HIF Oxygen Sensing Pathways in Lung Biology


1
Research Unit, Hospital of Santa Cristina, Research Institute Princesa (IP), Autonomous University of Madrid, 28009 Madrid, Spain
2
CIBER de Enfermedades Cardiovasculares, Carlos III Health Institute, 28029 Madrid, Spain
*
Authors to whom correspondence should be addressed.
Biomedicines 2018, 6(2), 68; https://doi.org/10.3390/biomedicines6020068
Submission received: 8 May 2018
/
Revised: 28 May 2018
/
Accepted: 30 May 2018
/
Published: 6 June 2018
(This article belongs to the Special Issue Hypoxia)
Abstract
:Cellular responses to oxygen fluctuations are largely mediated by hypoxia-inducible factors (HIFs). Upon inhalation, the first organ inspired oxygen comes into contact with is the lungs, but the understanding of the pulmonary HIF oxygen-sensing pathway is still limited. In this review we will focus on the role of HIF1α and HIF2α isoforms in lung responses to oxygen insufficiency. In particular, we will discuss novel findings regarding their role in the biology of smooth muscle cells and endothelial cells in the context of hypoxia-induced pulmonary vasoconstriction. Moreover, we will also discuss recent studies into HIF-dependent responses in the airway epithelium, which have been even less studied than the HIF-dependent vascular responses in the lungs. In summary, we will review the biological functions executed by HIF1 or HIF2 in the pulmonary vessels and epithelium to control lung responses to oxygen fluctuations as well as their pathological consequences in the hypoxic lung.
Keywords:
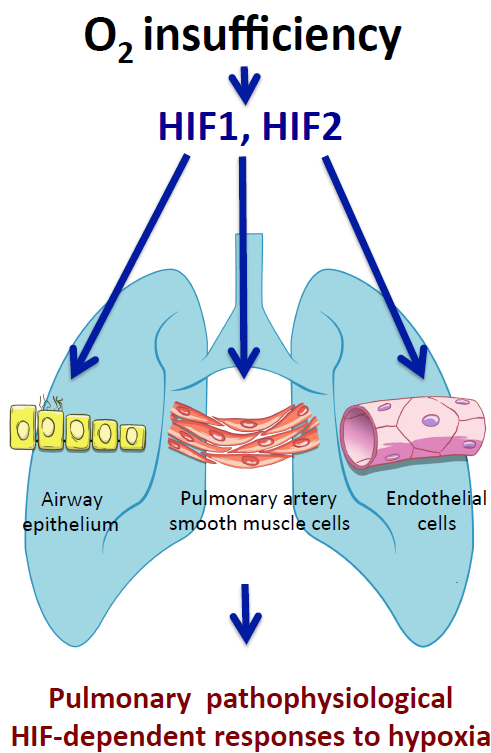
lungs; HIF; oxygen; pulmonary hypertension; hypoxia; airway; endothelium; pulmonary smooth muscle cells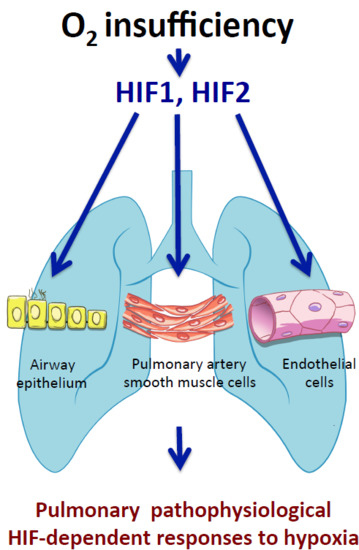
{kind=link}
{kind=link}
1. Introduction
The lungs are responsible for oxygen uptake and the pulmonary vascular responses that ensure adequate blood oxygenation in response to low oxygen tensions. These responses can be initiated in acute conditions of hypoxia (seconds to minutes), involving mechanisms such as membrane depolarization and the influx of calcium into pulmonary artery smooth muscle cells (PASMCs), which leads to elevated pulmonary vascular tone in poorly oxygenated alveoli [1,2,3]. In more prolonged hypoxic conditions (hours to days), the lungs also initiate a response through the hypoxia-inducible factors (HIFs) [4,5].
HIFs are heterodimeric transcription factors and comprised of one HIFα subunit (HIF1α, HIF2α or HIF3α) and a member of the HIFβ family, also known as the aryl hydrocarbon receptor nuclear translocator (ARNT) [6]. While the HIFβ subunit is stable, the stability of the HIFα subunits fluctuates in response to changes in oxygen tension through the prolyl 2-oxoglutarate-dependent Fe2+-dioxygenases PHD1, PHD2, and PHD3 [7,8,9]. In normoxic conditions, PHDs use oxygen to hydroxylate two conserved proline residues in the HIFα subunits and these hydroxylated prolyl residues can be recognized by the VHL (von Hippel-Lindau)/E3 ubiquitin ligase complex, which leads HIFα subunits to proteasomal degradation [9,10]. However, in hypoxic conditions, there is insufficient oxygen for PHDs to hydroxylate the HIFα subunits, which preclude their recognition by VHL leading to HIFα subunits stabilization. Consequently, HIFα subunits can enter the nucleus where they heterodimerize with HIFβ subunits and bind to DNA at hypoxia response elements (HREs), thereby driving a HIF-dependent transcriptional program [11,12].
While this molecular machinery is certainly operative in lung tissue [13,14,15], it has been less explored here than in other biological scenarios, which is surprising considering the fundamental role of the lungs in oxygen homeostasis. In conditions of hypoxia, both HIF1α and HIF2α are stabilized in the lungs [13,14,15]. In particular, Wiesener et al. showed that—in contrast to other tissues such as liver or kidney—pulmonary HIF2α expression is induced in hypoxia in a more larger extent than upon exposure to CO [14], which suggests that HIF activation in the lungs (possibly mainly in the airway epithelium) largely relies directly on the oxygen supply in the airway more than on red blood cell oxygen delivery.
In this review we focus on recent in vivo studies that have shed new light on the role of HIF1α and HIF2α isoforms in the response of the pulmonary vascular and airway epithelium to oxygen fluctuations (hypoxia).
2. The HIF Oxygen Sensing Pathways in the Pulmonary Vasculature
A well-recognized physiological response to acute or sustained hypoxia is the constriction of small pulmonary arteries, which is driven by hypoxia-dependent pathways in pulmonary artery smooth muscle cells (PASMCs) and endothelial cells [1,2,16,17,18]. In the lung, this physiological mechanism (termed hypoxic pulmonary vasoconstriction) has evolved to restrict blood flow to the alveoli that are less oxygenated, redirecting the blood towards the better oxygenated ones. However, when these hypoxic conditions become chronic and systemic, and when the oxygenation of virtually every alveolus is compromised, this physiological response becomes pathological and results in sustained pulmonary hypertension (PH). This pathological scenario is defined as an increase in the mean pulmonary arterial pressure above 25 mmHg at rest [19]. PH is characterized by increased smooth muscle coverage (muscularization) of the pulmonary arteries, with a subsequent increase in pulmonary vascular resistance that ultimately leads to right ventricular (RV) hypertrophy and heart failure [1]. PH can be initiated in different pathological settings and hypoxia-dependent PH has been specifically categorized as “pulmonary hypertension due to lung diseases and/or hypoxia” [20,21].
2.1. Chronic Activation of the HIF Pathways Leading to Pulmonary Hypertension
A large body of evidence has shown that sustained environmental hypoxia can induce PH through the HIF pathway. Indeed, the first evidence of the role of HIF in PH in vivo came from HIF-deficient animals. Mice carrying a heterozygous germline deletion of Hif1a or Hif2a exposed to chronic hypoxia showed impaired PH development, in part due to the more limited pulmonary vascular remodeling [16,17]. Further evidence in humans and mice about the role of HIF signaling in PH come from studies about the pathological inherited mutations resulting in HIF overactivation, which leads to PH. Indeed, homozygosity for the VHL allele harboring the mutation R200W, which degrades HIFα subunits less efficiently than the WT VHL, is associated with HIF2α-dependent polycythemia and PH [22]. Moreover, patients and mice carrying a gain of function HIF2α mutation (G537W in human or G536W in mice) develop severe PH, as well as exacerbated erythrocytosis [23,24].
These initial studies highlighted the contribution of the HIF pathway to hypoxia-induced hypertension. As discussed below, more recent in vivo studies have provided new insights into the relative contribution of HIF1α and HIF2α isoforms in different pulmonary cell types and their specific contribution to PH, as well as identifying the downstream HIF-dependent events relevant to this pathological scenario.
2.2. The HIF1α Activity in Pulmonary Smooth Muscle Cells Associated with Hypoxia-Induced Pulmonary Hypertension
Although the seminal studies indicated above provided evidence of the role HIF in PH, until recently the contribution of specific lung cell types to these responses remained unclear. In the last few years, transgenic animals targeting specific cell types has unveiled the role of HIF isoforms in PH. Pulmonary vascular remodeling is considered necessary for the development of hypoxia-induced PH and it involves the appearance of smooth muscle-like cells in vessels of the alveolar wall that is not normally muscularized, as well as the medial and adventitial thickening of the muscle and elastic vessels [25]. The main cell types involved in hypoxia driven vascular remodeling under are thought to be PASMCs and endothelial cells, although the role of other cells like pericytes, fibroblasts, macrophages or airway epithelial cells cannot be ruled out.
Although the role of HIF in PASMCs and endothelial cells has not been thoroughly explored in vivo, the current experimental data seems robust enough to conclude that HIF activity in endothelial and PASMC are key to understand hypoxia-induced vascular remodeling and PH (Figure 1). Interestingly, Yu et al. showed that cultured PASMCs can express some detectable HIF1α signal under normoxia, which can be further enhanced in hypoxia [13]. The role for HIF1 in PASMCs in the vascular remodeling, muscularization and PH has been found upon Hif1a gene inactivation in SMCs using Mhy11-CreERT2 mouse line [26]. Surprisingly, the attenuation of vascular remodeling and PH was not reflected by an attenuation of right ventricle cardiac hypertrophy, which is in line with previous data showing that increased PH might not fully explain right heart failure [27,28]. An additional study has used Pdgfrb-CreERT2 mice in order to inactivate Hif1a in previously identified pulmonary PDGFRβ+/αSMC+ progenitors that are primed in hypoxia-induced PH [29,30]. These mice show that distal arterioles are less covered by α-smooth muscle positive cells and a profound attenuation of PH and right ventricular hypertrophy [29]. The differences between these two studies can arise by the different Cre lines employed. In this line, Pdgfrb-CreERT2 can inactivate Hif1a not only in those pulmonary PDGFRβ+/αSMC+ progenitors but also in other mural populations such as heart PDGFR-β+ pericytes that could also contribute to HIF1α-dependent RV hypertrophy [29]. Finally, another study have revealed that Hif1a inactivation in PASMCs using SM22-Cre mice also shows a trend towards reduced artery muscularization when these mice are exposed to hypoxia [31]. Yet surprisingly, a parallel role of HIF1α in reducing vascular tone by repressing myosin light chain phosphorylation was also shown [31]. In summary, HIF1 is clearly a driver of hypoxia-induced PH and the size and features of this effect may vary depending on the Cre line use, and which other cells different than PASMC can be targeted. Finally, it will also be crucial to study whether HIF2 activity in PASMCs also contributes to hypoxia-induced PH, which as yet remains unexplored.
2.3. The Endothelial HIF2 Pathway in Hypoxia-Induced Pulmonary Hypertension
As indicated previously, global Hif2a inactivation supports its role in hypoxia-induced PH [17]. More recent studies revealed that the relevance of HIF2α in pulmonary vascular remodeling depends on its expression in endothelial cells [32,33,34,35] (Figure 1). Specific inactivation of Hif2a in endothelial cells using the Cdh5 (VE-Cadherin)-Cre mouse line was sufficient to block the development of hypoxia-induced PH [32]. Furthermore, inactivation of the Phd2 gene in endothelial cells using the same mouse Cre line (mimicking PHD2 inactivation in hypoxia) promoted HIF1α and HIF2α stabilization under normoxic conditions. More importantly, the loss of Phd2 was sufficient to enhance pulmonary muscularization and right ventricular systolic pressure (RVSP), leading to right ventricular hypertrophy and premature death [32]. While Hif1a inactivation had no effect over the aforementioned phenotype, simultaneous Hif2a inactivation rescued PH and right ventricular hypertrophy in endothelial Phd2 knockout mice. Similar results were obtained using a different mouse line expressing Cre recombinase under the control of the Tie2 promoter [33]. Furthermore, when Hif2a but not Hif1a is specifically deleted in pulmonary endothelial cells using the L1-Cre mouse line, the development of hypoxia-induced PH and RV hypertrophy was abrogated [34]. In summary all these studies point out an essential role of endothelial cell HIF2 activity in hypoxia-induced PH. However, a recent study have shown that Hif1a gene inactivation in endothelial cells using Cdh5 (VE-Cadherin)-Cre mouse line can compromise hypoxia-induced PH [29]. This study proposes that HIF1 activity induces PDGF-B in endothelial cells that ultimately can impact on PASMC cells to promote KLF14-dependent PASMC expansion (Figure 1). The reasons of this discrepancy of this study is unclear and possibly could be related differences between hypoxia exposure and PHD2 inactivation but this certainly warrants further investigation.
These studies also explored possible HIF2-target genes in endothelial cells that might be responsible for the role of endothelial HIF2 in hypoxia-induced PH. Several mechanisms have been proposed to explain the role of HIF2 in pulmonary vasculature remodeling and hypertension. A well-recognized HIF2-dependent gene in lung tissue is endothelin-1 (ET-1) [17,22,23,24], which is a potent vasoconstrictor [36]. Indeed, ET1 expression is markedly elevated in response to hypoxia in a HIF2-dependent manner, as well as in mice harboring a human HIF2α gain-of-function mutation and in a model of Chuvash disease [17,24,37]. More recently, endothelial HIF2 was shown to be responsible not only for an elevation in ET-1 but also for the concomitant downregulation of the apelin receptor that mediates apelin-dependent vasodilatation [32,38]. ET-1 and apelin receptors have been identified in PASMC [39,40], and therefore these data suggest that endothelial HIF2 triggers different mechanisms that—in a paracrine manner—potentially favor PASMC-mediated vasoconstriction. An additional proposed mechanism based on the crosstalk between endothelial cells and PASMCs is the secretion of endothelial CXCL12, which is released from endothelial cells upon HIF2-activation and promotes the expansion of PASMCs [33]. Importantly, increased RVSP and RV hypertrophy in endothelial Phd2-deficient mice (see above) is abrogated when Phd2 and Cxcl12 are simultaneously inactivated in endothelial cells. On the other hand, Cowburn et al. observed that mutant mice with a conditional deletion of Hif2a in pulmonary endothelial cells showed a decreased expression of the enzymes Arginase 1 and 2 [34]. As these enzymes have been implicated in promoting vascular remodeling by reducing airway nitric oxide availability [41,42], it was hypothesized that HIF2-dependent arginase activity was involved in hypoxia-induced PH. Indeed, Arginase 1 gene inactivation in the pulmonary endothelium of animals exposed to chronic hypoxia reduces RVSP and RV hypertrophy development [34]. Interestingly, Cowburn also analyzed endothelial progenitor cells from PAH patients, which show increased arginase expression levels that can be further enhanced by hypoxia [34,42,43], suggesting that PH patients can develop a more robust vascular response to hypoxia. Finally, a recent study also showed that HIF2 induces the thromboposdin-1 (TSP-1) expression in hypoxic lung tissue [44], which can also contribute to PH given that TSP1 deficient mice are partially protected from hypoxia-induced pulmonary artery muscularization [45,46]. As mentioned previously, these mechanisms are not necessarily mutually exclusive. Indeed, it is tempting to think that the most likely scenario is one where all of the aforementioned mechanisms are implicated together, and that they synergize and cooperate to promote HIF2-dependent PH.
In summary, preclinical data suggest that HIF1 is the main player in PASMCs, favoring hypoxia-induced PH, while the relevance of HIF2 in this pathological lung scenario is mediated by its effects on endothelial cells-induced vascular constriction.
3. Role of HIF Pathway in the Airway Epithelium
As can be seen above, the relevance of the HIF pathway in lung biology has largely been studied in the pulmonary vascular bed (endothelial cells and pulmonary vascular SMCs), although the relevance of this pathway in airway biology has also been shown. Seminal studies initially show that HIF2α is essential for lung development and for airway biology in particular. Indeed, Hif2a deficient mice succumb to a respiratory distress syndrome in which the mice become cyanotic as a consequence of failed lung expansion. At the molecular level, HIF2α governs airway surfactant expression by regulating the vascular endothelial growth factor (VEGF-A), which ultimately controls alveolar epithelium maturation [47]. However, the responses to HIF signaling in the hypoxic airway epithelium in adulthood remain largely unknown, although there is some evidence that HIF influences proliferation in the airway and provides protection from hypoxia.
The HIF Pathway in Airway Epithelium Proliferation
Although the role of HIF signaling has been mainly been explored in pulmonary vascular function, several studies have also highlighted the importance of HIF1α and HIF2α isoforms in the pulmonary epithelium. Immunohistological analysis showed nuclear HIF2α expression in the bronchial epithelial cells of hypoxic mice (10% O2) where induces the expression of well-recognized HIF2α-dependent genes in lung tissue, such as resistin-like α (RELMα, also named HIMF or FIZZ1) [48,49]. Human RELMβ (RELMα in mice) is a soluble factor that has been identified as a mitogenic factor for bronchial epithelial cells in vitro [50]. Indeed, HIF2 induces marked proliferation of bronchial epithelial cells after exposure to hypoxia (3–4 days), mainly in club cells [49] and possibly via RELMα. However, HIF2-dependent club cell proliferation may also be governed by cell autonomous mechanisms. Indeed, our own studies have shown that HIF2 acts as an activator of the mammalian target of rapamycin complex 1 (mTORC1) in clear cell renal cell carcinoma (ccRCC). The mTORC1 functions as a serine/threonine kinase that responds to amino acid availability and the energy status of the cell, playing a central role in cell growth and proliferation [51,52,53]. In fact, HIF2α-dependent mTORC1 activation is driven by the amino acid carrier SLC7A5 (also named LAT-1), provoking ccRCC proliferation [51]. We have also shown that this HIF2-Slc7a5-mTORC1 axis is operative in bronchial epithelial cells, suggesting its role in hypoxia-induced club cell proliferation [49,51]. Furthermore, HIF2 also induces the expression of FOXM1, a transcription factor that is critical for club cell proliferation and differentiation [54,55]. Overall, these data suggest that HIF2 fosters bronchial epithelial cell proliferation in hypoxia through cell autonomous mechanisms like mTORC1 and FOXM1 activation, as well as non-cell autonomous mechanisms such as those driven possibly by RELMα.
Cell proliferative responses to hypoxia are not restricted to club cells in the bronchial epithelium; they have also been found in the pulmonary neuroepithelial bodies (NEBs). These structures are presumed airway sensors, made-up of innervated clusters of amine (serotonin) and peptide-producing cells that reside in the bronchial epithelium [56,57]. In humans and in animal models, hypoxia leads to hyperplasia of NEBs, and PHD1 and PHD3 have been implicated in this response [56,58,59]. Indeed, Phd1- and Phd3-deficient mice show marked hypertrophia and NEB proliferation, in parallel to the induction of HIF1α in some of synaptophysin (SYP) positive cells typically found in NEBs [56,59]. However, it is unclear whether or not HIF1 contributes to NEB proliferation in response to hypoxia and whether NEB hyperproliferation is driven by HIF2, as in club cells. Nevertheless, NEBs are presumed to be polymodal airway sensors analogous to the carotid body (CB) glomus cells [3,60] and recently, the HIF2-mTORC1 proliferative pathway was shown to be involved in chronic hypoxia-induced enlargement of the carotid body (CB) [61,62]. Hence, the HIF2-mTORC1 pathway might not only drive hypoxia-dependent CB and club cell proliferation but also, hypoxia-dependent NEB hyperplasia. Finally, it should also be considered that CB enlargement and NEB hyperplasia in response to hypoxia are currently thought to be adaptive responses to hypoxia, potentiating ventilatory responses. Therefore, it will be relevant to understand the physiological meaning of this additional response involving HIF2α-dependent bronchial epithelium proliferation and how this might be integrated in the context of the adaptive response to systemic hypoxia. Moreover, further experimentation in human pathophysiological scenarios will be necessary to unveil clinically relevant aspects of the HIF oxygen-sensing pathway in human airway biology.
Finally, it might be considered that HIF2-dependent actions in bronchial epithelium could contribute not only for local airway adaptation to hypoxia, but also to more distant pulmonary vessel remodeling described above. In this line, hypoxia-induced RELM-α (largely produced in bronchial epithelium) causes PH characterized by an increase in mean pulmonary artery pressure, pulmonary vascular resistance, right heart hypertrophy, and vascular remodeling caused by chronic hypoxia as well as lung vascular inflammation [63,64]. At molecular level, RELMα also recruits bone marrow-derived macrophages, promotes IL-6 expression in PASMC and macrophages through HIF1α and favors vascular remodeling associated to PH [63,64]. These data suggest a possible HIF2α-dependent communication between airway epithelium and vascular bed, which can also contribute to HIF-dependent vascular remodeling and PH in the hypoxic lung.
4. Concluding Remarks and Perspectives
In this review we have focused on the HIF1 and HIF2-dependent responses that are confined to the pulmonary vascular bed and airway epithelium. However, it should be noted that some of the genes induced by the HIF pathway are soluble factors that may be implicated in vascular and epithelium communication. As such, HIF2-dependent soluble factors released by the airway epithelium (i.e., RELMα) may act in synergy with other effectors of HIF2 in endothelial cells to drive pathological vascular remodeling in PH. Inactivation of the Hif2a gene in different cell types in the airway epithelium will help to understand the contribution of local airway responses driven by HIF2 to distant vascular remodeling in PH. Moreover, many of the studies into the role of HIFs in lung biology have focused on pathological PH in chronic hypoxia (weeks and months). Thus, a greater effort to explore the short-term HIF-dependent responses to hypoxia (possibly non-pathological) will be necessary to define the novel molecular and cellular adaptive responses to pulmonary oxygen fluctuations.
Author Contributions
A.A.U. and J.A. conceived and wrote the manuscript.
Acknowledgments
This work was supported by grants from Ministerio de Educación y Ciencia (SAF2013-46058-R; SAF2016-76815).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 2016, 47, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.K.; López-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen-sensing mechanisms. N. Engl. J. Med. 2005, 353, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Schermuly, R.T.; Brandes, R.P.; Weissmann, N. Molecular mechanisms of hypoxia-inducible factor-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J. Physiol. 2016, 594, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.; Frid, M.G.; Shimoda, L.A.; Wiener, C.M.; Stenmark, K.; Semenza, G.L. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am. J. Physiol. 1998, 275, L818–L826. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: Implication of natural antisense HIF-1alpha. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Investig. 1999, 103, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Compernolle, V.; Tjwa, M.; Wiesener, M.S.; Maxwell, P.H.; Collen, D.; Carmeliet, P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Investig. 2003, 111, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Dean, H.P. The Pulmonary Circulation. J. Physiol. 1894, 16, 34–158. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; McLaughlin, V.V.; Dalaan, A.M.; Satoh, T.; Galiè, N. Treatment of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 323–336. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Smith, T.G.; Brooks, J.T.; Balanos, G.M.; Lappin, T.R.; Layton, D.M.; Leedham, D.L.; Liu, C.; Maxwell, P.H.; McMullin, M.F.; McNamara, C.J.; et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006, 3, e290. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; Harten, S.K.; Reid, C.D.; Tuddenham, E.G.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.; Lee, F.S. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.K.; Waypa, G.B.; Mungai, P.T.; Nielsen, J.M.; Czech, L.; Dudley, V.J.; Beussink, L.; Dettman, R.W.; Berkelhamer, S.K.; Steinhorn, R.H.; et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am. J. Respir. Crit. Care Med. 2014, 189, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Yuan, J.X. Hypoxia-inducible factor-1α in pulmonary arterial smooth muscle cells and hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Barnes, E.A.; Alvira, C.M.; Ying, L.; Reddy, S.; Cornfield, D.N. Hypoxia-inducible factor-1α in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ. Res. 2013, 112, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell Biol. 2016, 36, 1584–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaço, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zeng, H.; Xie, X.J.; Tao, Y.K.; He, X.; Roman, R.J.; Aschner, J.L.; Chen, J.X. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget 2016, 7, 58848–58861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, M.M.; Richardson, T.; Wang, T.; Mosqueira, M.; Arguiri, E.; Yu, H.; Yu, Q.C.; Solomides, C.C.; Morrisey, E.E.; Khurana, T.S.; et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J. Clin. Investig. 2010, 120, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Cruden, N.L.; Amer, D.A.; Li, V.K.; Goudie, E.B.; Johnston, N.R.; Sharma, S.; Neilson, I.; Webb, D.J.; Megson, I.L.; et al. Vascular effects of apelin in vivo in man. J. Am. Coll. Cardiol. 2008, 52, 908–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davie, N.; Haleen, S.J.; Upton, P.D.; Polak, J.M.; Yacoub, M.H.; Morrell, N.W.; Wharton, J. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 2002, 165, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Kleinz, M.J.; Davenport, A.P. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul. Pept. 2004, 118, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Dhaliwal, R.; Ivanovska, J.; Kantores, C.; McNamara, P.J.; Scott, J.A.; Belik, J.; Jankov, R.P. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L503–L510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; XiangLi, X.; Niu, H.; Wang, H.; Jia, P.; Gong, W.; Wu, D.; Qin, W.; Xing, C. Arginase inhibitor attenuates pulmonary artery hypertension induced by hypoxia. Mol. Cell. Biochem. 2016, 412, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Labrousse-Arias, D.; Castillo-González, R.; Rogers, N.M.; Torres-Capelli, M.; Barreira, B.; Aragonés, J.; Cogolludo, Á.; Isenberg, J.S.; Calzada, M.J. HIF-2α-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc. Res. 2016, 109, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.M.; Bauer, E.M.; Rogers, N.M.; Yao, M.; Feijoo-Cuaresma, M.; Pilewski, J.M.; Champion, H.C.; Zuckerbraun, B.S.; Calzada, M.J.; Isenberg, J.S. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc. Res. 2012, 93, 682–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, C.D.; Yu, L.; Al-Ansari, E.; Hales, C.A.; Quinn, D.A. Thrombospondin-1 null mice are resistant to hypoxia-induced pulmonary hypertension. J. Cardiothorac. Surg. 2010, 5, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compernolle, V.; Brusselmans, K.; Acker, T.; Hoet, P.; Tjwa, M.; Beck, H.; Plaisance, S.; Dor, Y.; Keshet, E.; Lupu, F.; et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002, 8, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.F.; Hellberg, A.K.; Balenger, S.; Depping, R.; Dodd-O, J.; Johns, R.A.; Li, D. Hypoxia-induced mitogenic factor has antiapoptotic action and is upregulated in the developing lung: Coexpression with hypoxia-inducible factor-2alpha. Am. J. Respir. Cell Mol. Biol. 2004, 31, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Torres-Capelli, M.; Marsboom, G.; Li, Q.O.; Tello, D.; Rodriguez, F.M.; Alonso, T.; Sanchez-Madrid, F.; García-Rio, F.; Ancochea, J.; Aragonés, J. Role Of Hif2α Oxygen Sensing Pathway In Bronchial Epithelial Club Cell Proliferation. Sci. Rep. 2016, 6, 25357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Meng, Q.; Wu, H.; Eid, G.; Zhang, G.; Zhang, X.; Yang, S.; Huang, K.; Lee, T.H.; Corrigan, C.J.; et al. Resistin-like molecule-β is a human airway remodelling mediator. Eur. Respir. J. 2012, 39, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Elorza, A.; Soro-Arnáiz, I.; Meléndez-Rodríguez, F.; Rodríguez-Vaello, V.; Marsboom, G.; de Cárcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordóñez, A.; Serrano-Oviedo, L.; et al. HIF2α acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schmelzle, T.; Hall, M.N. TOR, a central controller of cell growth. Cell 2000, 103, 253–262. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Ustiyan, V.; Wert, S.E.; Ikegami, M.; Wang, I.C.; Kalin, T.V.; Whitsett, J.A.; Kalinichenko, V.V. Foxm1 transcription factor is critical for proliferation and differentiation of Clara cells during development of conducting airways. Dev. Biol. 2012, 370, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.C.; Zhang, Y.; Snyder, J.; Sutherland, M.J.; Burhans, M.S.; Shannon, J.M.; Park, H.J.; Whitsett, J.A.; Kalinichenko, V.V. Increased expression of FoxM1 transcription factor in respiratory epithelium inhibits lung sacculation and causes Clara cell hyperplasia. Dev. Biol. 2010, 347, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Bishop, T.; Ratcliffe, P.J.; Yeger, H.; Cutz, E. Hyperplasia and hypertrophy of pulmonary neuroepithelial bodies, presumed airway hypoxia sensors, in hypoxia-inducible factor prolyl hydroxylase-deficient mice. Hypoxia 2016, 4, 69–80. [Google Scholar] [PubMed]
- Cutz, E.; Pan, J.; Yeger, H.; Domnik, N.J.; Fisher, J.T. Recent advances and contraversies on the role of pulmonary neuroepithelial bodies as airway sensors. Semin. Cell Dev. Biol. 2013, 24, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Gosney, J.R. Pulmonary neuroendocrine cells in species at high altitude. Anat. Rec. 1993, 236, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Livermore, S.; Pan, J.; Yeger, H.; Ratcliffe, P.; Bishop, T.; Cutz, E. Augmented 5-HT Secretion in Pulmonary Neuroepithelial Bodies from PHD1 Null Mice. Adv. Exp. Med. Biol. 2015, 860, 309–313. [Google Scholar] [PubMed]
- López-Barneo, J.; González-Rodríguez, P.; Gao, L.; Fernández-Agüera, M.C.; Pardal, R.; Ortega-Sáenz, P. Oxygen sensing by the carotid body: Mechanisms and role in adaptation to hypoxia. Am. J. Physiol. Cell Physiol. 2016, 310, C629–C642. [Google Scholar] [CrossRef] [PubMed]
- Hodson, E.J.; Nicholls, L.G.; Turner, P.J.; Llyr, R.; Fielding, J.W.; Douglas, G.; Ratnayaka, I.; Robbins, P.A.; Pugh, C.W.; Buckler, K.J.; et al. Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF-2 pathway. J. Physiol. 2016, 594, 1179–1195. [Google Scholar] [CrossRef] [PubMed]
- Macias, D.; Cowburn, A.S.; Torres-Torrelo, H.; Ortega-Sáenz, P.; López-Barneo, J.; Johnson, R.S. HIF-2α is essential for carotid body development and function. Elife 2018, 7, e34681. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Skinner, J.T.; Champion, H.C.; Crow, M.T.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L582–L593. [Google Scholar] [CrossRef] [PubMed]
- Johns, R.A.; Takimoto, E.; Meuchel, L.W.; Elsaigh, E.; Zhang, A.; Heller, N.M.; Semenza, G.L.; Yamaji-Kegan, K. Hypoxia-Inducible Factor 1α Is a Critical Downstream Mediator for Hypoxia-Induced Mitogenic Factor (FIZZ1/RELMα)-Induced Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 134–144. [Google Scholar] [CrossRef] [PubMed]
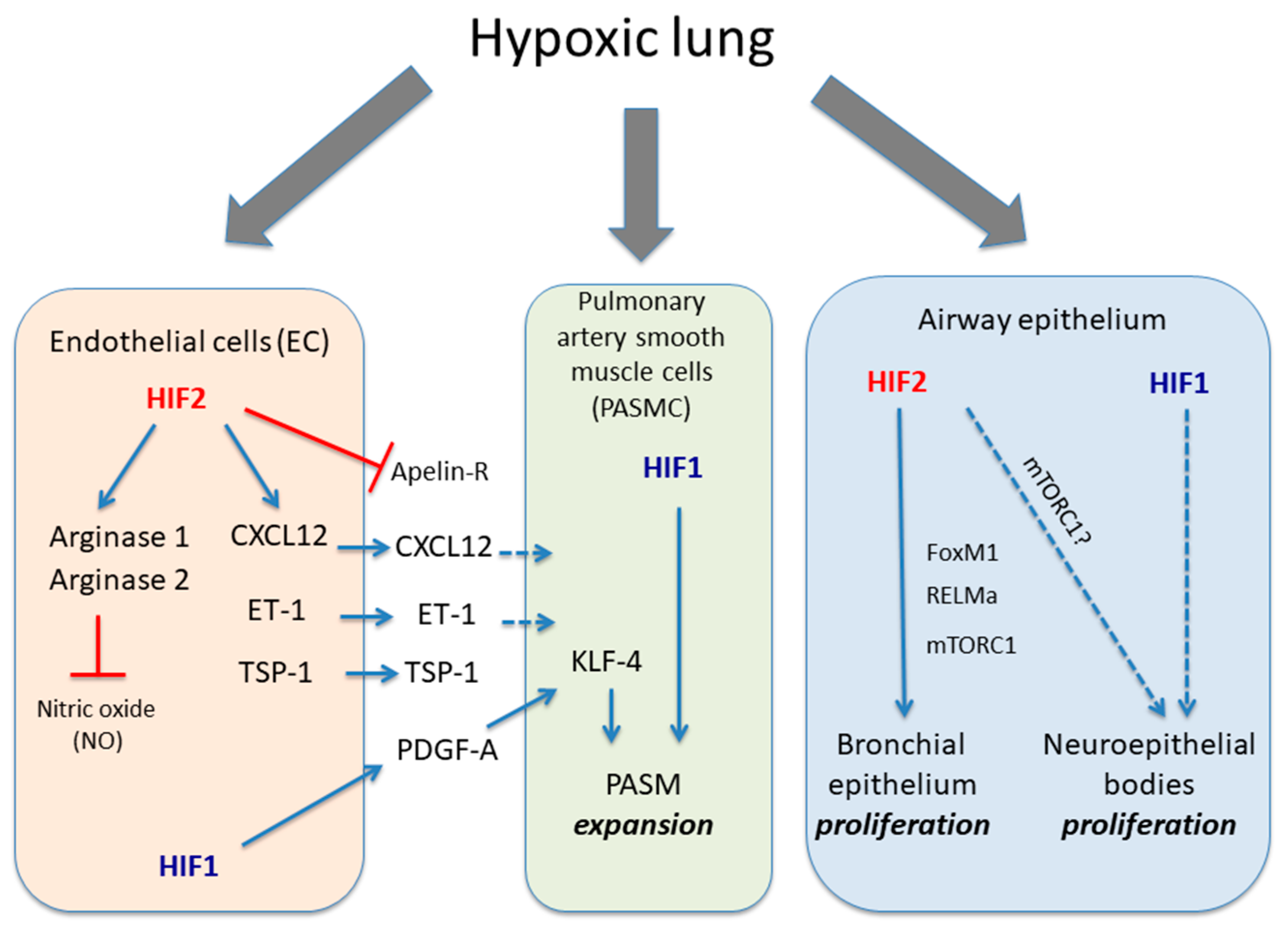
Figure 1.
Role of HIF1α and HIF2α isoforms in different lung cell types in response to hypoxia. The figure shows the biological actions executed of HIF1α and HIF2α isoforms in endothelial cells, PASMC and airway epithelium in response to hypoxia. Endothelial HIF2 induces a series of responses leading to vasoconstriction such as induction of arginase activity, which limit endothelial NO availability as well as release CXCL12, ET-1 and TSP1 in parallel with an inhibition of apelin receptor. HIF1 activity in endothelial cells can also release PDGF-B, which cooperates through KLF4 with PASMC HIF1 activity to promote their expansion. The figure also shows HIF2 activity in bronchial epithelial proliferation through potential mediators such as mTORC1, RELMα or FOXM1 as well as the potential role of HIF1α in neuroepithelial bodies proliferation located in pulmonary epithelium. Solid dark blue lines indicate activatory pathways; dashed dark blue lines indicate potential activatory actions and red lines indicate inhibitory actions.
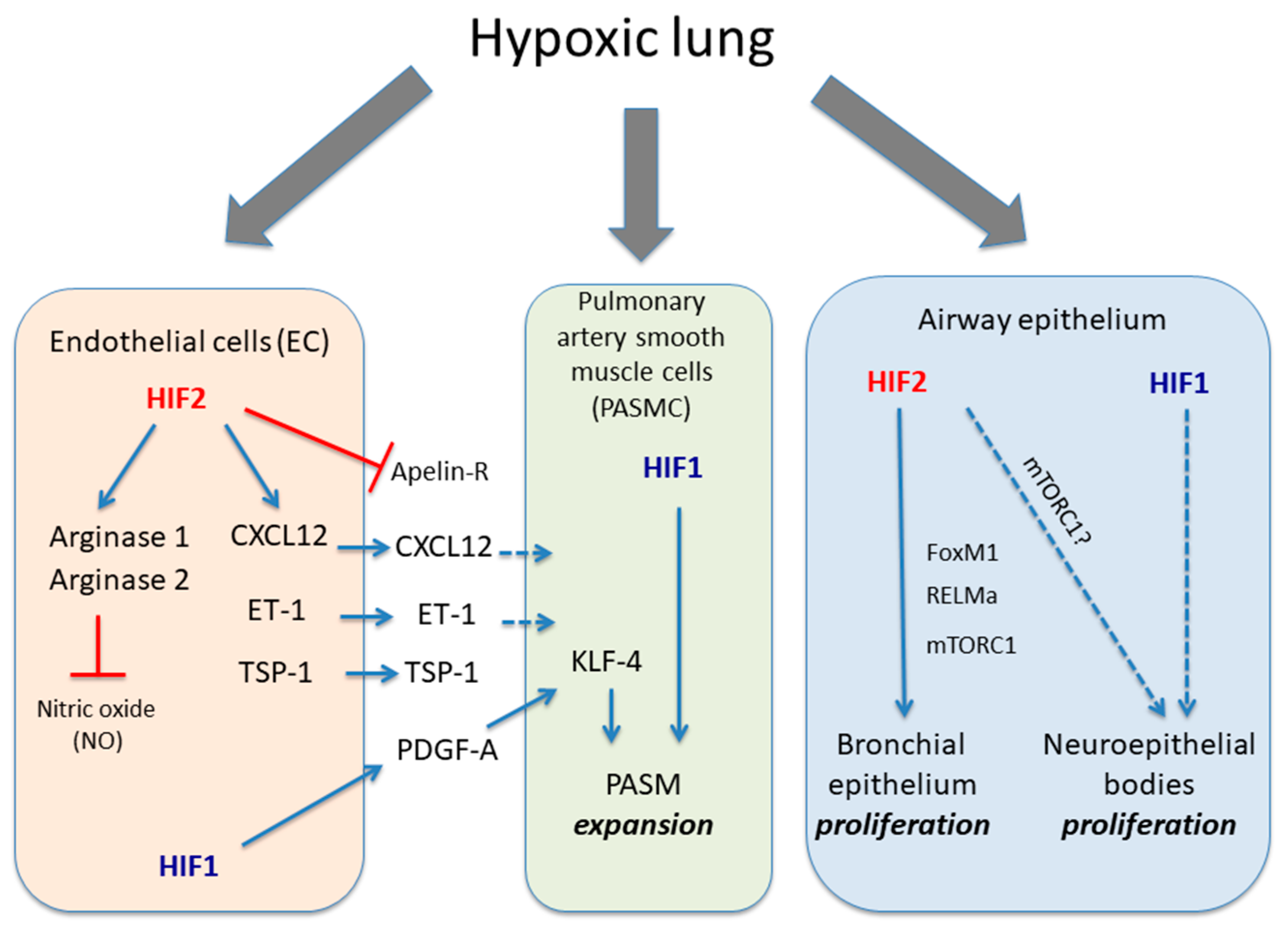
Figure 1.
Role of HIF1α and HIF2α isoforms in different lung cell types in response to hypoxia. The figure shows the biological actions executed of HIF1α and HIF2α isoforms in endothelial cells, PASMC and airway epithelium in response to hypoxia. Endothelial HIF2 induces a series of responses leading to vasoconstriction such as induction of arginase activity, which limit endothelial NO availability as well as release CXCL12, ET-1 and TSP1 in parallel with an inhibition of apelin receptor. HIF1 activity in endothelial cells can also release PDGF-B, which cooperates through KLF4 with PASMC HIF1 activity to promote their expansion. The figure also shows HIF2 activity in bronchial epithelial proliferation through potential mediators such as mTORC1, RELMα or FOXM1 as well as the potential role of HIF1α in neuroepithelial bodies proliferation located in pulmonary epithelium. Solid dark blue lines indicate activatory pathways; dashed dark blue lines indicate potential activatory actions and red lines indicate inhibitory actions.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Urrutia, A.A.; Aragonés, J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines 2018, 6, 68. https://doi.org/10.3390/biomedicines6020068
AMA Style
Urrutia AA, Aragonés J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines. 2018; 6(2):68. https://doi.org/10.3390/biomedicines6020068
Chicago/Turabian StyleUrrutia, Andrés A., and Julián Aragonés. 2018. "HIF Oxygen Sensing Pathways in Lung Biology" Biomedicines 6, no. 2: 68. https://doi.org/10.3390/biomedicines6020068
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.