
Surface Characterization of Some Novel Bonded Phase Packing Materials for HPLC Columns Using MAS-NMR Spectroscopy

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Sample Preparation
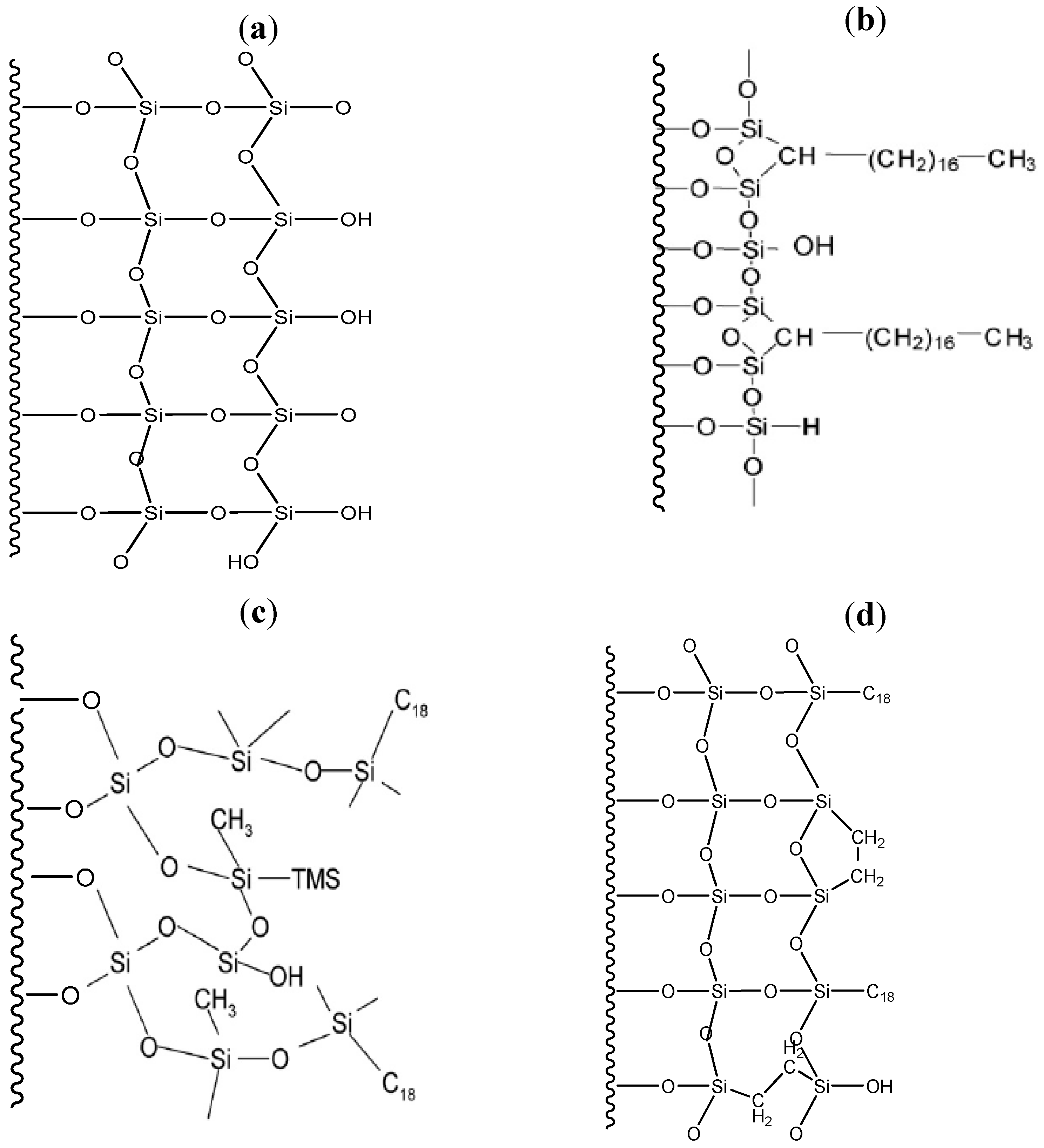

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Cogent bidentate C18 | XTerra | XTerra MS C18 | XBridge | XBridge Prep. C18 |
|---|---|---|---|---|---|
| Particle size (µm) | 4 | 5 | 5 | 5 | 5 |
| Pore size (Ǻ) | 100 | 120 | 120 | 135 | 135 |
| Pore volume (mL/g) | 0.92 | 0.64 | 0.64 | 0.70 | 0.70 |
| Surface area (m2/g) | 350 | 176 | 176 | 185 | 185 |
| Carbon load (%) | 16.0 | n/a | 15.5 | n/a | 18.0 |
| Ligand density (µmol/m2) | n/a | n/a | 2.40 | n/a | 3.10 |
| Endcapped | No | n/a | Yes | n/a | Yes |
2.2. Instruments and Experiments
3. Results and Discussion
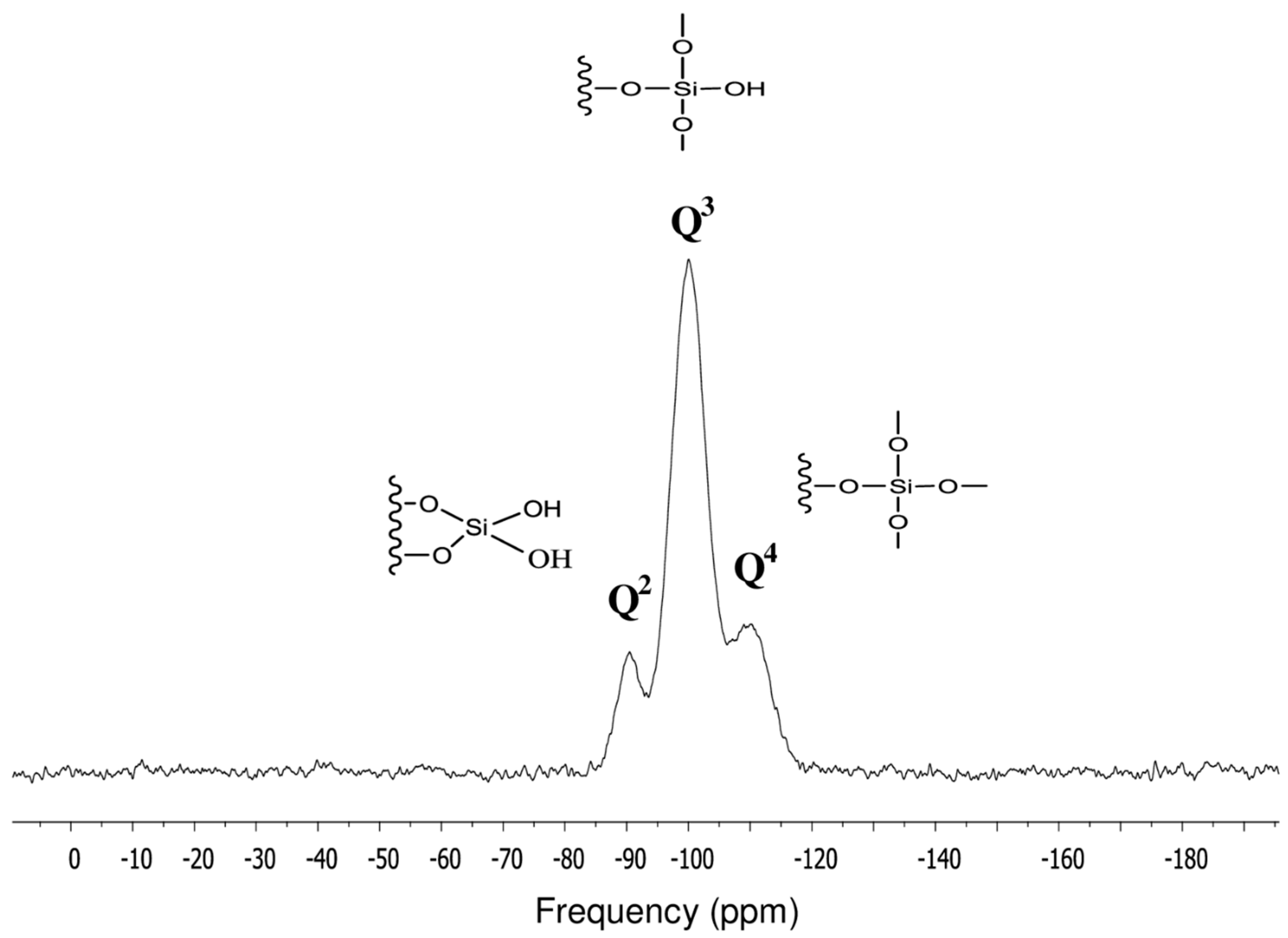
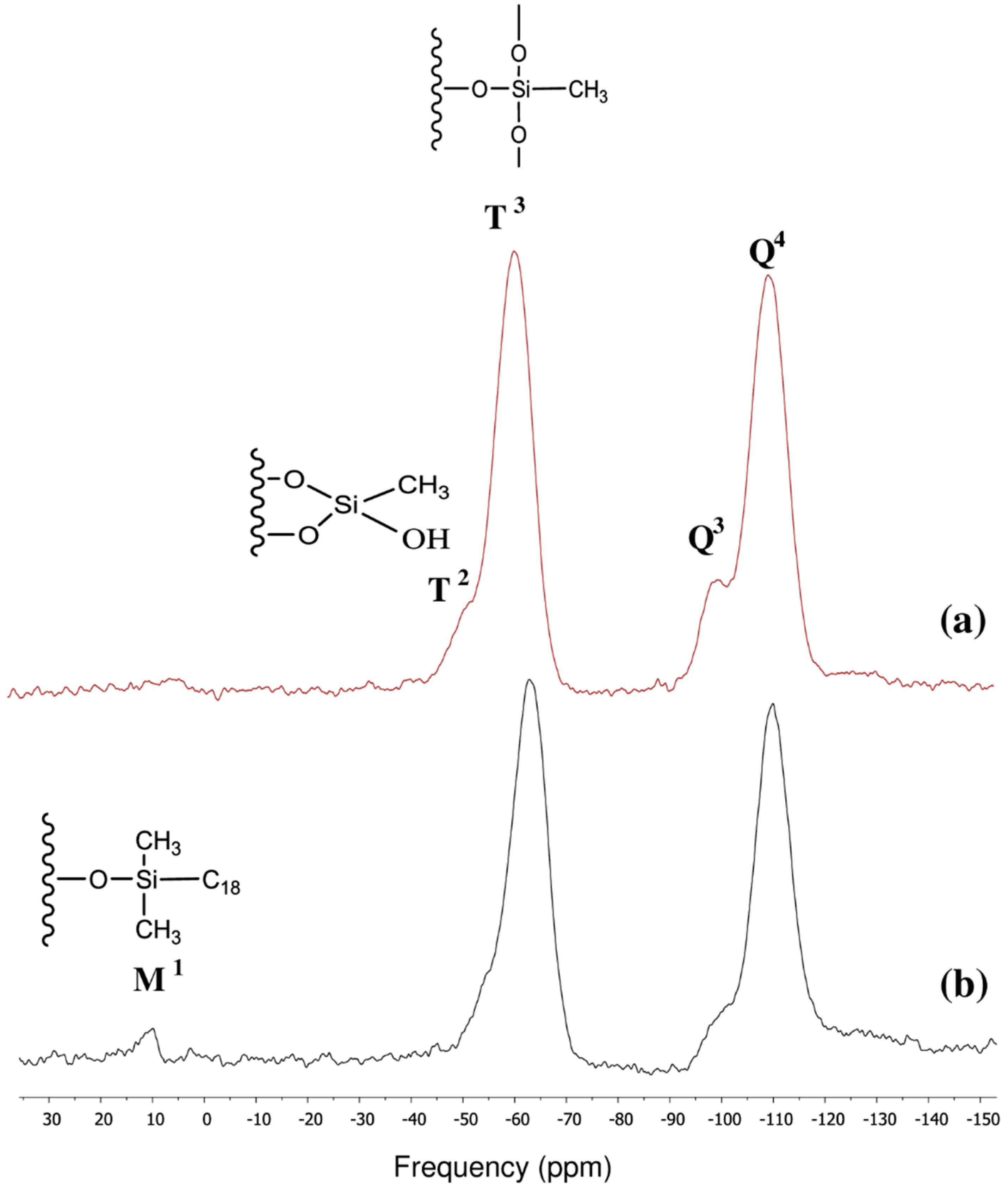
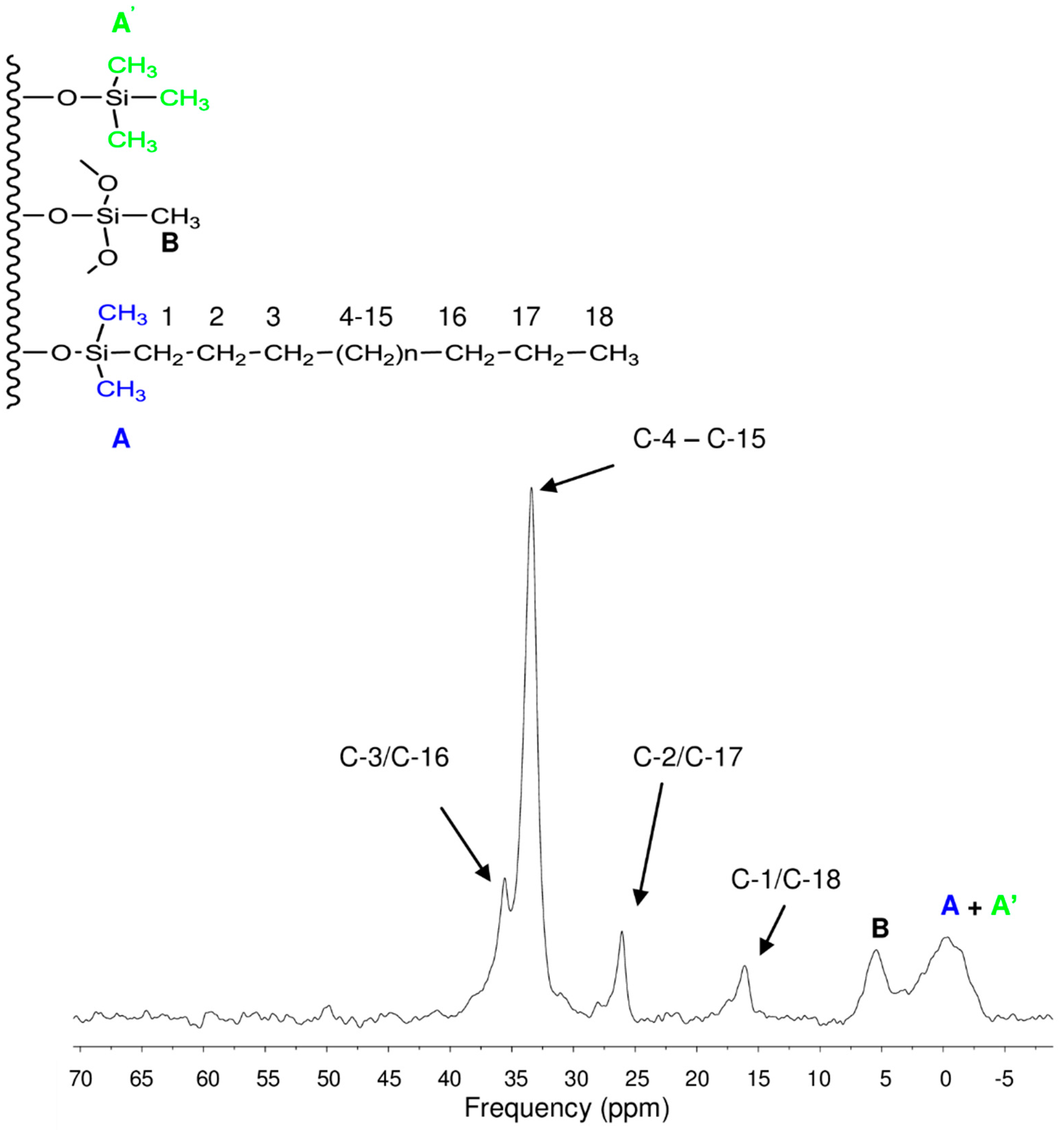
3.1. Surface Characterization of the Cogent Bidentate C18 on Type-C Silica

| Chemical moiety | Percentage (% by peak area) | Chemical shift (ppm) |
|---|---|---|
| Q2 | 12.95 | −90.80 |
| Q3 | 71.94 | −100.04 |
| Q4 | 15.11 | −110.03 |
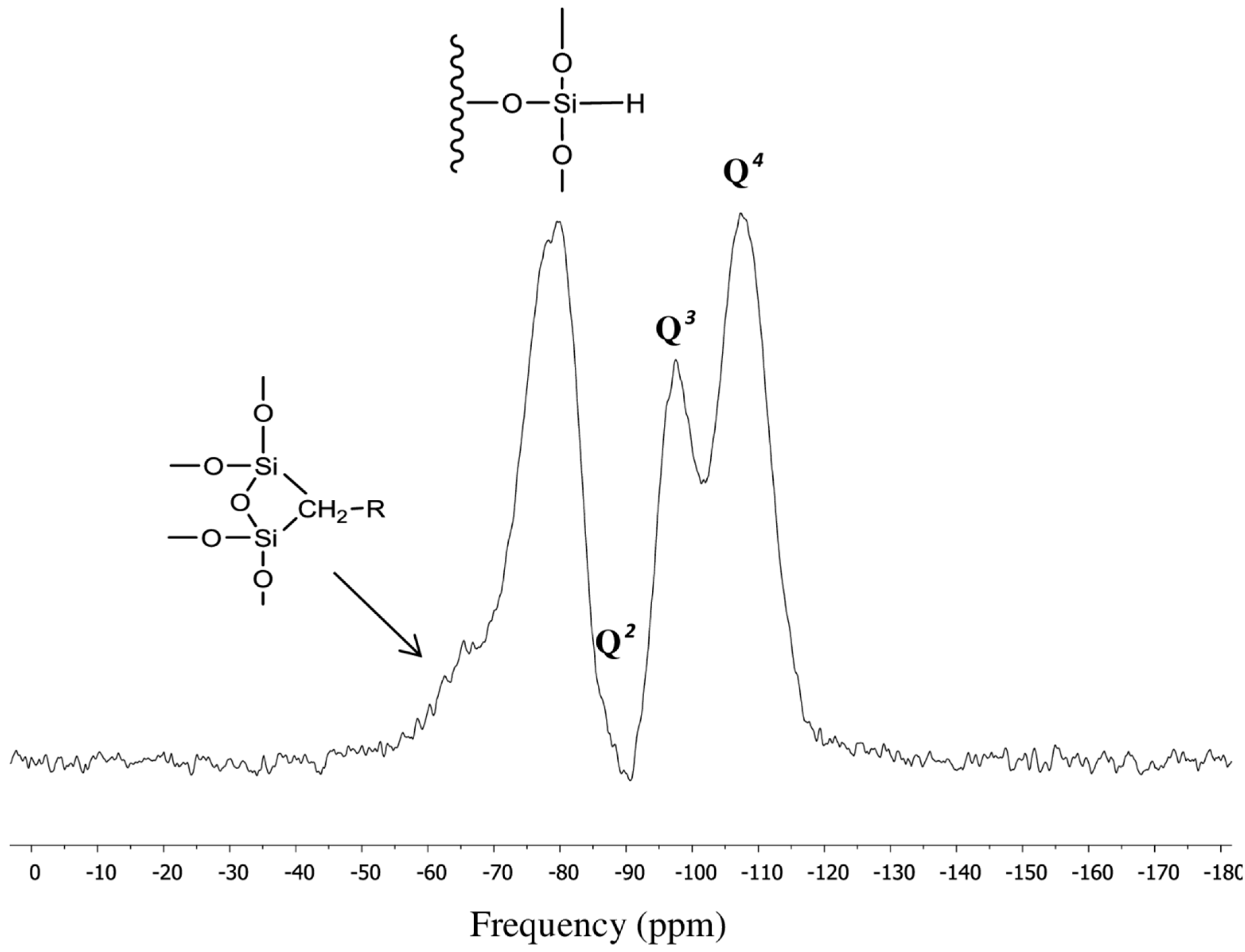
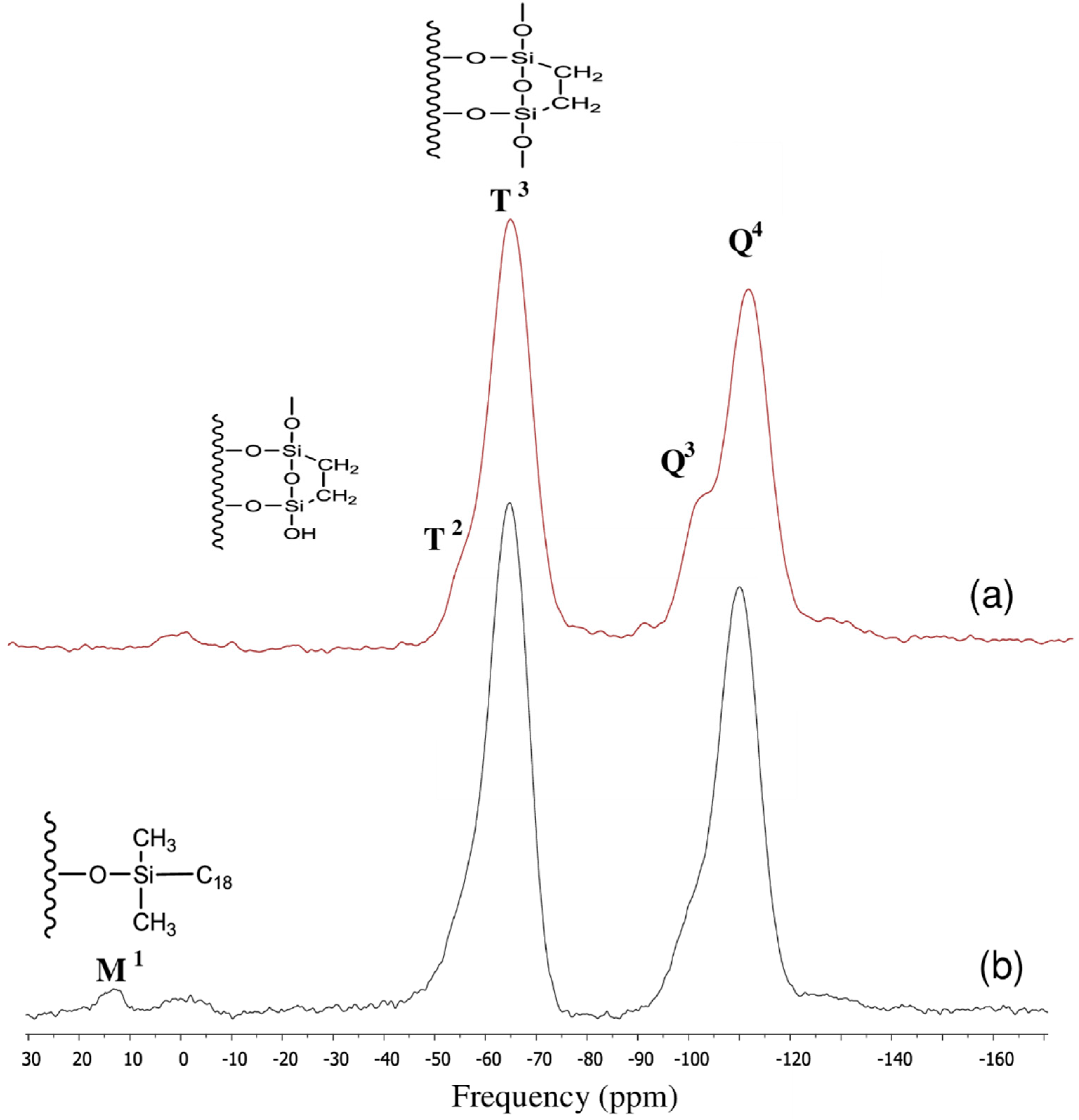
| Chemical moiety | Percentage (% by peak area) | Chemical shift (ppm) |
|---|---|---|
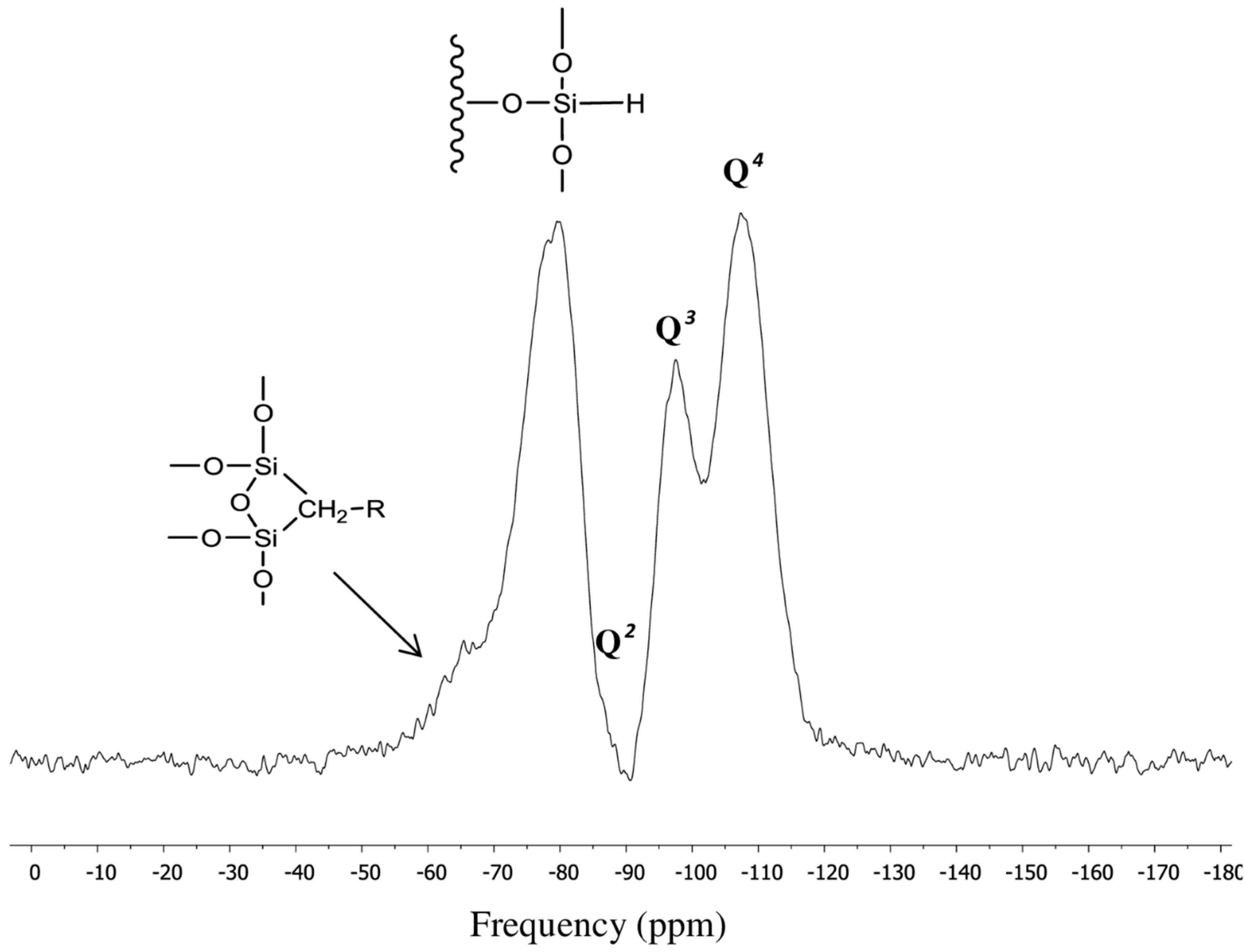
| Q2 | 2.84 | −89.90 |
| Q3 | 10.90 | −99.55 |
| Q4 | 36.99 | −110.00 |
 | 2.80 | −65.47 |
 | 46.47 | −80.72 |
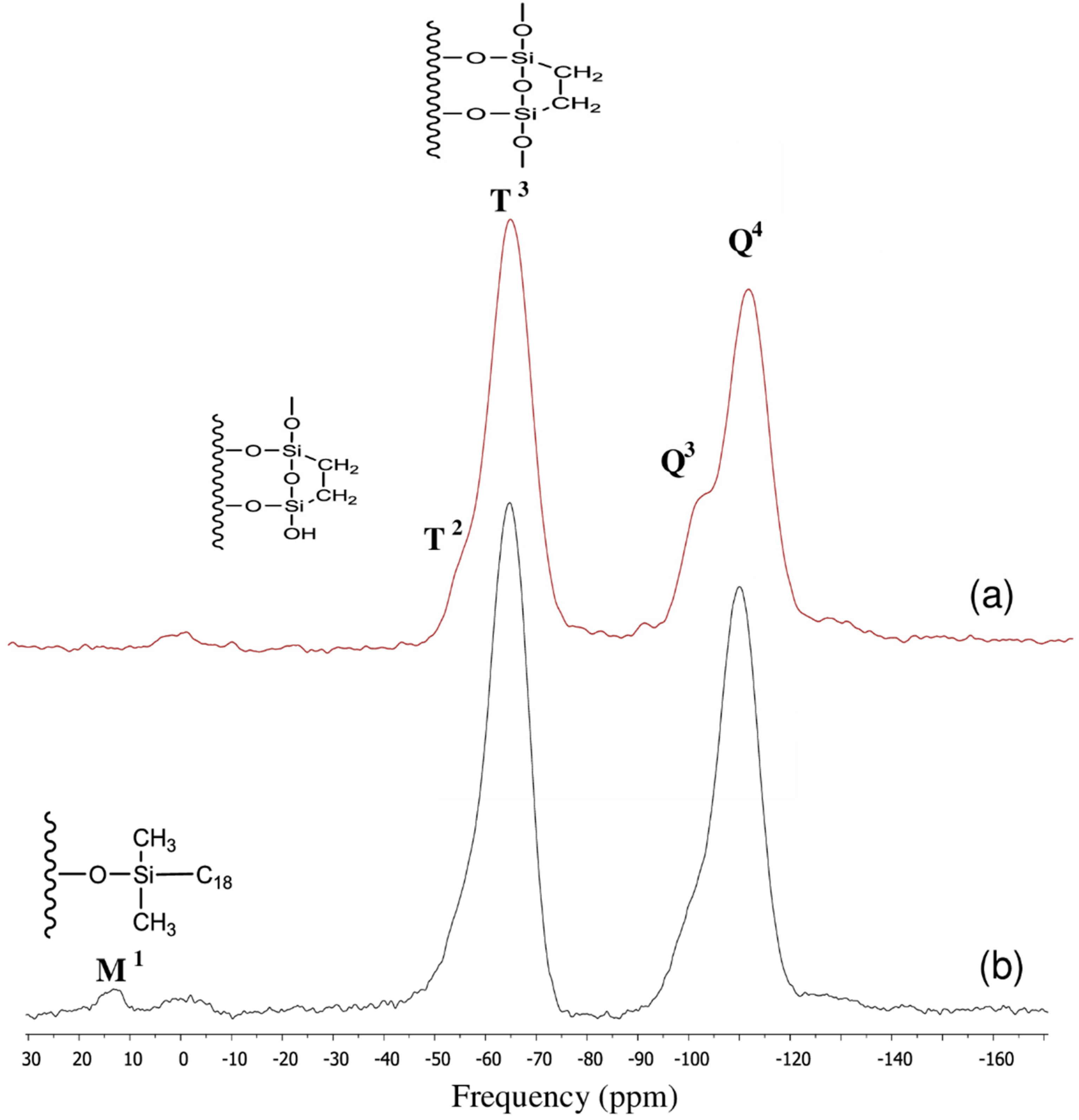
3.2. Surface Characterization of the XTerra Packing Material
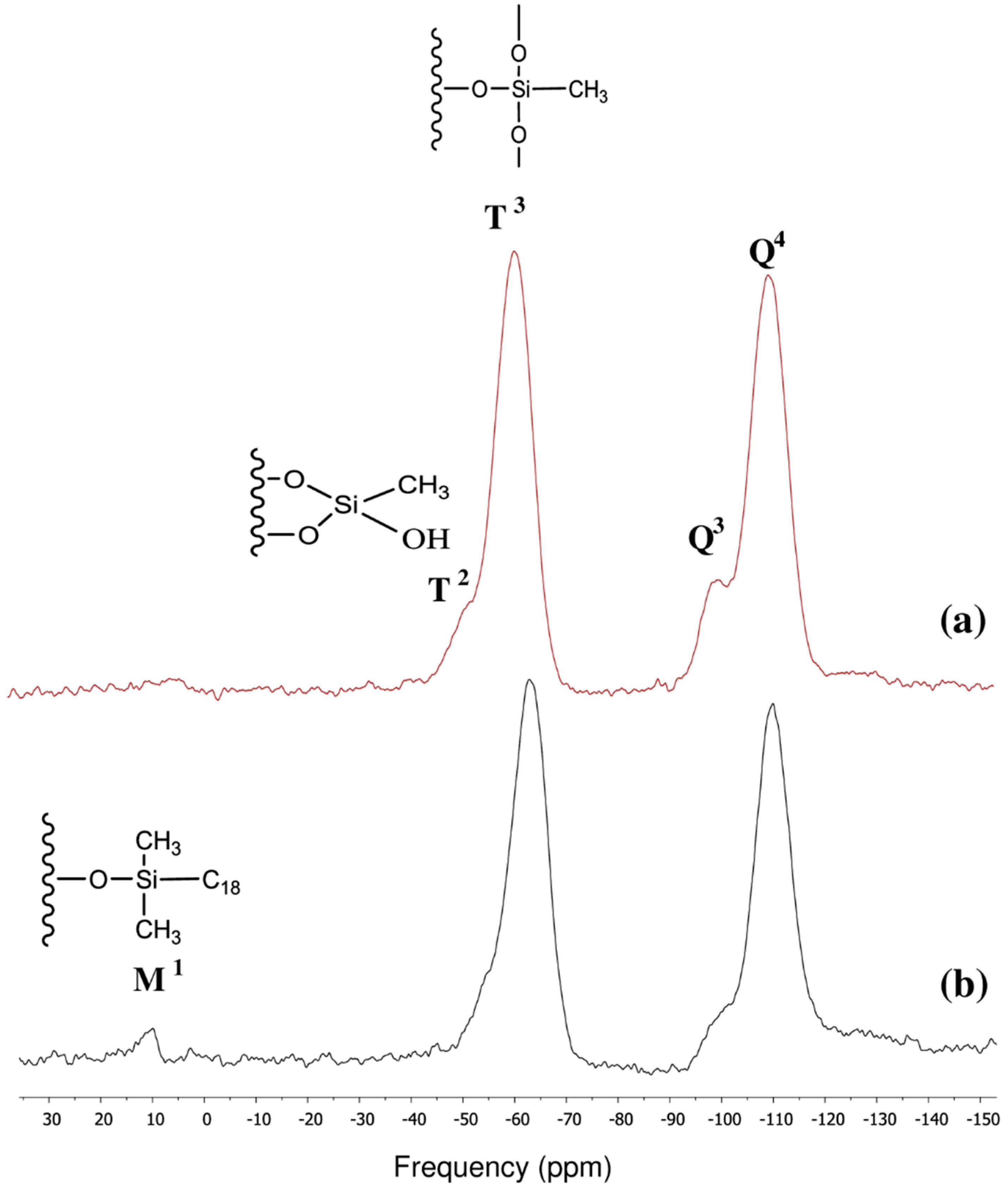
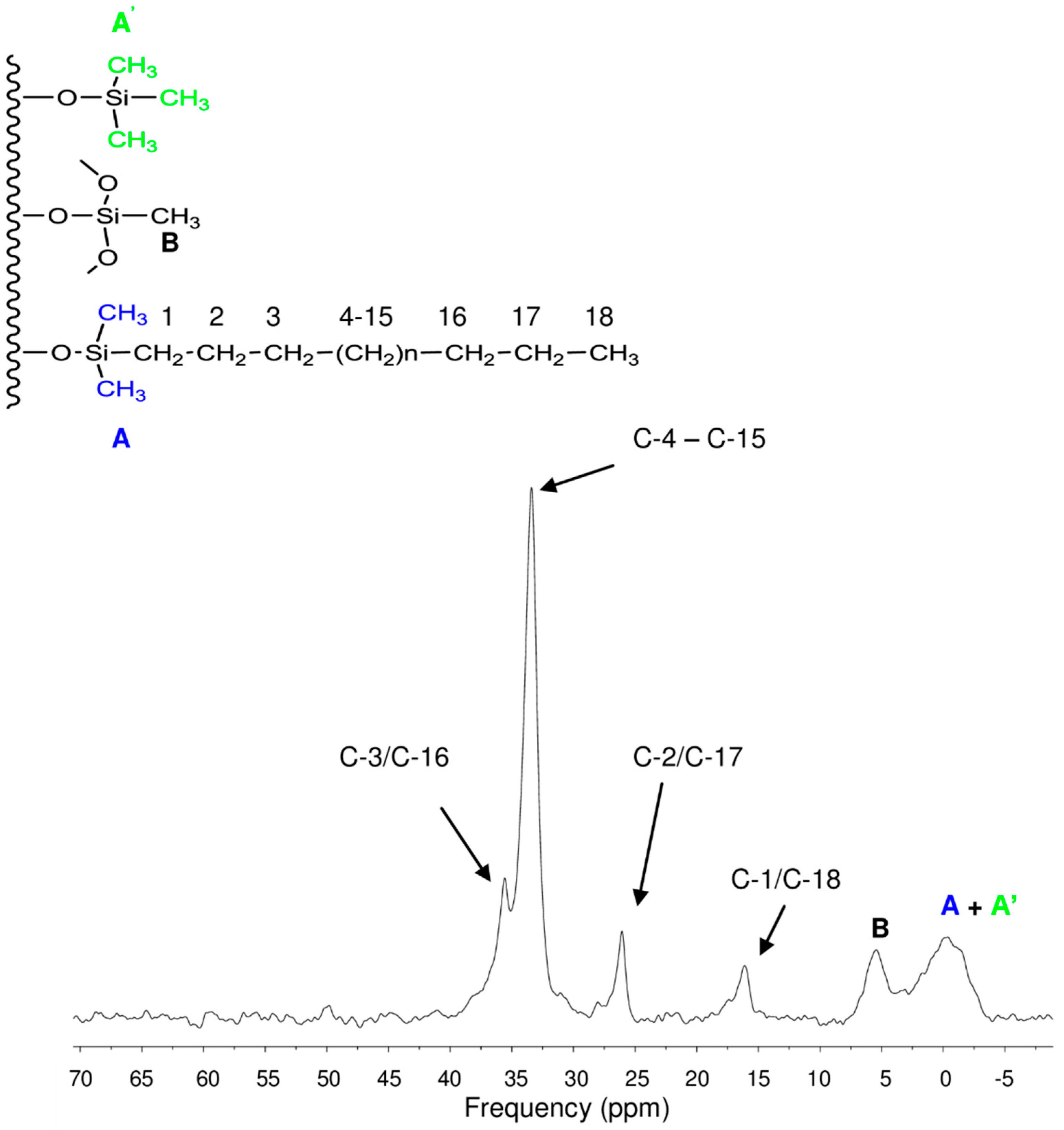
3.3. Surface Characterization of the XBridge Packing Material
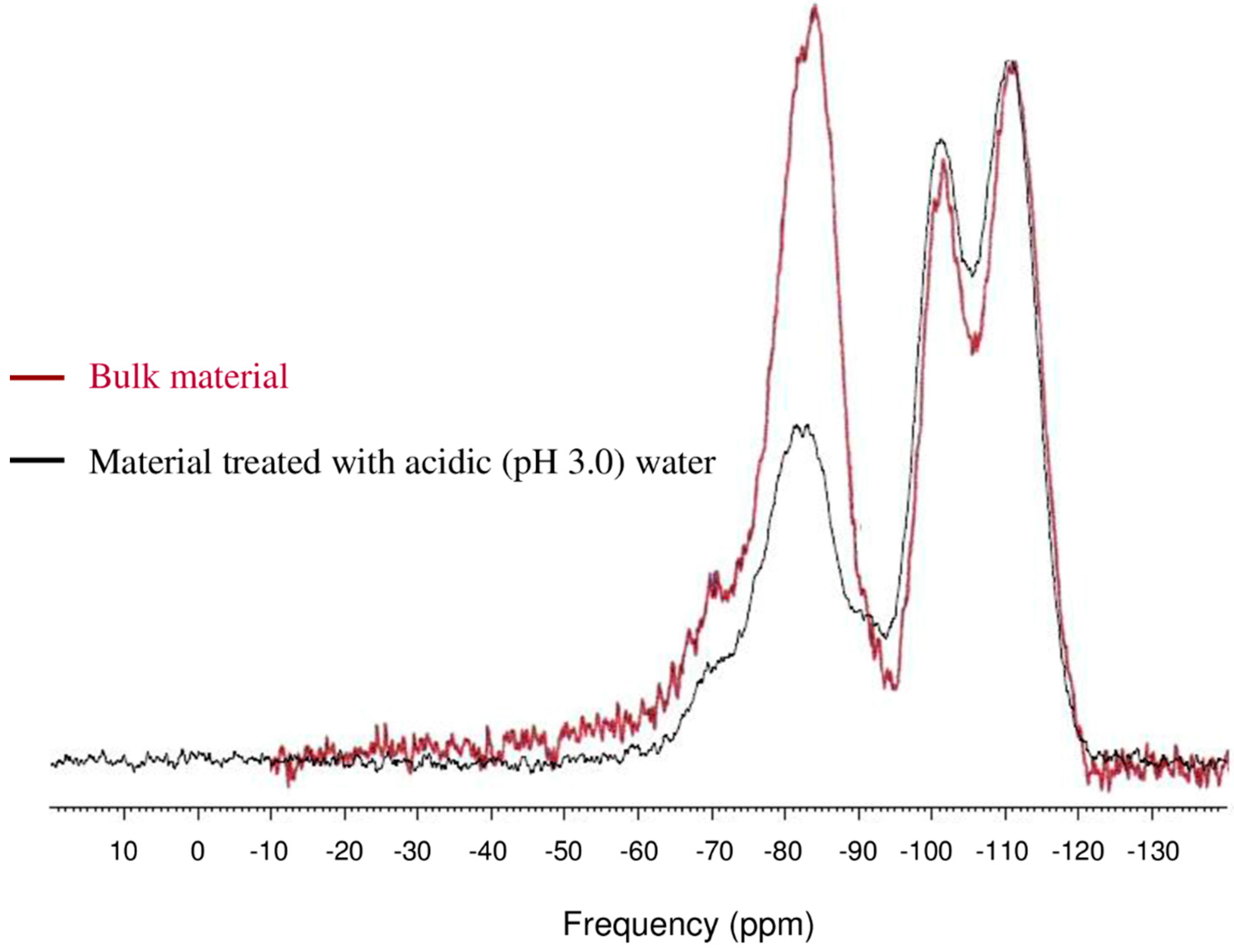
3.4. Hydrolytic Stability of Packing Materials in Aqueous Conditions
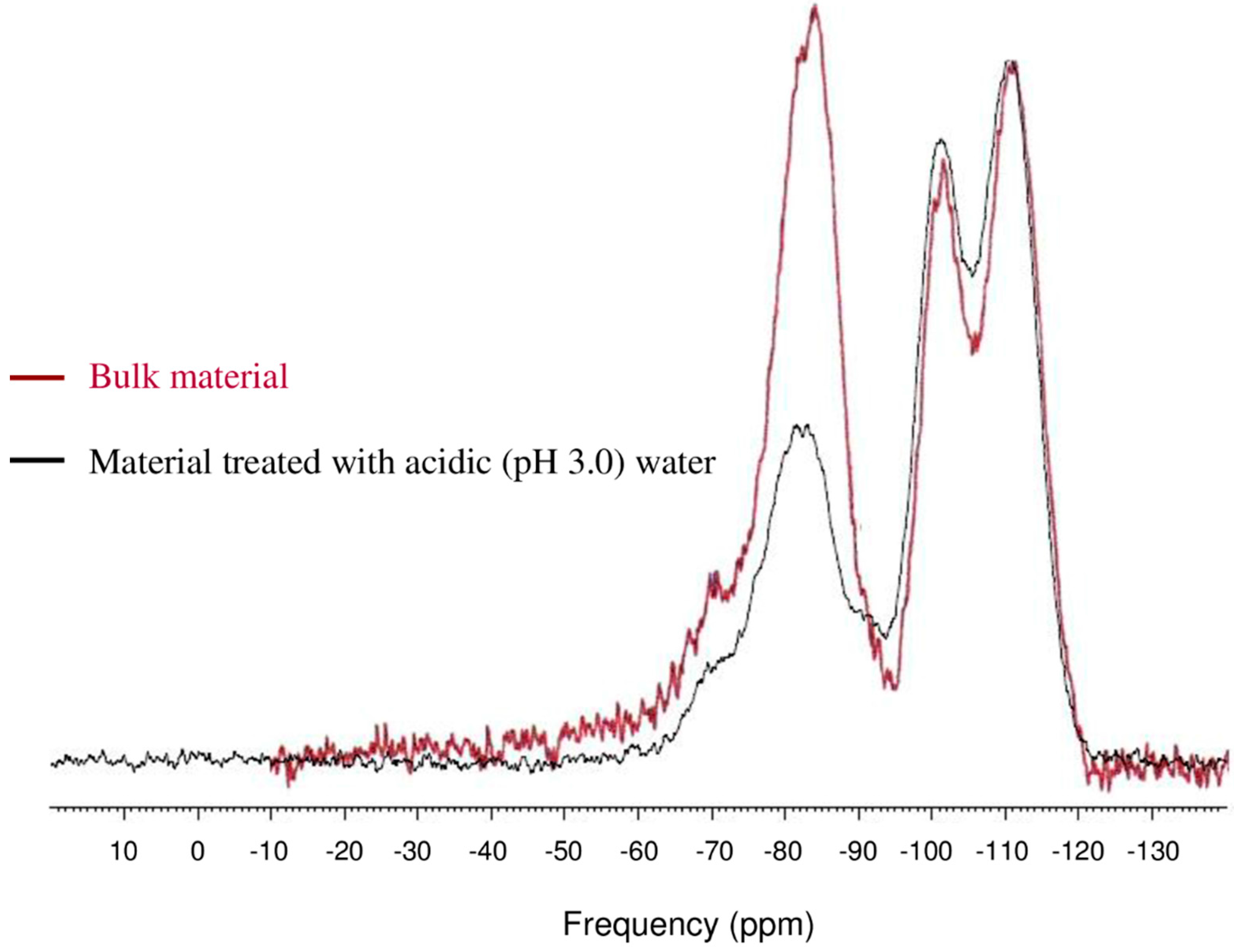
| Chemical moiety | Percentage (% by peak area) | ||
|---|---|---|---|
| Untreated bulk material | Water (pH 7.0) treatment | Water (pH 3.0) treatment | |
| Q2 | 2.84 | 2.84 | 4.88 |
| Q3 | 10.90 | 12.90 | 16.59 |
| Q4 | 36.99 | 37.70 | 40.98 |
| | 2.80 | 2.79 | 3.89 |
| | 46.47 | 43.77 | 33.66 |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Poole, C.F.; Poole, S.K. Chromatography Today; Elsevier: Amsterdam, The Netherlands, 1991; pp. 649–743. [Google Scholar]
- Pietrzyk, D.J.; Brown, P.R.; Hartwick, R.A. High Performance Liquid Chromatography; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Snyder, L.R.; Kirkland, J.J. Introduction to Modern Liquid Chromatography, 2nd ed.; Wiley: New York, NY, USA, 1977. [Google Scholar]
- Sander, L.C.; Wise, S.A. Recent advances in bonded phases for liquid chromatography. Crit. Rev. Anal. Chem. 1987, 18, 299–415. [Google Scholar] [CrossRef]
- Nawrocki, J.; Buszewski, B. Influence of silica surface chemistry and structure on the properties, structure and coverage of alkyl-bonded phases for high-performance liquid chromatography. J. Chromatogr. A 1988, 449, 1–24. [Google Scholar] [CrossRef]
- Gerber, F.; Krummen, M.; Potgeter, H.; Roth, A.; Siffrin, C.; Spoendlin, C. Practical aspects of fast reversed-phase high-performance liquid chromatography using 3μm particle packed columns and monolithic columns in pharmaceutical development and production working under current good manufacturing practice. J. Chromatogr. A 2014, 2, 127–133. [Google Scholar]
- Popp, M.; Sulyok, M.; Rosenberg, E. Chromatographic characterisation of a novel type of monolithic methylsilsesquioxane-based HPLC column. J. Sep. Sci. 2007, 30, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Buszewski, B.; Jezierska, M.; Wełniak, M.; Berek, D. Survey and trends in the preparation of chemically bonded silica phases for liquid chromatographic analysis. J. High Res. Chromatogr. 1998, 21, 267–281. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T. Hydride-Based Separation Materials for High Performance Liquid Chromatography and Open Tubular Capillary Electrochromatography. Chin. J. Chromatogr. 2005, 23, 595–608. [Google Scholar] [CrossRef]
- Bocian, S.; Buszewski, B. Residual silanols at reversed-phase silica in HPLC—A contribution for a better understanding. J. Sep. Sci. 2012, 35, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Unger, K.K.; Becker, N.; Roumeliotis, P. Recent developments in the evaluation of chemically bonded silica packings for liquid chromatography. J. Chromatogr. A 1976, 125, 115–127. [Google Scholar] [CrossRef]
- Khalid, A.M.; Thakur, M.M; Thompson, W.L.; Cao, C.; Schultz, W.J. Proton MAS NMR Analysis of Phenyl Silane Functionalized Silica Nanoparticles. NMR Spectrosc. Polym. 2011, 30, 495–508. [Google Scholar]
- Prado, A.G.; Moura, A.O.; Nunes, A.R. Nanosized Silica Modified with Carboxylic Acid as Support for Controlled Release of Herbicides. J. Agric. Food Chem. 2011, 59, 8847–8852. [Google Scholar] [CrossRef]
- Albert, K.; Bayer, E.J. Characterization of bonded phases by solid-state NMR spectroscopy. J. Chromatogr. A 1991, 544, 345–370. [Google Scholar] [CrossRef]
- Albert, K.; Brindle, R.; Schmid, J.; Buszewsky, R.; Bayer, E. CP/MAS NMR investigations of silica gel surfaces modified with aminopropylsilane. Chromatographia 1994, 38, 283–290. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T.; Soczewinski, E.; Christensen, P. Spectroscopic Studies of Butylphenyl, Mono-ol and Perfluorinated Bonded Phases. Chromatographia 1994, 39, 520–528. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T. Synthesis and Spectroscopic Characterization of a True Diol Bonded Phase. J. Chromatogr. A 1994, 687, 33–34. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T.; Williamsen, E.J.; Tam, R. Variable-Temperature Solid-State NMR Studies of Bonded Liquid Crystal Stationary Phases for HPLC. Chromatographia 1995, 41, 301–310. [Google Scholar] [CrossRef]
- Legrand, A.P. The Surface Properties of Silicas, 1st ed.; Wiley: Chichester, UK, 1998. [Google Scholar]
- Maciel, G.; Bronnimann, C.; Hawkins, B. Advances in Magnetic Resonance: The Waugh Symposium; Warren, W.S., Ed.; Academic Press: San Diego, CA, USA, 1990; Volume 14, p. 125. [Google Scholar]
- Chuang, I.S.; Kinney, D.R.; Bronnimann, C.E.; Zeigler, R.C.; Maciel, G.E. Effects of 1H-1H spin exchange in the 29Si CP-MAS NMR spectra of the silica surface. J. Phys. Chem. 1992, 96, 4027–4034. [Google Scholar] [CrossRef]
- Maciel, G.; Ellis, P. NMR Techniques in Catalysis; Bell, A.T., Pines, A., Eds.; Dekker: New York, NY, USA, 1994. [Google Scholar]
- Chuang, I.; Maciel, G. Probing Hydrogen Bonding and the Local Environment of Silanols on Silica Surfaces via Nuclear Spin Cross Polarization Dynamics. J. Am. Chem. Soc. 1996, 118, 401–406. [Google Scholar] [CrossRef]
- Haukka, S.; Lakornaa, E.; Root, A. An IR and NMR study of the chemisorption of titanium tetrachloride on silica. J. Phys. Chem. 1993, 97, 5085–5094. [Google Scholar] [CrossRef]
- Valtier, M.; Drujon, X.; Wilhelm, M.; Spiess, H. Quantitative Characterization of the Inner Structure of Core-Shell Latex Particles by 1H Solid-State NMR. Macromol. Chem. Phys. 2001, 202, 1262–1272. [Google Scholar] [CrossRef]
- Engelhardt, G.; Michel, D. High Resolution Solid-State NMR of Silicates and Zeolites; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Reimer, J.A.; Vaughan, R.W.; Knights, J.C. Proton magnetic resonance spectra of plasma-deposited amorphous Si:H films. Phys. Rev. Lett. 1980, 44, 193–196. [Google Scholar] [CrossRef]
- Pines, A.; Gibby, M.; Waugh, J. Proton enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973, 59, 569–590. [Google Scholar] [CrossRef]
- Albert, K. NMR investigations of stationaryg phases. J. Sep. Sci. 2003, 26, 215–224. [Google Scholar] [CrossRef]
- Pfleiderer, B.; Albert, K.; Bayer, E.; Van de Ven, L.; De Haan, J.; Cramers, C. A new approach to the silica gel surface: Characterization of different surface regions by silicon-29 magic angle spinning NMR relaxation parameters and consequences for quantification of silica gels by NMR. J. Phys. Chem. 1990, 94, 4189–419. [Google Scholar] [CrossRef]
- Maciel, G.E.; Sindorf, W.D. Silicon-29 NMR study of the surface of silica gel by cross polarization and magic-angle spinning. J. Am. Chem. Soc. 1980, 102, 7606–7607. [Google Scholar] [CrossRef]
- Wyndham, K.; O'Gara, J.; Walter, T.; Glose, K.; Lawrence, N.; Alden, B.; Izzo, G.; Hudalla, C.; Iraneta, P. Characterization and evaluation of C18 HPLC stationary phases based on ethyl-bridged hybrid organic/inorganic particles. Anal. Chem. 2003, 75, 6781–6788. [Google Scholar] [CrossRef] [PubMed]
- Challoner, R.; Harris, R.; Packer, K.; Taylor, M. Solid-state NMR studies of the high-silica zeolite Theta-1. Zeolites 1990, 10, 539–545. [Google Scholar] [CrossRef]
- Sindorf, W.D.; Maciel, G.E. Silicon-29 NMR study of dehydrated/rehydrated silica gel using cross polarization and magic-angle spinning. J. Am. Chem. Soc. 1983, 105, 1487–1493. [Google Scholar] [CrossRef]
- Gritti, F.; Perdu, C.; Guiochon, G.A. Comparison of the performance of a few packing materials designed to minimize the thermodynamic band tailing of basic compounds in reversed-phase liquid chromatography. J. Chromatogr. A 2008, 1180, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Yang, Q.; Jiang, D.; Yang, J.; Zhang, L.; Li, Y.; Li, C. Synthesis of bifunctionalized mesoporous organosilica spheres for high-performance liquid chromatography. J. Chromatogr. A 2006, 1103, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Bociana, S.; Rychlickib, G.; Matyskac, M.; Pesekc, J.; Buszewski, B. Study of hydration process on silica hydride surfaces by microcalorimetry and water adsorption. J. Colloid Interf. Sci. 2014, 416, 161–166. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abia, J. Surface Characterization of Some Novel Bonded Phase Packing Materials for HPLC Columns Using MAS-NMR Spectroscopy. Chromatography 2015, 2, 141-155. https://doi.org/10.3390/chromatography2020141
Abia J. Surface Characterization of Some Novel Bonded Phase Packing Materials for HPLC Columns Using MAS-NMR Spectroscopy. Chromatography. 2015; 2(2):141-155. https://doi.org/10.3390/chromatography2020141
Chicago/Turabian StyleAbia, Jude. 2015. "Surface Characterization of Some Novel Bonded Phase Packing Materials for HPLC Columns Using MAS-NMR Spectroscopy" Chromatography 2, no. 2: 141-155. https://doi.org/10.3390/chromatography2020141