New Development of Biomarkers for Gastrointestinal Cancers: From Neoplastic Cells to Tumor Microenvironment


1
Departments of Oncology and Surgery, the Sidney Kimmel Comprehensive Cancer Center, the Bloomberg-Kimmel Institute for Cancer Immunotherapy, the Pancreatic Cancer Precision Medicine Center of Excellence Program, the Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA
2
Merck Research Laboratory, Merck & Co., Kenilworth, NJ 07033, USA
*
Author to whom correspondence should be addressed.
Biomedicines 2018, 6(3), 87; https://doi.org/10.3390/biomedicines6030087
Submission received: 5 June 2018
/
Revised: 30 July 2018
/
Accepted: 10 August 2018
/
Published: 13 August 2018
(This article belongs to the Special Issue Cancer Biomarkers and Targets in Digestive Organs)
Abstract
:Biomarkers refer to a plethora of biological characteristics that can be quantified to facilitate cancer diagnosis, forecast the prognosis of disease, and predict a response to treatment. The identification of objective biomarkers is among the most crucial steps in the realization of individualized cancer care. Several tumor biomarkers for gastrointestinal malignancies have been applied in the clinical setting to help differentiate between cancer and other conditions, facilitate patient selection for targeted therapies, and to monitor treatment response and recurrence. With the coming of the immunotherapy age, the need for a new development of biomarkers that are indicative of the immune response to tumors are unprecedentedly urgent. Biomarkers from the tumor microenvironment, tumor genome, and signatures from liquid biopsies have been explored, but the majority have shown a limited prognostic or predictive value as single biomarkers. Nevertheless, use of multiplex biomarkers has the potential to provide a significantly increased diagnostic accuracy compared to traditional single biomarker. A comprehensive analysis of immune-biomarkers is needed to reveal the dynamic and multifaceted anti-tumor immunity and thus imply for the rational design of assays and combinational strategies.
1. Introduction
Biomarkers are defined as objective, quantifiable biological indicators of a normal or abnormal process, a condition or disease, or a response to treatment [1]. Prognostic biomarkers allow a selection of patients with a high risk for disease recurrence or rapid progression and help regarding decision making for a treatment regimen. Predictive biomarkers represent an array of indicators that project the patient’s response to the treatment. Currently, biomarkers are genetic, epigenetic, proteinic, or cellular alterations that are inherent to cancer cells, and have been an integral part of individualized cancer care.Glycoproteins, such as carcinoembryonic antigen (CEA) and cancer antigen 19-9 (CA19-9), are classical proteinic biomarkers for disease monitoring [2,3,4]. Biomarkers at the genome level, which are often driver mutations such as KRAS and epidermal growth factor receptor (EGFR), have been widely used as a guide for a selection of patients that might benefit from targeted therapies [4,5,6,7]. More recent literature has highlighted BRCA, a tumor suppressor gene involved in the repair of double-stranded DNA breaks, as a viable predictive biomarker for response to platinum agents and poly(ADP-ribose) polymerase (PARP) inhibitors [8]. Immunotherapy has revolutionized human cancer treatment by unleashing the potential of the antitumor immune response; however, only 15–20% of patients respond to immunotherapy [9,10]. This underpins the importance of identifying novel biomarkers that can select the patients for immunotherapy. Programmed death ligand 1 (PD-L1) positivity, T cell-inflamed phenotype, and high tumor mutational burden have been reported to enrich the patient populations that benefit from the treatment of immune checkpoint inhibitors (ICIs) [11,12,13,14,15]; however, these makers alone are insufficiently accurate for patient selection across different cancer types. Microsatellite instability (MSI), a pan-cancer biomarker, predicts the response of solid tumors to anti-PD-1/PD-L1 (Programmed death-1/Programmed death ligand 1) blockade, is only found in 1–2% of most of malignancies [12,16]. Preliminary clinical findings showed that the signatures that reflect the composition and metabolites of the gut microbiota would impact the antitumor immune response in patients receiving ICIs, including anti-cytotoxic T-lymphocyte antigen 4 [CTLA-4] and anti-PD-1/PD-L1 antibodies, and have the potential to predict durable clinical responses in non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), and melanoma; however, the role of the microbiome in predicting the benefits from the ICIs remains unclear for gastrointestinal (GI) cancer [17,18,19,20,21]. A few studies have also suggested the association of members of the gut microbiome with ICI toxicities, but evidence is lacking in the GI setting [21,22]. In addition, biomarkers that can guide the choice of combination immunotherapy are scarce [23]. Quantitative multiplexed approaches, which exert unique advantages in revealing the tumor-immune complexity, may represent a new avenue for biomarker discovery in the tumor microenvironment, especially for combinational therapies targeting the suppressive myeloid/stromal compartment [24,25]. This review summarizes the advances of biomarkers in gastrointestinal malignancies, with a focus on the development of new biomarkers that are of predictive and/or prognostic values in cancer therapies.
2. Current Clinical Application of Biomarkers in Gastrointestinal Malignancies
2.1. Tumor Markers
The tumor markers currently being used in the clinic are all surrogate markers of malignant diseases (Table 1). CEA is one of the most commonly used tumor markers for gastrointestinal malignancies and a member of the immunoglobulin superfamily [26]. It acts as a mediator for cell adhesion on cancer cells. The overexpression of CEA occurs in >90% of colorectal cancers (CRC) and 60% of other types of cancer, including gastric, lung, and pancreatic cancers [27]; thus, it has been widely used as a serum tumor marker. Its sensitivity and specificity are not high, particularly for the early stages of the disease [28]; therefore, it cannot be used as a biomarker for screening gastrointestinal cancers. However, in the patients with an established disease, the absolute level of the serum CEA correlates with the disease burden and has a prognostic value [29]. In CRC, CEA is the only laboratory test routinely recommended for surveillance. High levels of CEA after surgical resection imply the presence of a persistent disease and the need for further evaluation [30]. The serial measurement of the CEA levels after surgery in patients with colorectal cancer can detect recurrences earlier; nevertheless, this information does not lead to an improved treatment outcome [28]. Currently, the American Society of Clinical Oncology (ASCO) guidelines recommend that the serum CEA levels be obtained in most patients with CRC, so as to aid surgical treatment planning, posttreatment follow-up, and the assessment of prognosis [31]. Another common glycoprotein biomarker is CA19-9, which is used primarily to assess the disease response to therapy or to detect cancer recurrence in patients with gastric cancer, pancreatic cancer, gallbladder cancer, cholangiocarcinoma, or adenocarcinoma of the ampulla of Vater. It is the most useful tumor marker for pancreatic cancer, with sensitivity and specificity rates of 70–92% and 68–92%, respectively [32,33,34]. In addition, an elevated preoperative CA19-9 level is strongly associated with the presence of subradiographic unresectable diseases, and can be used for a selection of patients for staging the laparoscopy [35]. The lack of tumor specificity is the limitation of the currently used tumor markers. Tumor specific markers, particularly those reflecting the tumor biology, are highly demanded.
2.2. Targets of Matched Therapies
HER2, a tyrosine kinase receptor belonging to the epidermal growth factor receptor (EGFR) family, is an established prognostic factor and a therapeutic target for gastroesophageal adenocarcinoma [36,37,38,39]. Through the activation of downstream signaling pathways, including RAS/RAF/mitogen-activated protein kinase (RAS/RAF/MAPK) and phosphatidylinositol-3 kinase/protein kinase-B/mammalian target of rapamycin (PI3K/AKT/mTOR), aberrant HER2 amplification or overexpression can lead to uncontrolled cell-cycle progression, cell division, motility, survival, invasion, and adhesion [7]. Approximately 7–38% of gastroesophageal adenocarcinomas have an amplification and/or overexpression of HER2, with a slightly higher positivity in gastroesophageal junction (GEJ), intestinal-type, and well-moderately differentiated tumors [6,7,40,41]. The inhibition of HER2 with the monoclonal antibody trastuzumab in patients with HER2-amplified/overexpressed advanced-stage gastric or esophagogastric-junction adenocarcinomas, confers an improved response rate, progression-free survival, and overall survival when trastuzumab is combined with cisplatin and fluoropyrimidine [5]. Based on the above evidence from the phase III ToGA trial, the current guideline suggests that patients with advanced gastric cancer who are potential candidates for trastuzumab should be screened to determine their HER2 status.
Another biomarker for therapeutic target is c-MET, of which the aberrant expression has been reported in 18–100% of gastrointestinal tumors [42,43]. Activated c-MET signaling results in enhanced cancer cell proliferation, survival, and invasion, and is an independent prognostic factor for inferior survival [44,45,46]. Several monoclonal antibodies and small-molecule inhibitors of c-MET have been evaluated in clinical trials, however, most of the phase III trials of MET inhibitors showed negative results for gastric cancer. In hepatocellular carcinoma, although encouraging results were reported in phase II studies [47,48,49], the phase III trial failed to show an improvement in the overall survival compared with the placebo in patients with c-MET-positive advanced hepatocellular cancer (HCC), casting doubt on the role of MET inhibition as a viable therapeutic strategy [50,51]. Therefore, the development of biomarkers for therapeutic target is still a challenge.
In metastatic CRC, the KRAS mutation status has been widely reported as a prognostic and predictive biomarker [52]. KRAS mutations can be identified in 12–75% of colon cancers and are independently associated with a worse prognosis in the majority of the studies, albeit not all of the studies [53,54]. As the RAS oncogene is located at the downstream of the EGFR signaling pathway, the RAS mutations can lead to an activation of the pathway, even if the EGFR is blocked [55]. Thus, the KRAS mutation status is a biomarker for unresponsiveness to anti-EGFR therapy. Interestingly, there is a bias toward the right-sided CRC for the KRAS mutation, this may partially explain an inferior survival and poor response to targeted therapy with EGFR inhibitors for the right proximal CRC compared to the left colon CRC [56]. The characteristics of the HER2, c-MET, and KRAS expression in GI cancers are summarized in Table 2.
2.3. Mismatch Repair Genes
Mismatch repair (MMR) gene products function to repair the nucleotide base mispairings and small insertions or deletions that occur during DNA replication [57,58]. Thus, the MMR-deficient tumors could accumulate hundreds to thousands of somatic mutations, regardless of their cell of origin. It has been implicated in the pathogenesis of the hereditary nonpolyposis colorectal cancer syndromes, as well as a variety of different sporadic cancers. MMR-deficiency is present in 15–20% of all colorectal cancers (CRCs), 8.5–20% of gastric cancers, 3–7% in esophageal/GEJ adenocarcinomas, and 2–3% of pancreatic cancers [12,16,59,60]. MMR-deficiency has been shown to be positively prognostic for survival in patients with colon, gastric, and pancreatic cancers [57,60,61]. It could also serve as a potential predictive marker for a lack of efficacy of fluoropyrimidine based adjuvant chemotherapy in gastric cancer and colon cancer [62,63,64]. Importantly, MMR deficiency is a pan-cancer predictor for response to anti-PD-1/PD-L1 blockade therapies [65]. It is hypothesized that tumors with an MMR deficiency are enriched with missense mutations that are presented as neoepitopes to T cells, which are subsequently targets of anti-PD-1/PD-L1 therapies. Le et al. reported a phase II clinical trial of progressive metastatic carcinoma with or without MMR deficiency, and revealed significantly increased somatic mutations per tumor in the MMR–deficient tumors compared with the MMR-proficient tumors (mean, 1782 vs. 73). The MMR deficient patients had a remarkably increased immune-related objective response rate (40% vs. 0%) and prolonged immune-related progression-free survival rate (78% vs. 11%), compared to their counterparts [16]. In an expanded cohort of 86 patients with MMR-deficient tumors, the objective radiographic responses were noted in 53% of the patients (46 of 86 patients; 95% CI, 42–64%), with 21% (n = 18) achieving a complete radiographic response. Disease control (measured as partial response + complete response + stable disease) was achieved in 66 (77%) of the 86 patients (95% CI, 66–85%) [12]. This led to the approval from the Food and Drug Administration (FDA) for testing MMR-deficiency in order to identify the candidate patients who may benefit from a second-line PD-1 pathway blockade, regardless of the tumor types [66]. Of note, this is the first and only FDA approved pan-cancer biomarker for immune checkpoints blockade. Clinical trials investigating its role as predictive biomarkers in the first-line and (neo)adjuvant settings are ongoing [67,68].
3. New Development of Biomarkers
3.1. Biomarkers in Tumor Microenvironment
3.1.1. PD-L1 Expression
As above described, PD-1, which is expressed on activated lymphocytes, including T cells, B cells, and natural killer cells, limits the T cell effector functions within tissues. By upregulating the ligands for PD-1 (PD-L1), tumor cells induce the apoptosis of the effector T cells [69,70]. The reported incidence of PD-L1 expression ranges differently in the different tumor types (14–100%), whether or not these tumors respond to anti PD-1/PD-L1 treatment [71,72,73,74,75,76]. Early studies have suggested that PD-L1 positivity enriches the patient populations that can benefit from PD-1/PD-L1 axis inhibition [77,78], with the hypothesis that pre-existing immunity suppressed by PD-1/PD-L1 could be re-invigorated on antibody treatment with checkpoint blockade. However, more studies questioned the accuracy of PD-L1 as an effective predictive biomarker. In the recent phase III trials testing the adjuvant anti-PD-1 in resected stage III melanoma, the first-line anti-PD-1 antibody in combination with chemotherapy in metastatic NSCLC, and the combination of anti-PD-1 and anti-CTLA4 antibodies in NSCLC with a high mutational burden, the benefit of immunotherapy did not correlate with the PD-L1 expression level [79,80,81]. In the Keynote 059 trial, objective responses and complete responses (CRs) were observed in both the PD-L1-positive and negative gastric and gastroesophageal adenocarcinoma patients who had previously received at least two lines of treatment [82]. The PD-L1-positivity was defined as a combined positive score (CPS) ≥1%, where CPS is the number of PD-L1 staining tumor cells, lymphocytes, and macrophages divided by the total number of viable tumor cells multiplied by 100, using PD-L1 Immunohistochemistry (IHC) 22C3 pharmDx immunohistochemistry. Although the objective response rate (ORR) seemed higher in the patients with PD-L1–positive compared with the PD-L1–negative tumors (23 of 148 [15.5%] vs. 7 of 109 [6.4%], respectively), the patients with PD-L1–negative tumors also experienced objective responses, including CR in three patients (2.8%) [82]. Nevertheless, this study has gained the FDA approval of using PD-L1 positivity (Table 1) at the 1% cutoff as a biomarker to select patients with metastatic gastric and gastroesophageal adenocarcinoma for the treatment of pembrolizumab [83]. In the Asian population, Nivolulab was approved for the treatment of the unresectable, advanced, or recurrent gastric cancer that has progressed after using conventional chemotherapy, based on the results from the phase III ATTRACTION-2 trial, regardless of PD-L1 status. In hepatocellular cancer (HCC), the report from the phase I/II trial suggests that ICIs elicited a promising response rate of 16–19% (49/255) in advanced HCC, but the response rate to the ICIs did not differ according to the PD-L1 expression status [82,83,84,85]. In PDACs, reports of the PD-L1 expression vary from 12–90%, however, single agent anti-PD1 treatment has shown no efficacy, except for MMR deficient patients, regardless of PD-L1 status [86].
Therefore, evolving evidence suggests that PD-L1 testing alone is insufficiently accurate to predict patient response to immunotherapy, although it may be used in some GI cancers to enrich the patients that may more likely benefit from anti-PD-1/PD-L1 antibodies (Table 3). Several factors may explain the heterogeneity of the predictive values for the PD-L1 expression, including differences in the PD-L1 IHC assay platforms and detection antibodies, differing IHC cutoffs, tissue preparation, processing variability, primary versus metastatic lesions, oncogenic versus induced PD-L1 expression, and the staining of tumor versus immune cells [42,47,75,87,88]. It should be noted that the PD-L1 expression measured in the clinical assays may only represent a snapshot of the dynamic and multifaceted immune cells and their complex interaction with neoplastic cells. A comprehensive characterization of the tumor microenvironment is necessary to adequately assess the strength of PD-L1 in predicting the immune response to anti-PD-L1/PD-1 therapies.
3.1.2. Tumor Infiltrating Lymphocyte
Tumor infiltrating lymphocytes (TILs) represent a potent machinery of the adaptive immunity that has the antitumor potential. TILs have been shown to be associated with improved prognoses and response to immunotherapy in various cancer type (Table 3) [24,44,45,46,89,90,91]. In colorectal cancers, the type, density, and location of the immune cells, specifically the cytotoxic and memory T cells, has been reported to be a better predictor of survival than (Union for International Cancer Control ) UICC-TNM staging 89]. Among the tumors with similar degrees of T cell infiltration, those with the greatest proportion of CD103+ memory T cells have the best prognosis [92]. To standardize the method of evaluating TILs in CRC, a new method that measures the area occupied by mononuclear cells over the stromal area on hematoxylin and eosin (H-E)-stained sections was proposed. The results from such a method confirmed the density of TILs as a useful prognostic factor in CRC [93]. In Epstein–Barr virus (EBV)-associated gastric cancer an association between a high percentage of TILs, low intratumoral PD-L1 expression, and longer disease-free survival (DFS) was demonstrated [94]. A meta-analysis on 23 relevant studies of 3173 hepatocellular carcinoma (HCC) patients showed that high levels of CD8+ and CD3+ TILs had a better prognostic value on the overall survival (OS), yet high levels of FoxP3+ TILs had a worse prognostic value on OS and DFS/Relapse-free survival (RFS), implicating that TILs may serve as a prognostic biomarker in HCC [95]. A TIL density of ≥5% was reported to be associated with a better objective response as well as the progression free survival (PFS) in NSCLC patients treated with Nivolumab [96]. Recently, a T cell inflamed expression score utilizing 18 gene signatures was shown to be significantly associated with a Pembrolizumab response in gastric/GEJ cancer [97,98]. A significant but nonlinear association was found between the T cell-inflamed gene expression score and PD-L1 expression. These results suggest the potential for a T cell-inflamed gene expression profiling score in association with the PD-L1 expression as biomarkers for treatment selection in gastric/GEJ cancers. In pancreatic cancer, variable frequencies of endogenous CD8+ T cells, CD4+Foxp3− T cells, and CD4+Foxp3+ regulatory T cells (Treg) were reported. Notably, these T cells were enriched within CD20+ lymphoid aggregates, with a trend toward longer survival in those patients with tumoral Tertiary lymphoid structures [99]. In our cohort of 24 pancreatic ductal adenocarcinomas receiving neoadjuvant GVAX®vaccination, which is a tumor vaccine composed of autologous tumor cells genetically modified to secrete granulocyte–macrophage colony-stimulating factor (GM-CSF), the ratios of CD8+ T cell to CD68+ T cell are favorable predictors of survival, as reported in other malignancies [100,101]. Nevertheless, the above results need to be confirmed in future trials and more studies on whether and how TILs or effector T cells can be used to predict the response to immunotherapy in gastrointestinal malignancies are warranted.
3.1.3. Immunosuppressive Myeloid Cells
Tumor-associated myeloid cells not only create a suppressive or anergic environment by blocking T cell functions and proliferation, but also accelerate tumor growth by promoting cancer stemness, angiogenesis, stroma deposition, epithelial-to-mesenchymal transition, and metastasis [102]. The accumulation of the intratumoral and circulating myeloid derived suppressive cells (MDSCs) has been shown to be associated with disease progressiveness and prognosis in pancreatic adenocarcinoma, hepatocellular carcinoma, and gastric cancer (Table 3) [102,103,104,105,106]. In our study evaluating 24 pancreatic ductal adenocarcinomas from patients who received neoadjuvant GVAX vaccination, although, essentially all of the tumors have induction of TILs and PD-L1 expression, the survival of patients is correlated with the infiltration of myeloid cells [76]. Nevertheless, myeloid cells are also critical for an innate immune response. It is unlikely that a single myeloid marker would be able to predict the immune response. Multiplex biomarker assays will need to be developed for characterizing immunosuppressive myeloid cells before a clinical assay can be used for predicting their response to immunotherapy.
3.1.4. Intratumoral Stroma
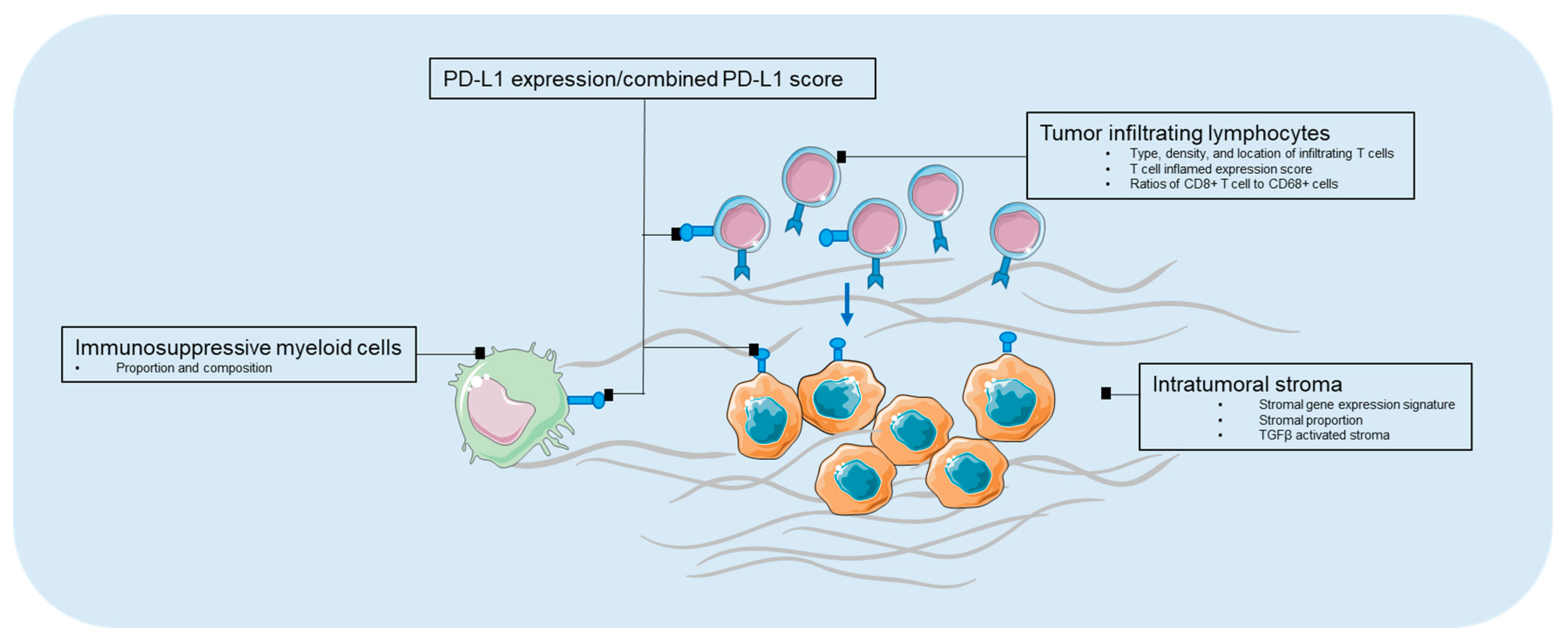
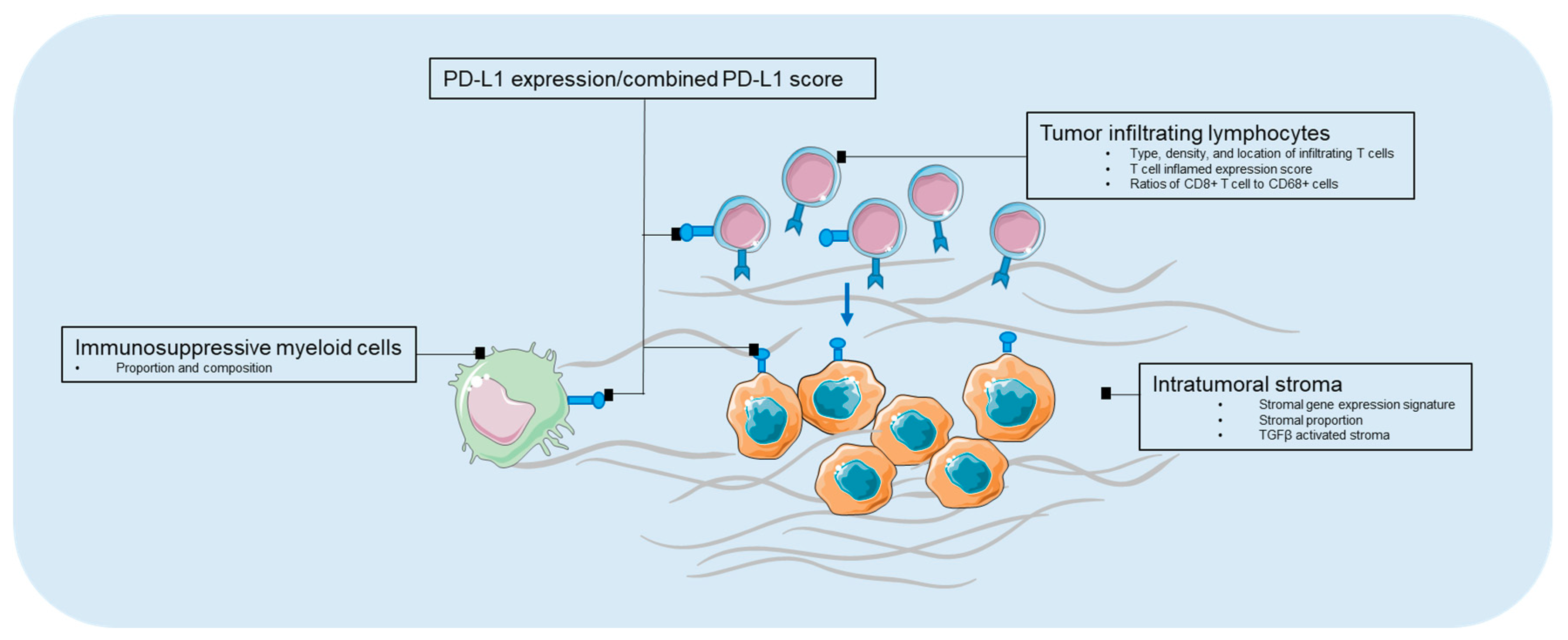
The intratumoral stromal (ITS) proportion, composition, and activation status represent another array of biomarkers for the disease prognosis. Stromal proportion, quantified by histopathological microscopy analysis of the conventional hematoxylin and eosinstained slides, has been reported to be independently associated with poor prognosis in several types of cancers, including gastric cancer, esophageal, and colon cancers (Table 3) [107,108,109,110,111]. Wu et al. showed that the stromal gene expression signature as well as the ITS proportion quantified by morphometry in tissue sections of patient samples, correlated with the survival of gastric cancer patients in multiple independent cohorts. Measuring the relative amount of ITS may enable the identification of subgroups of gastric cancer patients that benefit from stroma-directed therapies [112]. More recently, transforming growth factor beta (TGFβ) activated stroma was found to represent a primary mechanism of immune evasion that engenders T cell exclusion and primary resistance to anti-PD-1–PD-L1 therapy in microsatellite-stable (MSS) CRC. In murine models, the authors showed that the inhibition of TGFβ signaling in the stoma with a TGF-β receptor 1 (TGF-βR1) specific inhibitor could lead to a potent anti-tumour cytotoxic T cell response and prevent metastasis [113]. Admittedly, there are promising applications in immunotherapies targeting intratumoral stroma and in combination with immune checkpoint inhibitors, however, the identification of accompanying predictive features from intratumoral stroma to enrich for populations that can benefit from combinational therapies are crucial. A summary of biomarkers in the tumor microenvironment is depicted in Figure 1.
3.2. Biomarkers in Tumor Genomics
3.2.1. Targeted Gene Panels
Targeted gene sequencing is an emerging approach for identifying potentially targetable genomic biomarkers and matching them for treatments. However, a number of challenges remain. For example, the Memorial Sloan Kettering Cancer Center has developed an (IMPAC) targeted gene sequencing panel, which included 341 genes initially, and has expanded to 410 cancer-associated genes [114]. As reported, 10,945 tumors from 10,366 patients with advanced cancer were sequenced. Eleven percent of the patients were enrolled in a genomically matched clinical trial. Among the 10,366 patients, 338 patients had pancreatic cancer. Five of these pancreatic cancer patients died before the results were finalized. Potentially actionable findings were noted in 26% of these pancreatic cancer patients. Nevertheless, only three of the 225 patients (1%) who would need treatments received matched therapy based on the sequencing results [115]. Two had no benefit and one had an unknown response. Therefore, the practical application of molecular results to guide individual patient treatment is currently limited in patients with pancreatic adenocarcinoma.
3.2.2. Mutational Burden
The tumor mutational burden (TMB) has been shown to be significantly associated with a clinical benefit to immune checkpoint blockade in various cancer types [6,11,13,14,15,35,116]. However, most of the GI cancers have low mutation burdens [5], except those with MMR-deficiency. In a cohort of 1375 patients across various GI tumors, colon cancer was reported to have the highest TMB (mean: 11.6 and 9.9 mutations [mut]/megabase [MB]), whereas biliary cancers and pancreatic adenocarcinomas had the lowest TMB (mean: 5.7 and 4.9 mut/MB) [117]. Using a cut-off of 17 mut/MB to define high vs. low TMB, the high TMB was seen most frequently in right sided colon cancer (12%), gastric cancer (11%), and anal cancer (8%), and least frequently in pancreatic cancer (1.3%) and esophageal squamous cell carcinoma (0%). In addition, primary tumors, MSI-H and/or MSS with POLE mutations were observed to have a higher TMB. Those with a higher frequency of somatic mutations and tumor-specific neoantigens were found to have more abundant infiltration of CD8+ T lymphocytes and a higher expression of regulatory molecules (CTLA-4, PD-1, Lymphocyte-activation gene 3[LAG-3] and indolamine 2,3-dioxygenase 1 [IDO1]) [118].
3.3. Biomarkers in Liquid Biopsies
A tumor tissue biopsy would be necessary to establish the diagnosis; however, it would not be feasible for monitoring the treatment response [119]. The analysis of biomarkers from peripheral blood, including the circulating tumor DNA (ctDNA), circulating tumor cells (CTC), and exosomes, using a noninvasive approach known as liquid biopsy, has emerged as a way to overcome the restrictions of tumor tissue biopsies and has exhibited a great potential of being used to detect the recurrence and measure the treatment response [120].
3.3.1. ctDNA
ctDNAs are predominantly released as a result of the apoptosis or necrosis of actively growing cancer cell, but can also be secreted directly from the circulating tumor cells [121]. Notably, ctDNA can maintain tumor-specific genomic aberrations, including point mutations in tumor suppressors and oncogenes, copy number variants, DNA methylation patterns, and chromosomal rearrangements, providing a comprehensive genomic profiling for tumor evolution and dynamics disease monitoring [119]. In CRC, ctDNA has been shown to successfully gather the real-time molecular evolution in patients treated with EGFR targeted therapy [122,123]. The ctDNA analyses can not only identify genetic alterations that are likely to be responsible for resistance to EGFR blockade, but also can guide the selection of rare populations of patients who are likely to respond to targeted agents [124]. Changes of circulating tumor DNA (ctDNA) levels during therapy might also be an indicator of clinical efficacy with ICIs. In a small prospective pilot study (n = 15), the ctDNA levels at week eight showed synchronous changes with tumor size and as well as an association with PFS in various cancer types [125]. In the chemotherapy setting, the RAS/BRAF mutations detected in ctDNA correlated with a worse PFS in the metastatic CRC patients (n = 27) treated with first-line chemotherapy [126]. However, the role of ctDNA in predicting the response to treatment needs to be validated in a larger population.
3.3.2. CTC
CTCs are surrogates of tumor cells in the bloodstream. CTC prevalence differs with cancer type and stage. In patients with metastatic GI malignancies, CTC could be detected in 30–66% of patients [127], and its presence has been shown to correlate with decreased OS or decreased PFS [128]. The value of CTCs as a therapeutic target to monitor the treatment response and detect relapse has also been reported [129]. Nevertheless, the prognostic and predictive role of CTC is not established in a non-metastatic setting, given the scarcity of CTC in this patient population [130,131]. On the other hand, CTC recently demonstrated its value in monitoring the response to immunotherapy. In a prospective cohort of 49 metastatic melanoma patients treated with ICI, a decrease in an RNA signature score of CTC within seven weeks of therapy correlated with a marked improvement in PFS (hazard ratio [HR], 0.17; p = 0.008) and overall survival (OS) (HR, 0.12; p = 0.04) [132]. The promising results support the rationale to apply CTCs in monitoring the tumor burden in other cancer types such as GI cancers.
Thus far, despite the interesting and promising results from small cohorts of studies on liquid biopsy approaches as predictive or prognostic biomarkers, there is insufficient evidence of clinical utility of the majority of ctDNA/CTC assays in either advanced cancer or early-stage cancer [133]. Discordance exists between the results of different platforms [134]. Prospective trials in large populations will be required to establish the clinical utility of ctDNA and CTC.
3.3.3. Exosomes
Exosomes are endosome derived extracellular vesicles (EVs) ranging in 30–120 nm [135]. They carry a cargo of proteins, metabolites, RNAs (mRNA, miRNA, long non coding RNA), DNAs (mtDNA, ssDNA, and dsDNA), and lipids [136], and represent an important source as a biomarker from liquid biopsies. In pancreatic cancer, the glypican-1 (GPC1)+ endosomes were reported as a diagnostic biomarker to distinguish healthy subjects and patients with a benign pancreatic disease from patients with early- and late-stage pancreatic cancer, with absolute specificity and sensitivity. The levels of the GPC1(+) endosomes correlated with the tumor burden and the survival of pre- and post-surgical patients [137]. A rapid, highly sensitive, and widely usable detection method based on the amplified luminescent proximity homogeneous assay, using photosensitizer-beads for cancer cell-derived EVs was proposed, with the utilization of monitoring the circulating EVs with the antigen CD147 for the detection of colorectal cancer [138]. Although endosomes held great promise for non-invasive early detection and target for potential therapeutics, it has several limitations. One of the biggest challenges in exosome biology is how to accurately measure the quantity and purity of the exosomes. Only a small subset of EVs carry the relevant communication content, thus its actual efficiency may be difficult to detect. In addition, more knowledge of the specific markers of the EV subtypes and fundamental roles of each type of EV is required to better inform their utilization in various disease settings. The advantages and limitations of various liquid biopsy approaches are summarized in Table 4.
4. Prospective
There are only a handful of biomarkers used in clinics for the management of GI malignancies. Although many new biomarkers have been identified for GI malignancies, their clinical assays have not been validated. On the other hand, biomarker assays are highly demanded by a selection of patients for appropriate treatment. Nevertheless, such a biomarker is often not conceived until a clinical trial of experimental therapeutics fails to meet its endpoint because of a lack of patient selection. The development of experimental therapeutics will not be advanced until an adequate biomarker assay is established. Therefore, in the future, biomarker development should be done in parallel with drug development. Whenever a potential therapeutic target is identified, a companion biomarker assay should be developed. For immunotherapy, a single biomarker is often not sufficient in predicting the treatment response. A comprehensive analysis of immune-biomarkers can not only provide the rational design of combination immunotherapy, but can also identify multiple immune-biomarkers, and subsequently develop a multiplex assay to co-evaluate multiple immune-biomarkers. In addition, recent research on ion channels and aquaporins have suggested their function as possible modulators of important processes in gastrointestinal carcinogenesis, including colorectal, pancreatic, gastric, and esophageal cancers, as well as their potential as new cancer biomarkers once appropriately validated [141,142,143].
Acknowledgments
This study is partially funded by NIH grants R01 CA169702 (L.Z.); R01 CA197296 (L.Z.); NIH grant K23 CA148964 (L.Z.); the Commonwealth Foundation (L.Z.), the Bloomberg-Kimmel Institute for Cancer Immunotherapy(L.Z., J.Z.), the Viragh Foundation and the Skip Viragh Pancreatic Cancer Center at Johns Hopkins (L.Z.); the Sol Goldman Pancreatic Cancer Research Center (L.Z.); the Zhang Family Gift Fund (L.Z.); National Cancer Institute Specialized Programs of Research Excellence in Gastrointestinal Cancers grant P50 CA062924 (L.Z.); Sidney Kimmel Comprehensive Cancer Center grant P30CA006973 (L.Z., C.L.W.).
Conflicts of Interest
S.Q. is an employee of Merck and Co. L.Z. receives grant supports from Bristol-Meyer Squibb, Merck, iTeos, Amgen, Gradalis, and Halozyme. L.Z.’s laboratory receives the royalty for licensing GVAX to Aduro Biotech. L.Z. received the consultant fee from Biosynergies, NovaRock, Merck, and Astrozeneca.
References
- Institute National Cancer Biomarkers. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/biomarker (accessed on 27 July 2018).
- Badreddine, R.; Wang, K.K. Biomarkers in gastrointestinal cancers. Am. J. Gastroenterol. 2008, 103, 2106–2110. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, E.; Metaxa-Mariatou, V.; Tsaousis, G.; Tsoulos, N.; Tsirigoti, A.; Efstathiadou, C.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Bourkoula, E.; et al. Molecular predictive markers in tumors of the gastrointestinal tract. World J. Gastrointest. Oncol. 2016, 8, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Deschoolmeester, V.; Lardon, F.; Pauwels, P.; Peeters, M. Biomarkers in Gastrointestinal Cancer: Focus on Colon, Pancreas and Gastric Cancer; InTech: London, UK, 2012. [Google Scholar]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (toga): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Bang, Y.J.; Feng-Yi, F.; Xu, J.M.; Lee, K.W.; Jiao, S.C.; Chong, J.L.; Lopez-Sanchez, R.I.; Price, T.; Gladkov, O.; et al. HER2 screening data from toga: Targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer 2015, 18, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Okines, A.; Cunningham, D.; Chau, I. Targeting the human EGFR family in esophagogastric cancer. Nat. Rev. Clin. Oncol. 2011, 8, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.S.; Cheung, W.Y. A contemporary review of the treatment landscape and the role of predictive and prognostic biomarkers in pancreatic adenocarcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Murala, S.; Alli, V.; Kreisel, D.; Gelman, A.E.; Krupnick, A.S. Current status of immunotherapy for the treatment of lung cancer. J. Thorac. Dis. 2010, 2, 237–244. [Google Scholar] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.A.; Wolchok, J.D.; Snyder, A. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2015, 373, 1984. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–pd-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazama, S.; Tamada, K.; Yamaguchi, Y.; Kawakami, Y.; Nagano, H. Current status of immunotherapy against gastrointestinal cancers and its biomarkers: Perspective for precision immunotherapy. Ann. Gastroenterol. Surg. 2018, 2, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Gorris, M.A.J.; Halilovic, A.; Rabold, K.; van Duffelen, A.; Wickramasinghe, I.N.; Verweij, D.; Wortel, I.M.N.; Textor, J.C.; de Vries, I.J.M.; Figdor, C.G. Eight-color multiplex immunohistochemistry for simultaneous detection of multiple immune checkpoint molecules within the tumor microenvironment. J. Immunol. 2018, 200, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, K.; Wakabayashi-Nakao, K.; Ohshima, K.; Sakura, N.; Yamaguchi, K.; Mochizuki, T. Novel protein isoforms of carcinoembryonic antigen are secreted from pancreatic, gastric and colorectal cancer cells. BMC Res. Notes 2013, 6, 381. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, P.; Govindan, N.; Yusoff, M.M.; Ichwan, S.J.A. Proteins are potent biomarkers to detect colon cancer progression. Saudi J. Biol. Sci. 2017, 24, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, R.H. Carcinoembryonic antigen. Ann. Intern. Med. 1986, 104, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, J.P.; Koehl, C.; Adloff, M. Carcinoembryonic antigen (cea) in diagosis and prognosis of colorectal carcinoma. Dis. Colon Rectum 1980, 23, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.J.; Mitchell, E.P. Carcinoembryonic antigen in the staging and follow-up of patients with colorectal cancer. Cancer Investig. 2005, 23, 338–351. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Mangu, P.B.; Flynn, P.J.; Korde, L.; Loprinzi, C.L.; Minsky, B.D.; Petrelli, N.J.; Ryan, K.; Schrag, D.H.; Wong, S.L.; et al. Follow-up care, surveillance protocol, and secondary prevention measures for survivors of colorectal cancer: American society of clinical oncology clinical practice guideline endorsement. J. Clin. Oncol. 2013, 31, 4465–4470. [Google Scholar] [CrossRef] [PubMed]
- Pleskow, D.K.; Berger, H.J.; Gyves, J.; Allen, E.; McLean, A.; Podolsky, D.K. Evaluation of a serologic marker, ca19-9, in the diagnosis of pancreatic cancer. Ann. Intern. Med. 1989, 110, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Ćwik, G.; Wallner, G.; Skoczylas, T.; Ciechański, A.; Zinkiewicz, K. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch. Surg. 2006, 141, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Paganuzzi, M.; Onetto, M.; Marroni, P.; Barone, D.; Conio, M.; Aste, H.; Pugliese, V. Ca 19-9 and Ca 50 in benign and malignant pancreatic and biliary diseases. Cancer 1988, 61, 2100–2108. [Google Scholar] [CrossRef]
- Maithel, S.K.; Maloney, S.; Winston, C.; Gönen, M.; D’Angelica, M.I.; DeMatteo, R.P.; Jarnagin, W.R.; Brennan, M.F.; Allen, P.J. Preoperative ca 19-9 and the yield of staging laparoscopy in patients with radiographically resectable pancreatic adenocarcinoma. Ann. Surg. Oncol. 2008, 15, 3512–3520. [Google Scholar] [CrossRef] [PubMed]
- Bartley, A.N.; Washington, M.K.; Colasacco, C.; Ventura, C.B.; Ismaila, N.; Benson, A.B., 3rd; Carrato, A.; Gulley, M.L.; Jain, D.; Kakar, S.; et al. HER2 testing and clinical decision making in gastroesophageal adenocarcinoma: Guideline from the college of american pathologists, american society for clinical pathology, and the american society of clinical oncology. J. Clin. Oncol. 2017, 35, 446–464. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-B.; Rhim, J.S.; Park, S.-C.; Kimm, S.-W.; Kraus, M.H. Amplification, overexpression, and rearrangement of the erbB-2 protooncogene in primary human stomach carcinomas. Cancer Res. 1989, 49, 6605–6609. [Google Scholar] [PubMed]
- Yonemura, Y.; Ninomiya, I.; Ohoyama, S.; Kimura, H.; Yamaguchi, A.; Fushida, S.; Kosaka, T.; Miwa, K.; Miyazaki, I.; Endou, Y.; et al. Expression of c-erbB-2 oncoprotein in gastric carcinoma. Immunoreactivity for c-erbB-2 protein is an independent indicator of poor short-term prognosis in patients with gastric carcinoma. Cancer 1991, 67, 2914–2918. [Google Scholar] [CrossRef]
- Yonemura, Y.; Yamaguchi, A.; Fushida, S.; Kimura, H.; Ohoyama, S.; Miyazaki, I.; Endou, Y.; Tanaka, M.; Sasaki, T. Evaluation of immunoreactivity for erbB-2 protein as a marker of poor short term prognosis in gastric cancer. Cancer Res. 1991, 51, 1034–1038. [Google Scholar] [PubMed]
- Yoon, H.H.; Shi, Q.; Sukov, W.R.; Wiktor, A.E.; Khan, M.; Sattler, C.A.; Grothey, A.; Wu, T.T.; Diasio, R.B.; Jenkins, R.B.; et al. Association of HER2/ERBB2 expression and gene amplification with pathologic features and prognosis in esophageal adenocarcinomas. Clin. Cancer Res. 2012, 18, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Bandla, S.; Godfrey, T.E.; Tan, D.; Luketich, J.D.; Pennathur, A.; Qiu, X.; Hicks, D.G.; Peters, J.H.; Zhou, Z. HER2 amplification, overexpression and score criteria in esophageal adenocarcinoma. Mod. Pathol. 2011, 24, 899–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Renzo, M.F.; Olivero, M.; Giacomini, A.; Porte, H.; Chastre, E.; Mirossay, L.; Nordlinger, B.; Bretti, S.; Bottardi, S.; Giordano, S.; et al. Overexpression and amplification of the MET/HGF receptor gene during the progression of colorectal cancer. Clin. Cancer Res. 1995, 1, 147–154. [Google Scholar] [PubMed]
- Bradley, C.A.; Salto-Tellez, M.; Laurent-Puig, P.; Bardelli, A.; Rolfo, C.; Tabernero, J.; Khawaja, H.A.; Lawler, M.; Johnston, P.G.; Van Schaeybroeck, S.; et al. Targeting c-MET in gastrointestinal tumours: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 150. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Sawada, H.; Yamada, Y.; Watanabe, A.; Tatsumi, M.; Yamashita, J.; Matsuda, M.; Sakaguchi, T.; Hirao, T.; Nakano, H. The prognostic significance of amplification and overexpression of c-MET and c-ERB b-2 in human gastric carcinomas. Cancer 1999, 85, 1894–1902. [Google Scholar] [CrossRef]
- Yu, S.; Yu, Y.; Zhao, N.; Cui, J.; Li, W.; Liu, T. C-MET as a prognostic marker in gastric cancer: A systematic review and meta-analysis. PLoS ONE 2013, 8, e79137. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Fujimoto, J.; Suzuki, T.; Yamamoto, H.; Okamoto, E. Expression of hepatocyte growth factor and its receptor, the c-met proto-oncogene, in hepatocellular carcinoma. Hepatology 1997, 25, 619–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucali, P.; Santoro, A.; Rodriguez-Lope, C.; Simonelli, M.; Camacho, L.; Senzer, N.; Bolondi, L.; Lamar, M.; Abbadessa, G.; Schwartz, B. Final results from ARQ 197-114: A phase ib safety trial evaluating ARQ 197 in cirrhotic patients (PTS) with hepatocellular carcinoma (HCC). J. Clin. Oncol. 2010, 28, 4137. [Google Scholar] [CrossRef]
- Rimassa, L.; Porta, C.; Borbath, I.; Daniele, B.; Salvagni, S.; Van Laethem, J.L.; Van Vlieberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A. Tivantinib (ARQ 197) versus placebo in patients (PTS) with hepatocellular carcinoma (HCC) who failed one systemic therapy: Results of a randomized controlled phase II trial (RCT). Am. Soc. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the HGF/C-met pathway in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Weekes, C.D.; Clark, J.W.; Zhu, A.X. Tivantinib for advanced hepatocellular carcinoma: Is met still a viable target? Lancet Oncol. 2018, 19, 591–592. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of met-high, advanced hepatocellular carcinoma (metiv-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Souglakos, J.; Philips, J.; Wang, R.; Marwah, S.; Silver, M.; Tzardi, M.; Silver, J.; Ogino, S.; Hooshmand, S.; Kwak, E.; et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br. J. Cancer 2009, 101, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreyev, H.J.; Norman, A.R.; Cunningham, D.; Oates, J.R.; Clarke, P.A. Kirsten ras mutations in patients with colorectal cancer: The multicenter “rascal” study. J. Natl. Cancer Inst. 1998, 90, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.H.; Tougeron, D.; Shi, Q.; Alberts, S.R.; Mahoney, M.R.; Nelson, G.D.; Nair, S.G.; Thibodeau, S.N.; Goldberg, R.M.; Sargent, D.J.; et al. KRAS codon 12 and 13 mutations in relation to disease-free survival in braf–wild-type stage III colon cancers from an adjuvant chemotherapy trial (n0147 alliance). Clin. Cancer Res. 2014, 20, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of kras mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Tejpar, S.; Stintzing, S.; Ciardiello, F.; Tabernero, J.; Van Cutsem, E.; Beier, F.; Esser, R.; Lenz, H.J.; Heinemann, V. Prognostic and predictive relevance of primary tumor location in patients with ras wild-type metastatic colorectal cancer: Retrospective analyses of the crystal and fire-3 trials. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.; Nicolaides, N.C.; Wei, Y.F.; Ruben, S.M.; Carter, K.C.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; Adams, M.D.; et al. Mutation of a mutl homolog in hereditary colon cancer. Science 1994, 263, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.C.; Rustgi, A.K. DNA mismatch repair and cancer. Gastroenterology 1995, 109, 1685–1699. [Google Scholar] [CrossRef]
- Farris, A.B., III; Demicco, E.G.; Le, L.P.; Finberg, K.E.; Miller, J.; Mandal, R.; Fukuoka, J.; Cohen, C.; Gaissert, H.A.; Zukerberg, L.R. Clinicopathologic and molecular profiles of microsatellite unstable barrett esophagus-associated adenocarcinoma. Am. J. Surg. Pathol. 2011, 35, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Wotherspoon, A.; Peckitt, C.; Gonzalez, D.; Hulkki-Wilson, S.; Eltahir, Z.; Fassan, M.; Rugge, M.; Valeri, N.; Okines, A.; et al. Mismatch repair deficiency, microsatellite instability, and survival: An exploratory analysis of the medical research council adjuvant gastric infusional chemotherapy (magic) trial. JAMA Oncol. 2017, 3, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Nakata, B.; Wang, Y.Q.; Yashiro, M.; Nishioka, N.; Tanaka, H.; Ohira, M.; Ishikawa, T.; Nishino, H.; Hirakawa, K. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin. Cancer Res. 2002, 8, 2536–2540. [Google Scholar] [PubMed]
- De la Chapelle, A. Microsatellite instability. N. Engl. J. Med. 2003, 349, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Viale, G.; Trapani, D.; Curigliano, G. Mismatch repair deficiency as a predictive biomarker for immunotherapy efficacy. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First Cancer Treatment for Any Solid Tumor with a Specific Genetic Feature. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm560167.htm (accessed on 27 July 2018).
- Diaz, L.A.; Le, D.T.; Yoshino, T.; Andre, T.; Bendell, J.C.; Koshiji, M.; Zhang, Y.; Kang, S.P.; Lam, B.; Jaeger, D. Phase 3, open-label, randomized study of first-line pembrolizumab (pembro) vs. investigator-choice chemotherapy for mismatch repair-deficient (dmmr) or microsatellite instability-high (MSI-H) metastatic colorectal carcinoma (MCRC): Keynote-177. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Frank, A.S.; Qian, S.; Andrew, B.N.; Mody, K.; Levasseur, A.; Dueck, A.C.; Asha, R.D.; Christopher, H.L.; Deirdre, J.C.; Federico, I.R.; et al. Randomized trial of folfox alone or combined with atezolizumab as adjuvant therapy for patients with stage III colon cancer and deficient DNA mismatch repair or microsatellite instability (atomic, alliance a021502). J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zhu, Y.; Jiang, J.; Zhao, J.; Zhang, X.G.; Xu, N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-l1) in gastric carcinoma and its clinical significance. Acta Histochem. 2006, 108, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.-L. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L. PD-L1 expression in pancreatic cancer. JNCI J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Perez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (keynote-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. First-line nivolumab in stage iv or recurrent non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage iii melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 clinical keynote-059 trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Accelerated Approval to Pembrolizumab for Advanced Gastric Cancer. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm577093.htm (accessed on 27 July 2018).
- Mahalingam, D. The coming of age: Immunotherapy in gastrointestinal malignancies. J. Gastrointest. Oncol. 2018, 9, 140–142. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (checkmate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhang, J.; Wolfgang, C.L.; Zheng, L. Precision immuno-oncology: Prospects of individualized immunotherapy for pancreatic cancer. Cancers (Basel) 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Z.; Tian, H.; Li, Y.; Li, M.; Zhao, W.; Zhang, C.; Wang, T.; Liu, J.; Zhang, A.; et al. Circulating MIC-1/GDF15 is a complementary screening biomarker with cea and correlates with liver metastasis and poor survival in colorectal cancer. Oncotarget 2017, 8, 24892–24901. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.; Kerr, K.; Yatabe, Y.; Hirsch, F.R. PL 03.03 blueprint 2: PD-L1 immunohistochemistry comparability study in real-life, clinical samples. J. Thorac. Oncol. 2017, 12, S1606. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; van Gisbergen, K.P.J.M.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Iseki, Y.; Shibutani, M.; Maeda, K.; Nagahara, H.; Fukuoka, T.; Matsutani, S.; Kashiwagi, S.; Tanaka, H.; Hirakawa, K.; Ohira, M. A new method for evaluating tumor-infiltrating lymphocytes (TILs) in colorectal cancer using hematoxylin and eosin (he)-stained tumor sections. PLoS ONE 2018, 13, e0192744. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.N.; Kang, B.W.; Kwon, O.K.; Park, K.B.; Lee, S.S.; Chung, H.Y.; Yu, W.; Bae, H.I.; Jeon, S.W.; Kang, H. Intratumoural PD-L1 expression is associated with worse survival of patients with epstein–barr virus-associated gastric cancer. Br. J. Cancer 2017, 117, 1753. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; He, J.-C.; Yang, Y.; Wang, J.-M.; Qian, Y.-W.; Yang, T.; Ji, L. The prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 7525. [Google Scholar] [CrossRef] [PubMed]
- Gataa, I.; Mezquita, L.; Auclin, E.; Le Moulec, S.; Alemany, P.; Kossai, M.; Massé, J.; Caramella, C.; Remon Masip, J.; Lahmar, J.; et al. 112PPathological evaluation of tumor infiltrating lymphocytes and the benefit of nivolumab in advanced non-small cell lung cancer (NSCLC). Ann. Oncol. 2017, 28. [Google Scholar] [CrossRef] [Green Version]
- Wallden, B.; Pekker, I.; Popa, S.; Dowidar, N.; Sullivan, A.; Hood, T.; Danaher, P.; Mashadi-Hossein, A.; Lunceford, J.K.; Marton, M.J.; et al. Development and analytical performance of a molecular diagnostic for anti-PD1 response on the ncounter dx analysis system. J. Clin. Oncol. 2016, 34. [Google Scholar]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. Ifn-γ–related mrna profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Hulbert, A.; Pierce, R.H.; Greenberg, P.D.; Hingorani, S.R. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol. Res. 2017, 5, 978–991. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, E.W.; Wong, S.C.; Ong, S.M.; Chong, D.Q.; Ling, K.L. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma s100a8/a9 proinflammatory proteins. J. Immunol. 2013, 190, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.-S.; Lee, H.-M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the TH2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Kondo, Y.; Kimura, O.; Morosawa, T.; Fujisaka, Y.; Umetsu, T.; Kogure, T.; Inoue, J.; Nakagome, Y.; Shimosegawa, T. PD-L1+mdscs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci. Rep. 2016, 6, 39296. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Iwasa, Y.; Lengauer, C.; Nowak, M.A. Dynamics of colorectal cancer. Semin. Cancer Biol. 2005, 15, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Richards, C.H.; McMillan, D.C.; Horgan, P.G.; Roxburgh, C.S.D. The relationship between tumour stroma percentage, the tumour microenvironment and survival in patients with primary operable colorectal cancer. Ann. Oncol. 2014, 25, 644–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Ham, I.-H.; Son, S.Y.; Han, S.-U.; Kim, Y.-B.; Hur, H. Intratumor stromal proportion predicts aggressive phenotype of gastric signet ring cell carcinomas. Gastric Cancer 2017, 20, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, A.; Tollenaar, R.A.E.M.; Pelt, G.W.; Zeestraten, E.C.M.; Dutton, S.; McConkey, C.C.; Domingo, E.; Smit, V.T.H.B.M.; Midgley, R.; Warren, B.F.; et al. The proportion of tumor-stroma as a strong prognosticator for stage II and III colon cancer patients: Validation in the victor trial. Ann. Oncol. 2013, 24, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ma, W.; Wang, J.; Yu, L.; Zhang, X.; Wang, Z.; Tan, B.; Wang, N.; Bai, B.; Yang, S.; et al. Tumor-stroma ratio is an independent predictor for survival in esophageal squamous cell carcinoma. J. Thorac. Oncol. 2012, 7, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Grabsch, H.; Ivanova, T.; Tan, I.B.; Murray, J.; Ooi, C.H.; Wright, A.I.; West, N.P.; Hutchins, G.G.A.; Wu, J.; et al. Comprehensive genomic meta-analysis identifies intra-tumoural stroma as a predictor of survival in patients with gastric cancer. Gut 2013, 62, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFB drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.F.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H.; et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Xiu, J.; Weinberg, B.A.; El-Deiry, W.S.; Weiner, L.M.; Gatalica, Z.; Liu, Z.; El Ghazaly, H.; Xiao, N.; Hwang, J.J.; et al. Characterization of tumor mutation burden (TMB) in gastrointestinal (GI) cancers. J. Clin. Oncol. 2017, 35, 530. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wick, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, L.; Fan, H.; Wang, H.; Luber, B.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Forbes, M.E.; Bitting, R.L.; O’Neill, S.S.; Chou, P.C.; Topaloglu, U.; Miller, L.D.; Hawkins, G.A.; Grant, S.C.; DeYoung, B.R.; et al. Incorporating blood-based liquid biopsy information into cancer staging: Time for a tnmb system? Ann. Oncol. 2018, 29, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.-A.; Bethel, K.; Králíčková, M.; Kuhn, P. Circulating tumor cells: Fluid surrogates of solid tumors. Annu. Rev.Pathol. 2017, 12, 419–447. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Scholer, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Chan, C.W.; Chan, K.C.; Cheng, S.H.; Wong, J.; Wong, V.W.; Wong, G.L.; Chan, S.L.; Mok, T.S.; Chan, H.L.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating tumor DNA changes for early monitoring of anti-pd1 immunotherapy: A proof-of-concept study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zang, W.; Ge, Y.; Weygant, N.; Yu, P.; Li, L.; Rao, G.; Jiang, Z.; Yan, R.; He, L.; et al. RAS/BRAF circulating tumor DNA mutations as a predictor of response to first-line chemotherapy in metastatic colorectal cancer patients. Can. J. Gastroenterol. Hepatol. 2018, 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Dandachi, N.; Hofmann, G.; Samonigg, H.; Loibner, H.; Obwaller, A.; van der Kooi, A.; Tibbe, A.G.; Doyle, G.V.; Terstappen, L.W.; et al. Comparison of two methods for enumerating circulating tumor cells in carcinoma patients. Cytom. B Clin. Cytom. 2005, 68, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiroya, T.; Yuko, K. Circulating tumor cells in gastrointestinal cancer. J. Hepato-Biliary-Pancreat. Sci. 2010, 17, 577–582. [Google Scholar]
- Li, Y.; Gong, J.; Zhang, Q.; Lu, Z.; Gao, J.; Li, Y.; Cao, Y.; Shen, L. Dynamic monitoring of circulating tumour cells to evaluate therapeutic efficacy in advanced gastric cancer. Br. J. Cancer 2016, 114, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotelo, M.J.; Sastre, J.; Maestro, M.L.; Veganzones, S.; Viéitez, J.M.; Alonso, V.; Grávalos, C.; Escudero, P.; Vera, R.; Aranda, E.; et al. Role of circulating tumor cells as prognostic marker in resected stage iii colorectal cancer. Ann. Oncol. 2015, 26, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.J.; Uhr, J.W.; Terstappen, L.W.M.M. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sullivan, R.J.; Kalinich, M.; Kwan, T.T.; Giobbie-Hurder, A.; Pan, S.; LiCausi, J.A.; Milner, J.D.; Nieman, L.T.; Wittner, B.S.; et al. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 2467–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating tumor DNA analysis in patients with cancer: American society of clinical oncology and college of american pathologists joint review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Ignatiadis, M.; Lee, M.; Jeffrey, S.S. Circulating tumor cells and circulating tumor DNA: Challenges and opportunities on the path to clinical utility. Clin. Cancer Res. 2015, 21, 4786–4800. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using exoscreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Kidess-Sigal, E.; Liu, H.E.; Triboulet, M.M.; Che, J.; Ramani, V.C.; Visser, B.C.; Poultsides, G.A.; Longacre, T.A.; Marziali, A.; Vysotskaia, V.; et al. Enumeration and targeted analysis of kras, BRAF and PIK3CA mutations in ctcs captured by a label-free platform: Comparison to ctdna and tissue in metastatic colorectal cancer. Oncotarget 2016, 7, 85349–85364. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, J.E.; Grover, P.; Winter, M.; Hewett, P.J.; Price, T.J.; Thierry, B. Detection and clinical significance of circulating tumor cells in colorectal cancer—20 years of progress. Mol. Med. 2015, 21, S25–S31. [Google Scholar] [CrossRef] [PubMed]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta BBA 2015, 1848, 2685–2702. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Basha, R.; Rajitha, B.; Alese, O.B.; Alam, A.; Pattnaik, S.; El-Rayes, B. Aquaporins: Their role in gastrointestinal malignancies. Cancer Lett. 2016, 373, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Pelagalli, A.; Squillacioti, C.; Mirabella, N.; Meli, R. Aquaporins in Health and Disease: An Overview Focusing on the Gut of Different Species. Int. J. Mol. Sci. 2016, 17, 1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Biomarkers in tumor microenvironment. PD-L1—programmed death-1 ligand-1.

{kind=link}
Table 1.
Major molecular markers in clinical application.
| Molecule | Tumor Type | Implication |
|---|---|---|
| Tumor markers | ||
| CEA | Colorectal, gastric, and pancreatic cancers | Indicating residual disease, progressive, or recurrent disease |
| Measuring treatment response | ||
| CA19-9 | Pancreatic cancer | Indicating residual disease, progressive, or recurrent disease |
| Measuring treatment response | ||
| Targets of matched therapies | ||
| HER2 | Gastric or esophagogastric-junction cancers | Selecting for targeted therapy |
| KRAS | Colorectal, gastric, and pancreatic cancers | Predicting for treatment unresponsiveness |
| Mismatch repair Genes | ||
| MMR | Solid tumors | Predicting for treatment responsiveness |
| Biomarkers in tumor microenvironment | ||
| PD-L1 expression | Gastric cancer | Enriching patient population responding to anti-PD-1/PD-L1 therapies |
CEA—carcinoembryonic antigen; MMR—mismatch repair; PD-L1—programmed death ligand 1; PD-1—programmed death-1.
Table 2.
Characteristics of HER2, c-Met, and KRAS expression in gastrointestinal (GI) cancers.
| Molecule | Genomic Alterations | Pathways Involved | Cancer types | Treatment |
|---|---|---|---|---|
| HER2 | Amplification/ overexpression | Activation of the MAPK and the PI3K/AKT axis | Gastric or esophagogastric-junction cancers | Monoclonal antibodies (e.g., cetuximab and trastuzumab) |
| c-MET | Amplification/ overexpression | Activation of GRB2-SOS–RAS–MAPK, the PI3K/AKT axis, and STAT3 pathway | Colorectal cancer, gastric cancer, pancreatic cancers and hepatocellular carcinoma | Monoclonal antibodies (e.g., rilotumumab, ficlatuzumab, and TAK-701); Tyrosine kinase inhibitors (e.g., tivantinib, cabozantinib, and crizotinib) |
| KRAS | Activating mutation within catalytic RAS domain | RAS–RAF–MEK | Colorectal cancer | Downstream pathway inhibitors (e.g., MEK inhibitors selumetinib and trametinib) |
MAPK—mitogen-activated protein kinase; GRB2—growth factor receptor-bound protein 2; STAT—signal transducer and activator of transcription; PI3K—the p85 subunit of phosphatidylinositol 3-kinase; SOS—son of sevenless homologue 1.
Table 3.
New development of biomarkers.
| Molecule | Tumor Type | Implication |
|---|---|---|
| Biomarkers in tumor microenvironment | ||
| PD-L1 expression | Other cancer types, except gastric cancer | Enriching patient population responding to anti-PD-1/PD-L1 therapies |
| Tumor infiltrating lymphocyte | Colon and gastric cancers | Indicating good prognosis |
| Immunosuppressive myeloid cells | Pancreatic, hepatocellular, and gastric cancers | Indicating poor prognosis |
| Intratumoral stroma | Gastric, pancreatic, esophageal, and colon cancers | Indicating poor prognosis |
| Biomarkers in tumor genomics | ||
| Targeted gene panels | Pan-cancer | Selecting patients for targeted therapies |
| Mutational burden | Pan-cancer | Enriching patient population responding to anti-PD-1/PD-L1 therapies |
| Biomarkers in liquid biopsies | ||
| ctDNA/CTC/Exosomes | Pan-cancer | Indicating residual disease, progressive, or recurrent disease |
| Measuring treatment response | ||
CTC—circulating tumor cells.
Table 4.
Advantages, disadvantages of ctDNA, CTC, and exosome as biomarkers.
| Approaches | Advantages | Disadvantages | References |
|---|---|---|---|
| ctDNA | Higher sensitivity; quick renew/short half-life; maintain tumor-specific genomic aberrations | Not suitable for functional assay, noises from normal cell-free DNA, challenges in methods’ standardization | [134,138,139] |
| CTC | Allow morphological/molecular/functional study; potentials for therapeutic targets | Low specificity, particularly in early stage setting; challenges in methods’ standardization limited capture techniques | [121,127,128,134,140] |
| Exosomes | Higher sensitivity; higher serum concentration; diverse EV contents; Potential for therapeutic targets | Isolation and purification of exosomes; specific exosome marker to identify subset of EVs; not suitable for functional assay; challenges in methods’ standardization | [135,136,137] |
EV—extracellular vesicles.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, J.; Quadri, S.; Wolfgang, C.L.; Zheng, L. New Development of Biomarkers for Gastrointestinal Cancers: From Neoplastic Cells to Tumor Microenvironment. Biomedicines 2018, 6, 87. https://doi.org/10.3390/biomedicines6030087
AMA Style
Zhang J, Quadri S, Wolfgang CL, Zheng L. New Development of Biomarkers for Gastrointestinal Cancers: From Neoplastic Cells to Tumor Microenvironment. Biomedicines. 2018; 6(3):87. https://doi.org/10.3390/biomedicines6030087
Chicago/Turabian StyleZhang, Jiajia, Shafat Quadri, Christopher L. Wolfgang, and Lei Zheng. 2018. "New Development of Biomarkers for Gastrointestinal Cancers: From Neoplastic Cells to Tumor Microenvironment" Biomedicines 6, no. 3: 87. https://doi.org/10.3390/biomedicines6030087
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.