An Isocratic Toxic Chemical-Free Mobile Phase HPLC-PDA Analysis of Malachite Green and Leuco-Malachite Green

Abstract
:1. Introduction
2. Experimental Section
2.1. Chemicals and Reagents
2.2. Equipments

{kind=link}
{kind=link}
| Column | Pore Diameter | Pore Volume | Surface Area | Carbon Load | HPLC Target Compounds | ||||
|---|---|---|---|---|---|---|---|---|---|
| Silica Type | Trade Name | Ethanol-Water | Ethanol-OSA c | ||||||
| Separation | Peak Forms | Separation | Peak Forms | ||||||
| (nm) | (mL/g) | (m2/g) | (%) | ||||||
| (a) C18 | Inertsil ODS-4 | 10 | 1.05 | 450 | 11 | N-LMG d | - | N-LMG | - |
| (b) C18 | InertSustain C18 | 10 | 0.85 | 350 | 14 | N-LMG | - | N-LMG | - |
| (c) diol | Inertsil HILIC | 10 | 1.05 | 450 | 20 | N-LMG | - | N-LMG | - |
| (d) C4 | Inertsil WP300 C4 | 30 | 1.05 | 150 | 3 | N-MG e | - | Separated | Symmetrical/ Sharp |
2.3. Operating Conditions

2.4. Preparation of Stock Standards and Working Mixed Solutions
2.5. HPLC Validation
2.5.1. Linearity
2.5.2. Detection Limit
2.5.3. Robustness
2.5.4. System Suitability Test
3. Results and Discussion
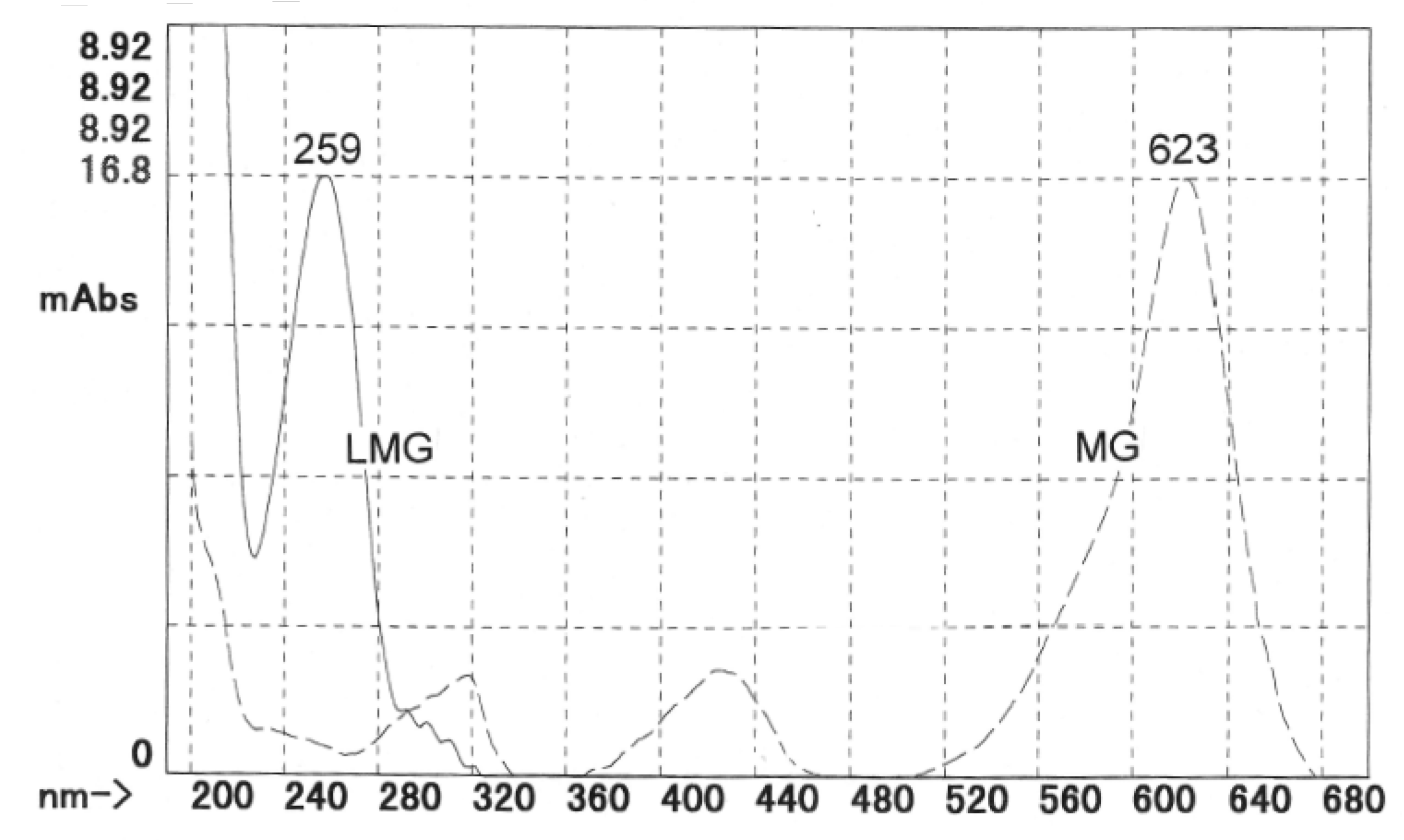
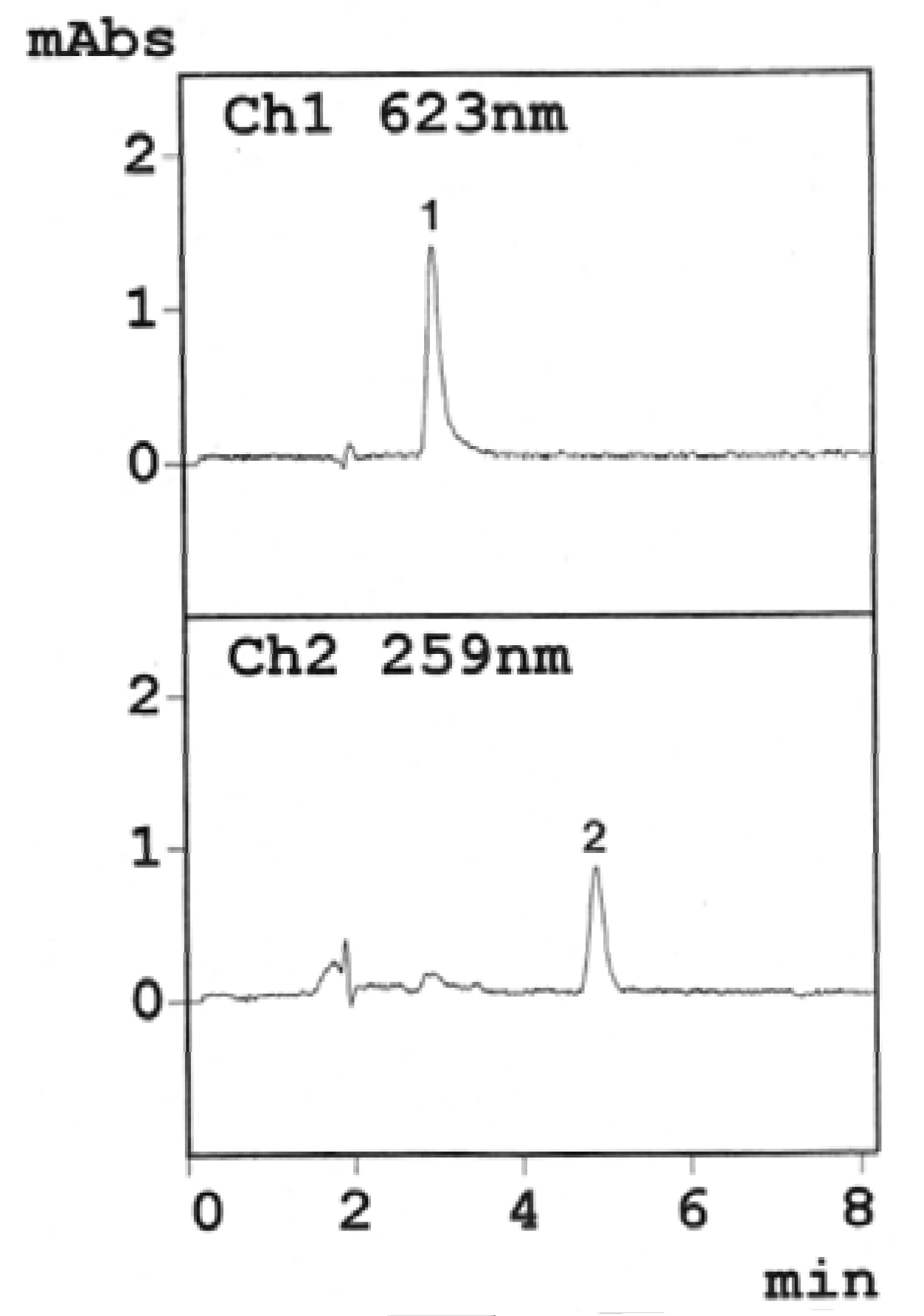
3.1. Optimum HPLC Conditions

3.2. HPLC Validation
3.2.1. Main Validation Data
| MG | LMG | Acceptance Limit a | ||
|---|---|---|---|---|
| Linearity (r) b | 0.9995 | 0.9993 | ≥0.999 | |
| Range (ng/mL) | 10–200 | |||
| Detection limit c (ng/mL) | 1.94 | 1.56 | ||
| System suitability | ||||
| Injection precision(%) d | ||||
| retention time | 0.32 | 0.69 | ≤1 | |
| peak area | 0.66 | 0.15 | ≤1 | |
| Tailing factor (TF) | 1.33 | 0.96 | ≤2 | |
3.2.2. Robustness
4. Conclusions
Conflicts of Interest
References
- BfR. Collection and Pre-Selection of Available Data to be Used for the Risk Assessment of Malachite Green Residues by JECFA, Updated * BfR Expert Opinion No. 007/2008 of 24 August 2007. Available online: http://www.bfr.bund.de/cm/349/collection_and_pre_selection_of_available_data_to_be_used_for_the_risk_assessment_of_malachite_green_residues_by_jecfa.pdf#search='leucomalachite+green+residue' (accessed on 15 January 2014).
- Brown, L. Aquaculture for Veterinarians—Fish Husbandry and Medicine; Pergamon Press: New York, NY, USA, 1993. [Google Scholar]
- Sudova, E.; Machova, J.; Svobodova, Z.; Vesely, T. Negative effects of malachite green and possibilities of its replacement in the treatment of fish eggs and fish: A review. Vet. Med. 2007, 52, 527–539. [Google Scholar]
- Veterinary Residues Committee (2001–2003). Annual Report on Surveillance for Veterinary Residues in Food in the UK for 2001, 2002, and 2003; Veterinary Residues Committee: Surrey, UK, 2003. [Google Scholar]
- Veterinary Residues Committee (2001–2010). Annual Report on surveillance for Veterinary Residues in Food in UK for 2001 to 2010; Veterinary Residues Committee: Surrey, UK, 2010. [Google Scholar]
- Olesen, P.T. Risk Assessment Malachite Green in Food; National Food Institute, Technical University of Denmark: Soeborg, Denmark, 2007. [Google Scholar]
- Hurtaud-Pessel, D.; Couedor, P.; Verdon, E.; Dowell, D. Determination of residues of three triphenylmethane dyes and their metabolites (malachite green, leuco malachite green, crystal violet, leuco crystal violet, and brilliant green) in aquaculture products by LC/MS/MS: First action 2012.25. J. AOAC Int. 2013, 96, 1152–1157. [Google Scholar] [CrossRef]
- Hidayah, N.; Abu Bakar, F.; Mahyudin, N.A.; Faridah, S.; Nur-Azura, M.S.; Zaman, M.Z. Detection of malachite green and leuco-malachite green in fishery industry. Int. Food Res. J. 2013, 20, 1511–1519. [Google Scholar]
- Hurtaud-Pessel, D.; Couedor, P.; Verdon, E. Liquid chromatography-tandem mass spectrometry method for the determination of dye residues in aquaculture products: Development and validation. J. Chromatogr. A 2011, 1218, 1632–1645. [Google Scholar] [CrossRef]
- Agilent Technologies. The Literature Library. Available online: http://www.chem-agilent.com/appnote/applinote.php?pubno=LCMS-200902TK-002 (accessed on 8 January 2014).
- Allen, J.L.; Meinertz, J.R.; Ofus, J.E. Determination malachite green and its lecuo form in water. J. AOAC Int. 1992, 75, 646–649. [Google Scholar]
- Rushing, L.G.; Hansen, E.B. Confirmation of malachite green, gentian violet and their leuco analogs in catfish and trout tissue by high-performance liquid chromatography utilizing electrochemistry with ultraviolet-visible diode array detection and fluorescence detection. J. Chromatogr. B 1997, 700, 223–231. [Google Scholar] [CrossRef]
- Tang, H.P.O.; Choi, J.Y.Y. Analysis of Malachite Green in Fish Sample; Hong Kong Service and Animal Research of Government Laboratory: Hongkong, China, 2005. [Google Scholar]
- Classification (The Dangerous Substances Directive 67/548/EEC). Available online: https://osha.europa.eu/en/legislation/directives/exposure-to-chemical-agents-and-chemical-safety/osh-related-aspects/58 (accessed on 10 May 2014).
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Yoshimura, T.; Nishinomiya, T.; Homda, Y.; Murabayashi, M. Green Chemistry—Aim for the Zero Emission-Chemicals; Sankyo Publishing Co.,Ltd. Press: Tokyo, Japan, 2001. [Google Scholar]
- FDA. Reviewer Guidance, Validation of Chromatographic Methods; Center for Drug Evaluation and Research (CFDER): Silver Spring, MD, USA, 1994.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Furusawa, N. An Isocratic Toxic Chemical-Free Mobile Phase HPLC-PDA Analysis of Malachite Green and Leuco-Malachite Green. Chromatography 2014, 1, 75-81. https://doi.org/10.3390/chromatography1020075
Furusawa N. An Isocratic Toxic Chemical-Free Mobile Phase HPLC-PDA Analysis of Malachite Green and Leuco-Malachite Green. Chromatography. 2014; 1(2):75-81. https://doi.org/10.3390/chromatography1020075
Chicago/Turabian StyleFurusawa, Naoto. 2014. "An Isocratic Toxic Chemical-Free Mobile Phase HPLC-PDA Analysis of Malachite Green and Leuco-Malachite Green" Chromatography 1, no. 2: 75-81. https://doi.org/10.3390/chromatography1020075