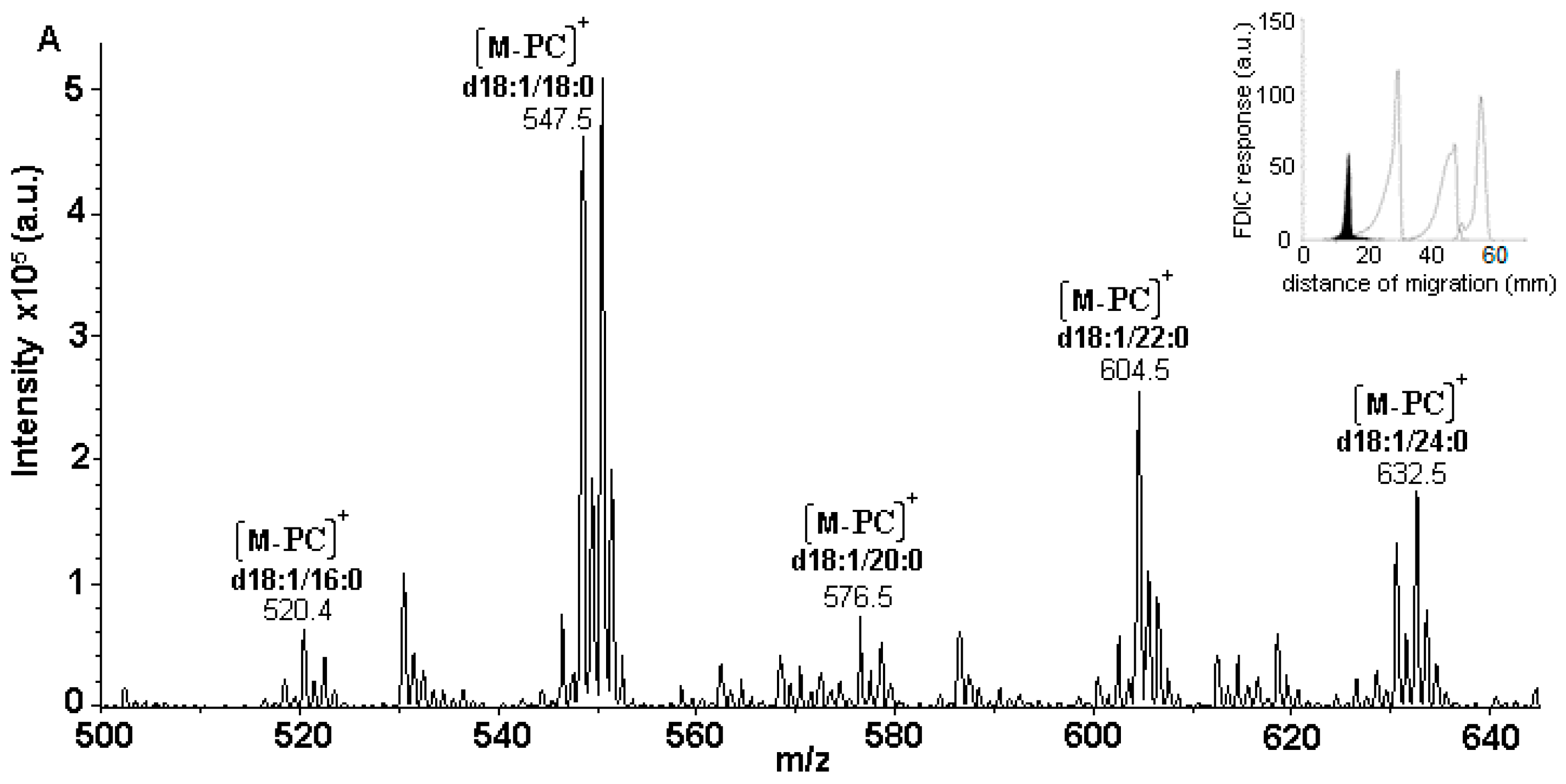
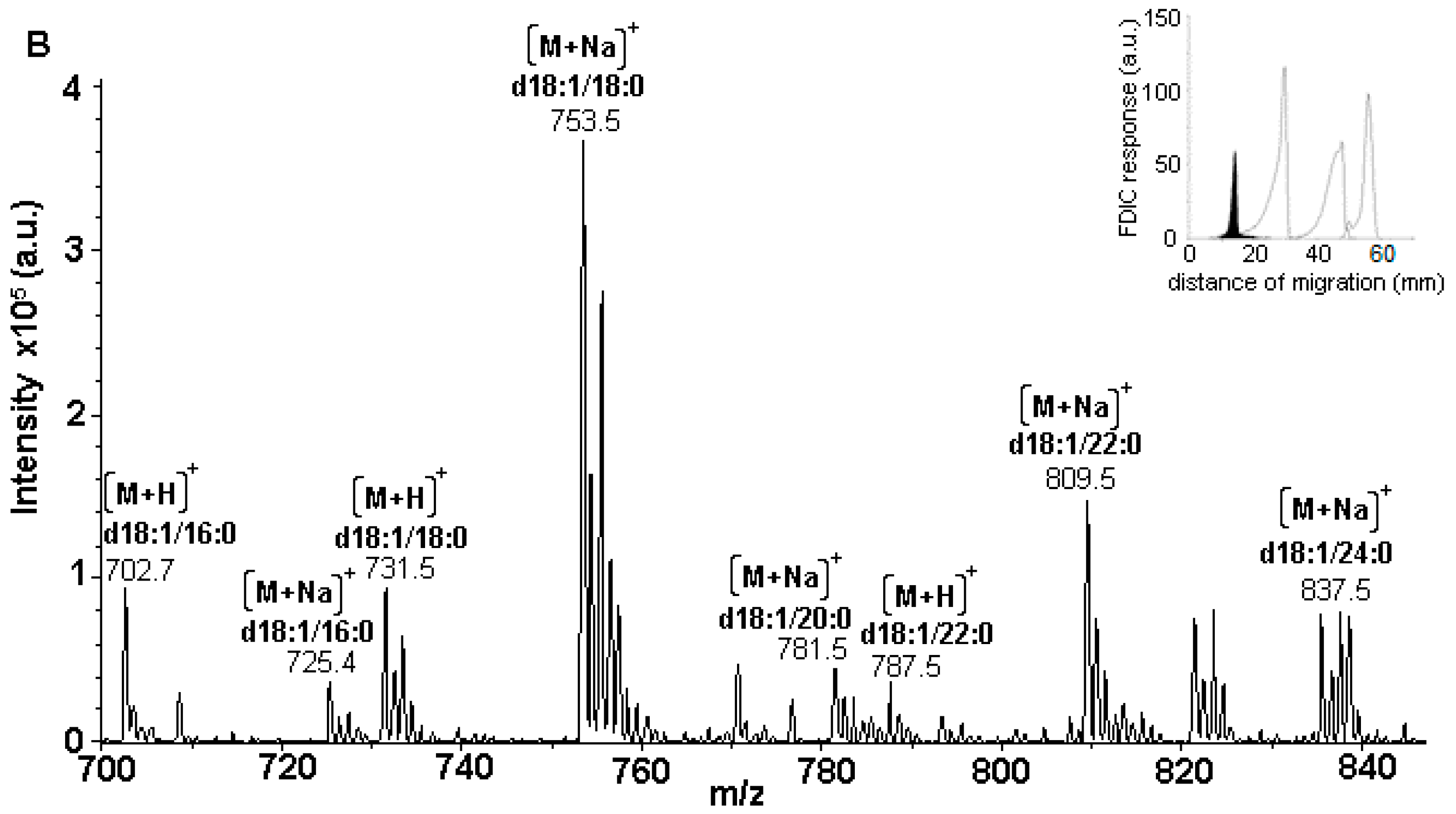
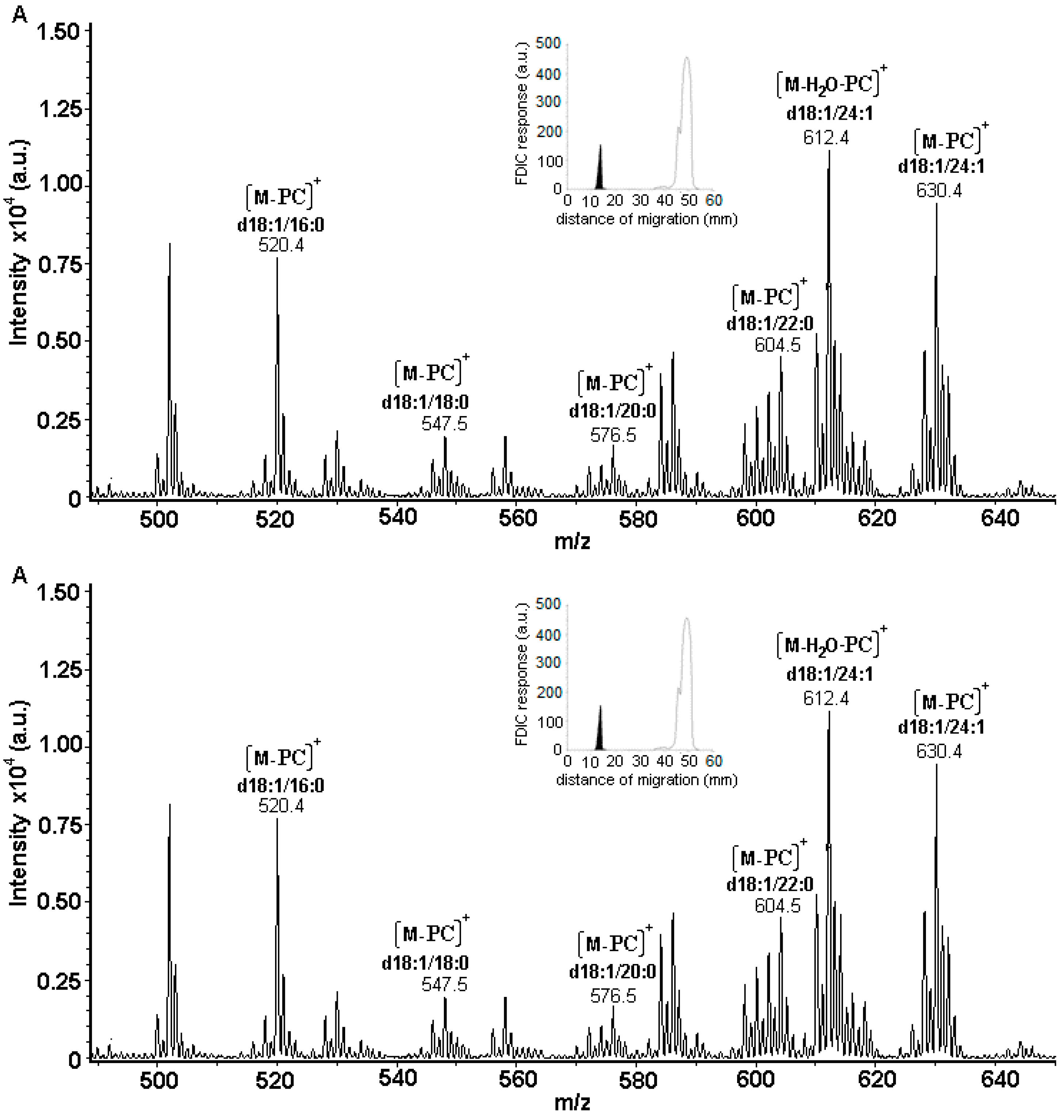
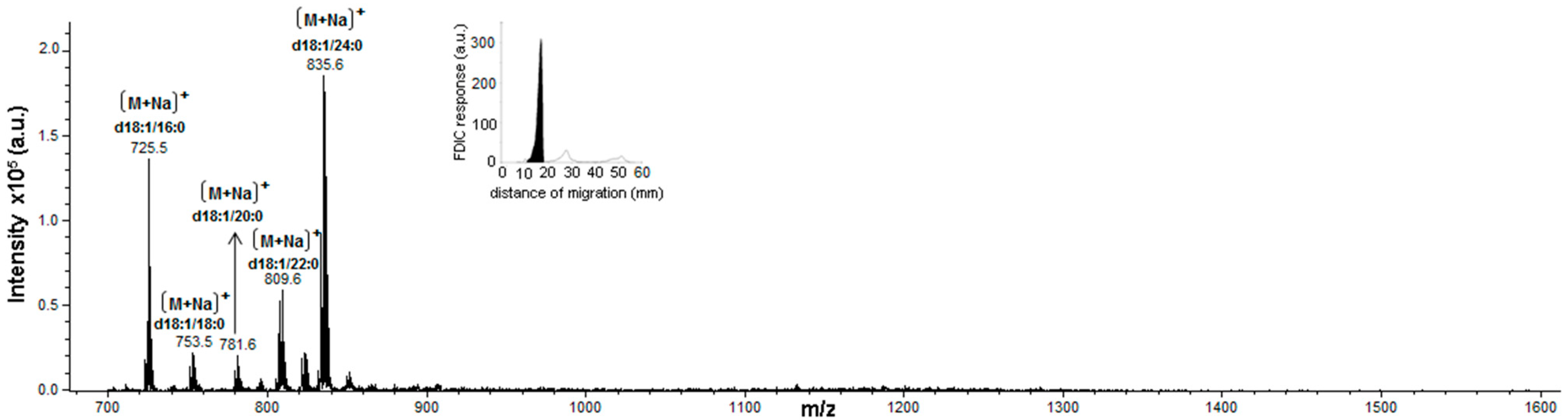
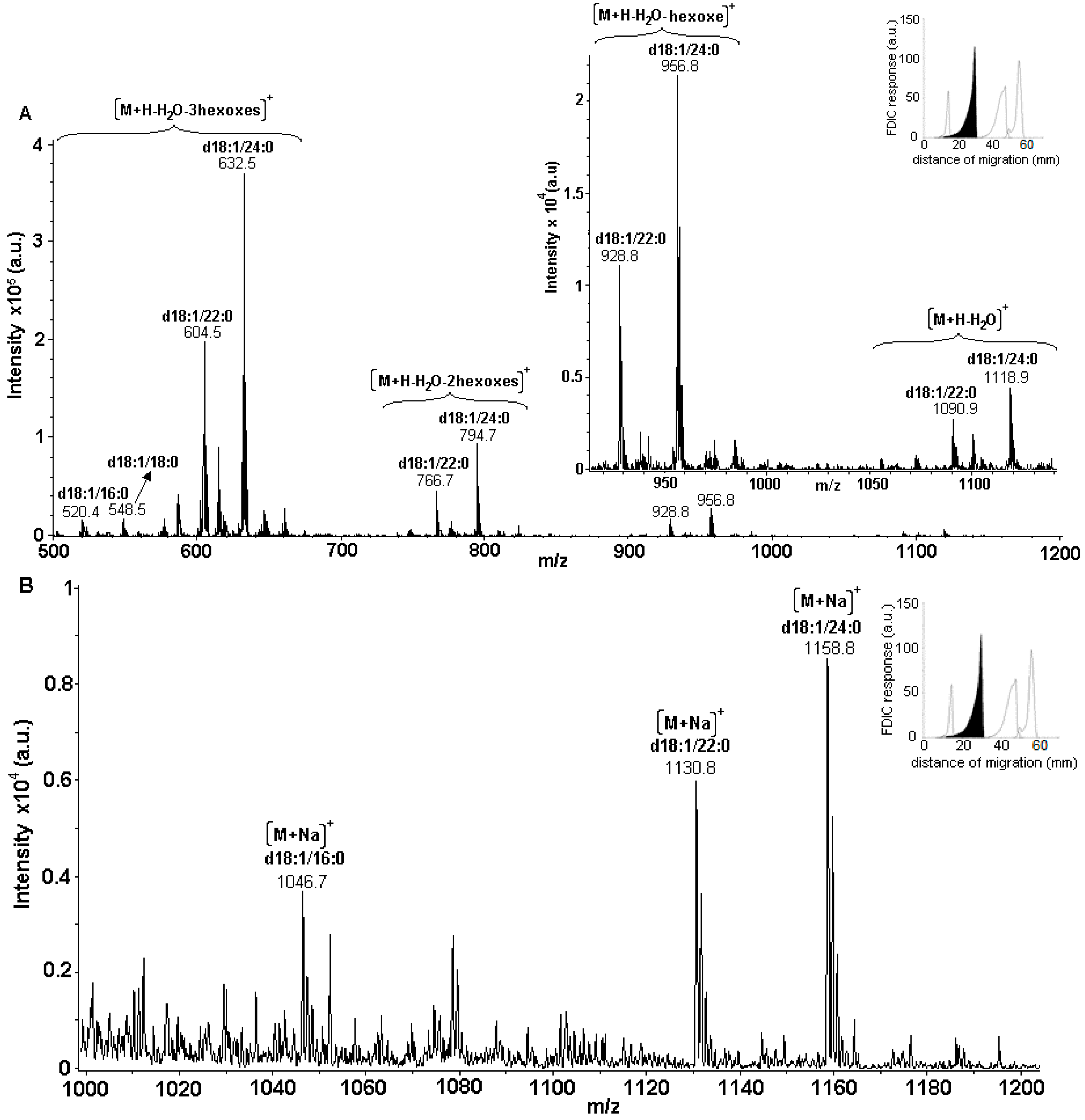
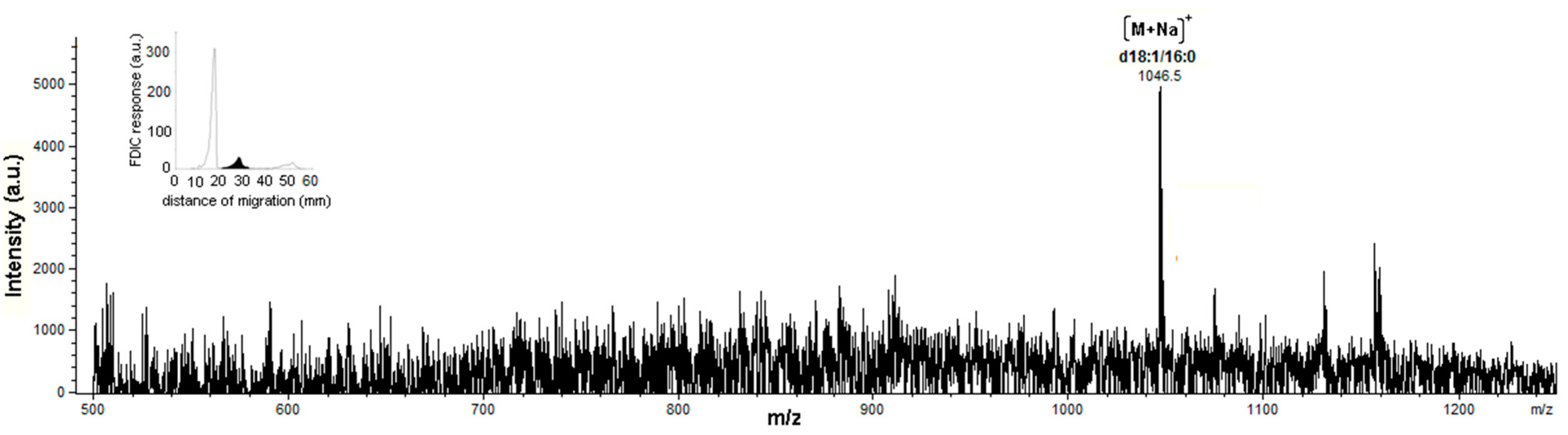
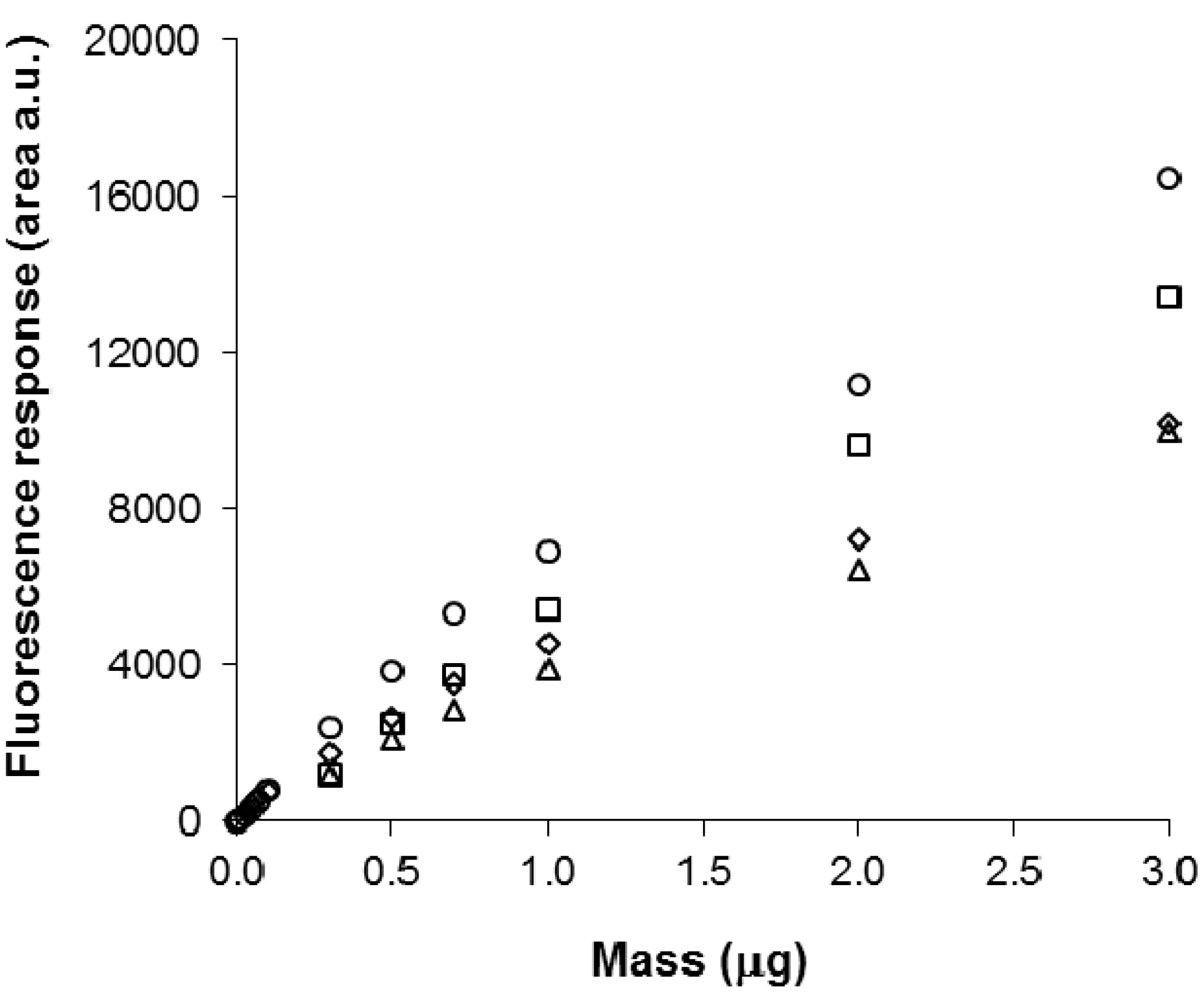
3.6. Quantitative Analysis of SLs by FDIC
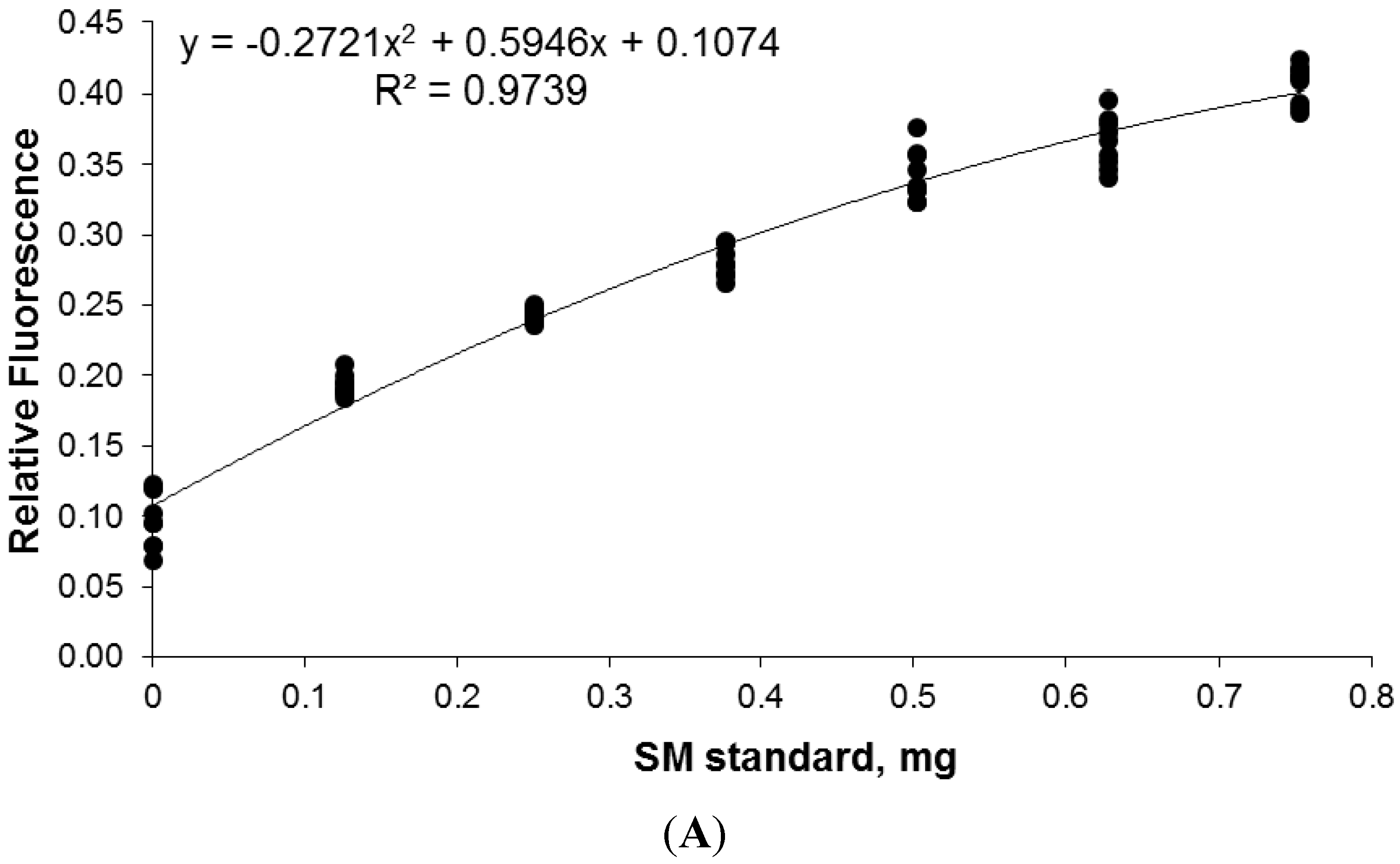

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Aerts, J.M.; Kallemeijn, W.W.; Wegdam, W.; Joao Ferraz, M.; van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.M.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids and inhibodies. J. Inherit. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Altarescu, G.; Beck, M. Fabry Disease; Springer: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Kooperman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Nat. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Manwaring, V.; Boutin, M.; Auray-Blais, C. A metabolomic study to identify new globotriaosylceramide related biomarkers in the plasma of Fabry disease patients. Anal. Chem. 2013, 85, 9039–9048. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Auray-Blais, C. Multiplex tandem mass spectrometry analysis of novel plasma lyso-Gb3-related analogues in Fabry disease. Anal. Chem. 2014, 86, 3476–3483. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine Is a Highly Sensitive and Specific Biomarker for Primary Diagnostic and Follow-Up Monitoring in Gaucher Disease in a Non-Jewish, Caucasian Cohort of Gaucher Disease Patients. PLoS One 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, R.; Forni, S.; Swift, C.; Brignol, N.; Wu, X.; Lockhart, D.J.; Blankenship, D.; Wang, X.; Grayburn, P.A.; Taylor, M.R.G.; et al. Risk of Death in Heart Disease is Associated With Elevated Urinary Globotriaosylceramide. J. Am. Heart Assoc. 2014, 3, e000394. [Google Scholar] [CrossRef] [PubMed]
- Gold, H.; Mirzaian, M.; Dekker, N.; Ferraz, M.J.; Lugtenburg, J.; Codée, J.D.C.; van der Marel, G.A.; Overkleeft, H.S.; Linhorst, G.; Groener, J.E.M.; et al. Quantification of globotriaosylsphingosine in plasma and urine of Fabry patients by stable isotope ultraperformance liquid chromatography-tandem mass spectrometry. Clin. Chem. 2013, 59, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Morlock, G.; Schwack, W. Hyphenations in planar chromatography. J. Chromatogr. A 2010, 1217, 6600–6609. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.; Süβ, R.; Teuber, K.; Eibish, M.; Schiller, J. Lipid analysis by thin-layer chromatography- A review of the current state. J. Chromatogr. A 2011, 1218, 2754–2774. [Google Scholar] [CrossRef] [PubMed]
- Reich, E.; Schibli, A. High-Performance Thin-Layer Chromatography for the Analysis of Medicinal Plants; Thieme: New York, NY, USA, 2007. [Google Scholar]
- Cebolla, V.L.; Jarne, C.; Domingo, M.P.; Domínguez, A.; Delgado-Camón, A.; Garriga, R.; Galbán, J.; Membrado, L.; Gálvez, E.M.; Cossío, F.P. Fluorescence detection by intensity changes for high-performance thin-layer chromatography separation of lipids using automated multiple development. J. Chromatogr. A 2011, 1218, 2668–2675. [Google Scholar] [CrossRef] [PubMed]
- Müthing, J.; Distler, U. Advances on the compositional analysis of glycosphingolipids combining thin-layer chromatography with mass spectrometr. Mass Spectrom. Rev. 2010, 29, 425–479. [Google Scholar] [PubMed]
- Ochalek, M.; Heissler, S.; Wohlrab, J.; Neubert, R.H.H. Characterization of model lipid membranes designed for studying impact of ceramide species on drug diffusion and penetration. Eur. J. Pharm. Biopharm. 2012, 81, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Kabrodt, K.; Lüttich, J.; Dittler, I.; Schellenberg, I. Improved HPTLC separation of lipids using Automated Multiple Development (AMD) and identification with the TLC-MS-Interface. In Proceedings of the HPTLC International Symposium 2011, Basel, Switzerland, July 2011; p. 63.
- Opitz, A.; Wirtz, M.; Melchior, D.; Mehling, A.; Kling, H.-W.; Neubert, R.R.H. Improved method for stratum corneum lipid analysis by automated multiple development HPTLC. Chromatographia 2011, 73, 559–565. [Google Scholar] [CrossRef]
- Farwanah, H.; Raith, K.; Neubert, R.H.H.; Wohlrab, J. Ceramide profiles of uninvolved skin in atopic dermatitis and psoriasis are comparable to those of healthy skin. Arch. Dermatol. Res. 2005, 296, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Raith, K.; Zellmer, S.; Lasch, J.; Neubert, R.H.H. Profiling of human stratum corneum ceramides by liquid chromatographyelectrospray mass spectrometry. Anal. Chim. Acta 2000, 418, 167–173. [Google Scholar] [CrossRef]
- Zellmer, S.; Lasch, J. Individual variation of human plantar stratum corneum lipids, determined by automated multiple development of high-performance thin-layer chromatography plates. J. Chromatogr. B 1997, 691, 321–329. [Google Scholar] [CrossRef]
- Farwanah, H.; Neubert, R.; Zellmer, S.; Raith, K. Improved procedure for the separation of major stratum corneum lipids by means of automated multiple development thin-layer chromatography. J. Chromatogr. B 2002, 780, 443–450. [Google Scholar] [CrossRef]
- Gildenast, T.; Lasch, J. Isolation of ceramide fractions from human stratum corneum lipid extracts by high-performance liquid chromatography. Biochim. Biophys. Acta 1997, 1346, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Bonté, F.; Saunois, A.; Pinguet, P.; Meybeck, A. Existence of a lipid gradient in the upper stratum corneum and its possible biological significance. Arch. Dermatol. Res. 1997, 289, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Müthing, J. Improved thin-layer chromatographic separation of gangliosides by automated multiple development. J. Chromatogr. B 1994, 657, 75–81. [Google Scholar] [CrossRef]
- Müthing, J.; Ziehr, H. Enhanced thin-layer chromatographic separation of GM1b-type gangliosides by automated multiple development. J. Chromatogr. B 1996, 687, 357–362. [Google Scholar] [CrossRef]
- Suzuki, A.; Miyazaki, M.; Matsuda, J.; Yoneshige, A. High-performance thin-layer chromatography/mass spectrometry for the analysis of neutral glycosphingolipids. Biochim. Biophys. Acta 2011, 1811, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, H.; Jonas, U.; Ohashi, M.; Klaiber, I.; Rahman, H. Direct electrospray-ionization mass spectrometric analysis of the major ganglioside from crucian carp liver after thin-layer chromatography. Comp. Biochem. Phys. B 1999, 122, 83–88. [Google Scholar] [CrossRef]
- Dreisewerd, K.; Müthing, J.; Rohlfing, A.; Meisen, I.; Vukelić, Z.; Peter-Katalinic, J.; Hillenkamp, F.; Berkenkamp, S. Analysis of gangliosides directly from thin-layer chromatography plates by infrared matrix-assisted laser desorption/ionization orthogonal time-of-flight mass spectrometry with a glycerol matrix. Anal. Chem. 2005, 77, 4098–4107. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Suzuki, Y.; Goto-Inoue, N.; Yoshida-Noro, C.; Suzuki, A. Structural characterization of neutral glycosphingolipids by thin-layer chromatography coupled to matrix-assisted laser desorption/ionization quadrupole ion trap time-of-flight MS/MS. Anal. Chem. 2006, 78, 5736–5743. [Google Scholar] [CrossRef] [PubMed]
- Meisen, I.; Peter-Katalinic, J.; Müthing, J. Direct analysis of silica gel extracts from inmunostained glycosphingolipids by nanoelectrospray ionization quadrupole time-of-flight mass spectrometry. Anal. Chem. 2004, 76, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Cansell, M.; Gouygou, J.P.; Jozefonvicz, J.; Letourneur, D. Lipid composition of cultured endothelial cells in relation to their growth. Lipids 1997, 32, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Cossío, F.P.; Arrieta, A.; Cebolla, V.L.; Membrado, L.; Garriga, R.; Vela, J.; Domingo, M.P. Berberinecation: A fluorecentchemosensor for alkanes and other low-polarity compounds. A theoretical explanation of this phenomenon. Org. Lett. 2000, 2, 2311–2313. [Google Scholar] [CrossRef] [PubMed]
- Cebolla, V.L.; Mateos, E.; Garriga, R.; Membrado, L.; Cossío, F.P.; Gálvez, E.M.; Matt, M.; Delgado-Camón, A. Changes in fluorescent emission induced by non-covalent interactions as a general detection procedure in thin-layer chromatography. ChemPhysChem 2012, 13, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Cossío, F.P.; Arrieta, A.; Cebolla, V.L.; Membrado, L.; Domingo, M.P.; Henrion, P.; Vela, J. Enhancement of fluorescence in thin-layer chromatography induced by the interaction between n-alkanes and an organic cation. Anal. Chem. 2000, 72, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, E.M.; Matt, M.; Cebolla, V.L.; Fernandes, F.; Membrado, L.; Cossío, F.P.; Garriga, R.; Vela, J.; Guermouche, M.H. General contribution of nonspecific interactions to fluorescence intensity enhancements. Anal. Chem. 2006, 78, 3699–3705. [Google Scholar] [CrossRef] [PubMed]
- Mateos, E.M.; Cebolla, V.L.; Membrado, L.; Vela, J.; Gálvez, E.M.; Matt, M.; Cossío, F.P. Coralynecation, a fluorescent probe for general detection in planar chromatography. J. Chromatogr. A 2007, 1146, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Cebolla, V.L.; Membrado, L.; Domingo, M.P.; Henrion, P.; Garriga, R.; González, P.; Cossío, F.P.; Arrieta, A.; Vela, J. Quantitative applications of fluorescence and ultraviolet scanning densitometry for compositional analysis of petroleum products in thin-layer chromatography. J. Chromatogr. Sci. 1999, 37, 219–226. [Google Scholar] [CrossRef]
- Luftmann, H. A simple device for the extraction of TLC spots: Direct coupling with an electrospray mass spectrometer. Anal. Bioanal. Chem. 2004, 378, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Luftmann, H.; Aranda, M.; Morlock, G.E. Automated interface for hyphenation of planar chromatography with mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 3772–3776. [Google Scholar] [CrossRef] [PubMed]
- Aranda, M.; Morlock, G. New method for caffeine quantification by planar chromatography coupled with electrospray ionization mass spectrometry using stable isotope dilution analysis. Rapid Commun. Mass Spectrom. 2007, 21, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Morlock, G.; Schwack, W. Coupling of planar chromatography to mass spectrometry. TrAC 2010, 29, 1157–1171. [Google Scholar]
- Farwanah, H.; Wirtz, J.; Kolter, T.; Raith, K.; Neubert, R.H.H.; Sandhoff, K. Normal phase liquid chromatography coupled to quadrupole time of flight atmospheric pressure chemical ionization mass spectrometry for separation, detection and mass spectrometric profiling of neutral sphingolipids and cholesterol. J. Chromatogr. B 2009, 877, 2976–2982. [Google Scholar] [CrossRef]
- Li, K.; Frankowski, K.J.; Belon, C.A.; Neuenswander, B.; Ndjomou, J.; Hanson, A.M.; Shanahan, M.A.; Schoenen, F.J.; Blagg, B.S.J.; Aubé, J.; et al. Optimization of potent hepatitis C virus NS3 helicase inhibitors isolated from the yellow dyes thioflavine S and primuline. J. Med. Chem. 2012, 55, 3319–3330. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomic reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Koscielniak, P. Nonlinear calibration by the standard addition method. Chemometr. Intell. Lab. 1999, 47, 275–287. [Google Scholar] [CrossRef]
- He, X.; Chen, F.; Gatt, S.; Schuchman, E.H. An enzymatic assay for quantifying spingomyelin in tissues and plasma from humans and mice with Niemann-Pick disease. Anal. Biochem. 2001, 293, 204–211. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Chen, F.; McGovern, M.M.; Schuchman, E.H. A fluorescence-based high-throughput sphingomyelin assay for the analysis of Niemann-Pick disease and other disorders of sphingomyelin metabolism. Anal. Biochem. 2002, 306, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, H.; Yamauchi, K.; Ohta, H.; Akamatsu, T.; Honda, T.; Katsuyama, T. Specific, rapid and sensitive enzymatic measurement of sphingomyelin, phosphatidylcholine and lysophosphatidylcholine in serum and lipid extracts. Clin. Biochem. 2008, 41, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez, A.; Jarne, C.; Cebolla, V.L.; Galbán, J.; Savirón, M.; Orduna, J.; Membrado, L.; Lapieza, M.-P.; Romero, E.; Sanz Vicente, I.; et al. A Hyphenated Technique based on High-Performance Thin Layer Chromatography for Determining Neutral Sphingolipids: A Proof of Concept. Chromatography 2015, 2, 167-187. https://doi.org/10.3390/chromatography2020167
Domínguez A, Jarne C, Cebolla VL, Galbán J, Savirón M, Orduna J, Membrado L, Lapieza M-P, Romero E, Sanz Vicente I, et al. A Hyphenated Technique based on High-Performance Thin Layer Chromatography for Determining Neutral Sphingolipids: A Proof of Concept. Chromatography. 2015; 2(2):167-187. https://doi.org/10.3390/chromatography2020167
Chicago/Turabian StyleDomínguez, Andrés, Carmen Jarne, Vicente L. Cebolla, Javier Galbán, María Savirón, Jesús Orduna, Luis Membrado, María-Pilar Lapieza, Elena Romero, Isabel Sanz Vicente, and et al. 2015. "A Hyphenated Technique based on High-Performance Thin Layer Chromatography for Determining Neutral Sphingolipids: A Proof of Concept" Chromatography 2, no. 2: 167-187. https://doi.org/10.3390/chromatography2020167