Roles of NF-κB Signaling in the Regulation of miRNAs Impacting on Inflammation in Cancer

 ,
, 
Abstract
:
1. Introduction
2. NF-κB Signaling Pathway Activation and Its Multifaceted Functional Role in Cancer and Inflammation
2.1. Oncogenic Functions of NF-κB: A Link between Inflammation and Cancer
2.2. Tumor Suppressor Function of NF-κB
3. MiRNAs: Epigenetic Regulators in Inflammation and Cancer
4. General Concept: NF-κB Meets miRNAs
4.1. MiRNAs Regulated by NF-κB
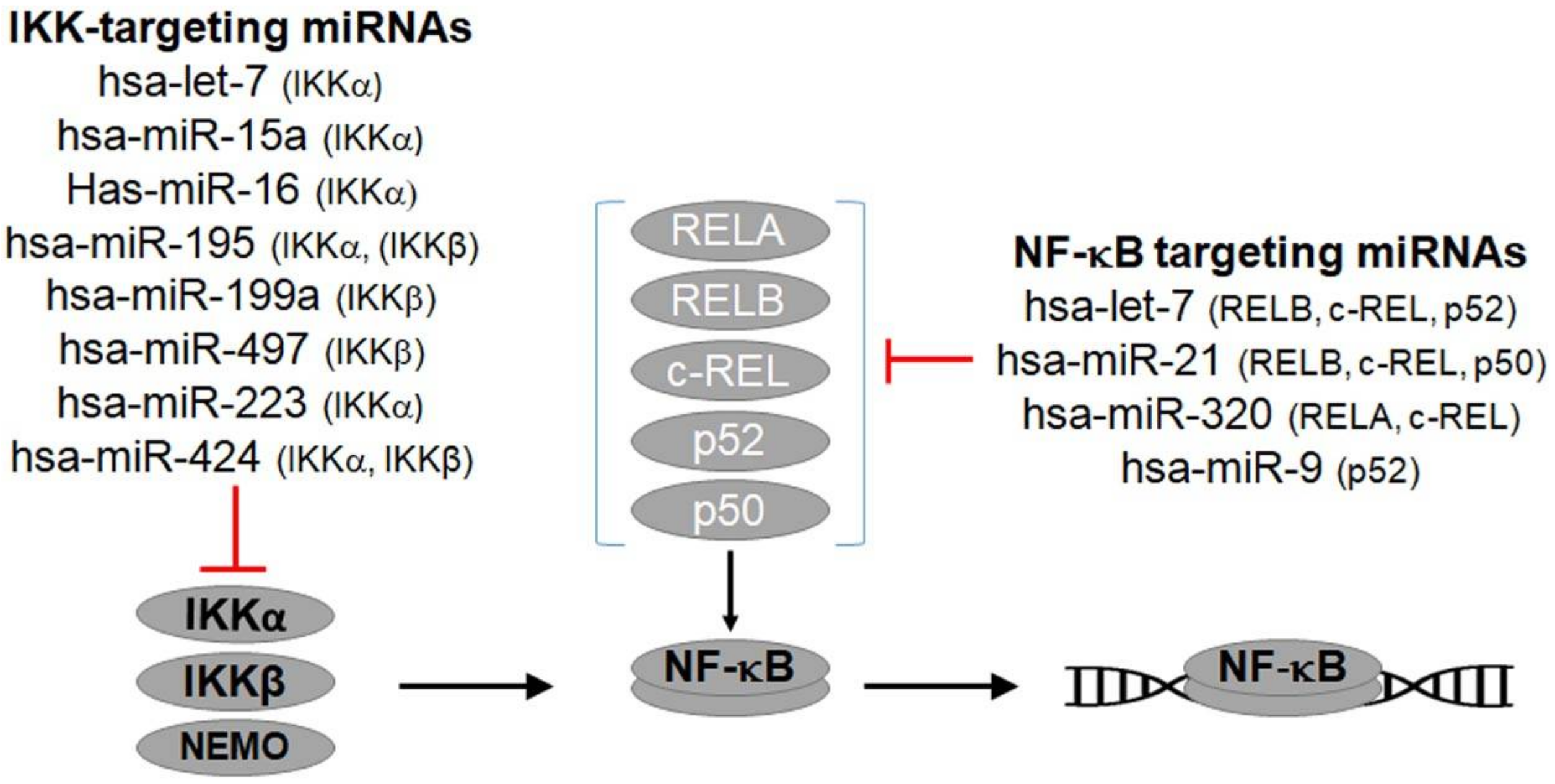
4.2. NF-κB-Regulating miRNAs
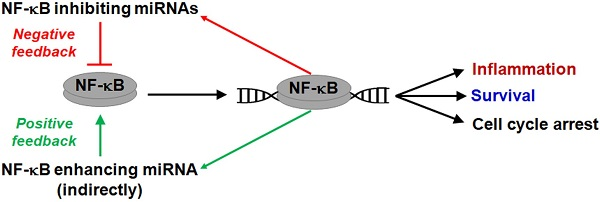
5. NF-κB-miRNA Feedback Loops and Transcriptional Regulatory Networks
6. Final Thoughts: Possible Therapeutic Approaches
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Massa, P.E.; Olivotto, E.; Bianchi, F.; Borzi, R.M.; Hanidu, A.; Li, X.; Li, J.; Marcu, K.B. Sustained NF-κB activation produces a short-term cell proliferation block in conjunction with repressing effectors of cell cycle progression controlled by E2F or FoxM1. J. Cell. Physiol. 2009, 218, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Z.; Bai, L.; Lin, Y. NF-κB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front. Biosci. (Landmark Ed.) 2011, 16, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. Nf-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Nf-κB: Tumor promoter or suppressor? Trends Cell Biol. 2004, 14, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Chavdoula, E.; Hatziapostolou, M.; Polytarchou, C.; Marcu, K.B.; Papavassiliou, A.G.; Sandaltzopoulos, R.; Kolettas, E. A step-by-step microRNA guide to cancer development and metastasis. Cell. Oncol. 2017, 40, 303–339. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Vartholomatos, G.; Tzavaras, T.; Hatziapostolou, M.; Fackelmayer, F.O.; Sandaltzopoulos, R.; Polytarchou, C.; Kolettas, E. Senescence-associated microRNAs target cell cycle regulatory genes in normal human lung fibroblasts. Exp. Gerontol. 2017, 96, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Baltimore, D. MicroRNAs and immunity: Tiny players in a big field. Immunity 2007, 26, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Baltimore, D. MicroRNAs, new effectors and regulators of NF-κB. Immunol. Rev. 2012, 246, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Mehta, A.; Zhao, J.L.; Lee, K.; Marinov, G.K.; Garcia-Flores, Y.; Baltimore, D. An NF-κB-microRNA regulatory network tunes macrophage inflammatory responses. Nat. Commun. 2017, 8, 851. [Google Scholar] [CrossRef] [PubMed]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.J.; Walhout, A.J. The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays 2009, 31, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Chariot, A. The NF-κB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009, 19, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.W.; Baldwin, A.S. Chapter three-IKK/nuclear factor-κB and oncogenesis: Roles in tumor-initiating cells and in the tumor microenvironment. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 121, pp. 125–145. [Google Scholar]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKβ-and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Zaynagetdinov, R.; Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.-S.; McLoed, A.G.; Ausborn, J.A.; Polosukhin, V.V.; Connelly, L.; Zhou, W.; Fingleton, B. Epithelial nuclear factor-κB signaling promotes lung carcinogenesis via recruitment of regulatory T lymphocytes. Oncogene 2012, 31, 3164. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. Nf-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.S.; Scoggins, R.M.; Han, W.; Polosukhin, V.V.; Connelly, L.; Yull, F.E.; Fingleton, B.; Blackwell, T.S. Epithelial NF-κB activation promotes urethane-induced lung carcinogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18514–18519. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-κB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Wullaert, A.; Sasaki, Y.; Schmidt-Supprian, M.; Rajewsky, K.; Roskams, T.; Pasparakis, M. Constitutive IKK2 activation in intestinal epithelial cells induces intestinal tumors in mice. J. Clin. Investig. 2011, 121, 2781–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Cogswell, P.C.; Baldwin, A.S. IKK is a therapeutic target in Kras-induced lung cancer with disrupted p53 activity. Genes Cancer 2014, 5, 41–55. [Google Scholar] [PubMed]
- Kim, C.; Pasparakis, M. Epidermal p65/NF-κB signalling is essential for skin carcinogenesis. EMBO Mol. Med. 2014, 6, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Pasparakis, M.; Kollias, G. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 2015, 212, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. P53 reulates glucose metabolism through an IKK-Nf-κB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Witzel, I.-I.; Perkins, N.D. P53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-κB. Cancer Res. 2011, 71, 5588–5597. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, L.; Thotakura, A.K.; Bennett, J.; Moretti, M.; Franzoso, G. The nuclear factor κB signaling pathway: Integrating metabolism with inflammation. Trends Cell Biol. 2012, 22, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Mantovani, A. Cancer-Related Inflammation: Common Themes and Therapeutic Opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sung, B. Nf-κB in cancer: A matter of life and death. Cancer Discov. 2011, 1, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. Nf-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhou, Y.; Shen, P. NF-κB and its regulation on the immune system. Cell Mol. Immunol. 2004, 1, 343–350. [Google Scholar] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of Nf-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is Nf-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and Nf-κB: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, T.; Perkins, N.D.; Wilson, C.L. NFKB1: A suppressor of inflammation, ageing and cancer. FEBS J. 2016, 283, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, S.J.; Li, H.; Cho, S.-N.; Dishop, M.K.; Wistuba, I.I.; Ji, L.; Kurie, J.M.; Dickey, B.F.; DeMayo, F.J. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease—Like airway inflammation in a k-ras–induced mouse model. Am. J. Respir. Cell Mol. Biol. 2009, 40, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.-W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. Il6 blockade reprograms the lung tumor microenvironment to limit the development and progression of k-ras-mutant lung cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. Il-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.J.; Ratnam, N.M.; Byrd, J.C.; Guttridge, D.C. Nf-κB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 2014, 9, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, H.; Schroering, A.; Ding, J.L.; Lane, W.S.; McGill, G.; Fisher, D.E.; Ding, H.-F. Nf-κB2 p100 is a pro-apoptotic protein with anti-oncogenic function. Nat. Cell Biol. 2002, 4, 888. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Billot, K.; Authier, H.; Bordereaux, D.; Baud, V. Relb inhibits cell proliferation and tumor growth through p53 transcriptional activation. Oncogene 2013, 32, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- De Donatis, G.M.; Le Pape, E.; Pierron, A.; Cheli, Y.; Hofman, V.; Hofman, P.; Allegra, M.; Zahaf, K.; Bahadoran, P.; Rocchi, S.; et al. Nf-κB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene 2016, 35, 2813. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Gao, G.; Li, J.; Huang, H.; Jin, H.; Zhu, J.; Che, X.; Huang, C. Tumor-suppressor NfκB2 p100 interacts with ERK2 and stabilizes PTEN mRNA via inhibition of mir-494. Oncogene 2016, 35, 4080–4090. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Jiang, Q.; Willette-Brown, J.; Xi, S.; Zhu, F.; Burkett, S.; Back, T.; Song, N.Y.; Datla, M.; Sun, Z.; et al. The pivotal role of IKKα in the development of spontaneous lung squamous cell carcinomas. Cancer Cell 2013, 23, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xie, K.; Gou, Q.; Chen, N. IκB kinase α functions as a tumor suppressor in epithelial-derived tumors through an NF-κB-independent pathway (review). Oncol. Rep. 2015, 34, 2225–2232. [Google Scholar] [CrossRef] [PubMed]
- Song, N.-Y.; Zhu, F.; Wang, Z.; Willette-Brown, J.; Xi, S.; Sun, Z.; Su, L.; Wu, X.; Ma, B.; Nussinov, R.; et al. IKKα inactivation promotes Kras-initiated lung adenocarcinoma development through disrupting major redox regulatory pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E812–E821. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. IKKα limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef]
- Liu, B.; Willette-Brown, J.; Liu, S.; Chen, X.; Fischer, S.M.; Hu, Y. IKKα represses a network of inflammation and proliferation pathways and elevates c-myc antagonists and differentiation in a dose-dependent manner in the skin. Cell Death Differ. 2011, 18, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wu, X.; Holzer, R.G.; Lee, J.H.; Todoric, J.; Park, E.J.; Ogata, H.; Gukovskaya, A.S.; Gukovsky, I.; Pizzo, D.P.; et al. Loss of acinar cell IKKα triggers spontaneous pancreatitis in mice. J. Clin. Investig. 2013, 123, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, Y.; Chernishof, V.; Loo, R.R.; Jang, H.; Tahk, S.; Yang, R.; Mink, S.; Shultz, D.; Bellone, C.J.; et al. Proinflammatory stimuli induce IKKα-mediated phosphorylation of pias1 to restrict inflammation and immunity. Cell 2007, 129, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cui, H.; Wang, Z.; Zhang, B.; Ding, J.; Liu, L.; Ding, H.F. Loss of negative feedback control of nuclear factor-κB2 activity in lymphocytes leads to fatal lung inflammation. Am. J. Pathol. 2010, 176, 2646–2657. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G.; De Santa, F. Shaping alternative NF-κB-dependent gene expression programs: New clues to specificity. Cell Death Differ. 2006, 13, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.Y.; Huang, W.; Asagiri, M.; Spann, N.; Hoffmann, A.; Glass, C.; Ghosh, G. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep. 2012, 2, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Kolovos, P.; Georgomanolis, T.; Koeferle, A.; Larkin, J.D.; Brant, L.; Nikolicc, M.; Gusmao, E.G.; Zirkel, A.; Knoch, T.A.; van Ijcken, W.F.; et al. Binding of nuclear factor κB to noncanonical consensus sites reveals its multimodal role during the early inflammatory response. Genome Res. 2016, 26, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Jin, J.; Xiao, Y.; Chang, J.H.; Yu, J.; Hu, H.; Starr, R.; Brittain, G.C.; Chang, M.; Cheng, X.; Sun, S.C. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-κB signaling. Nat. Immunol. 2012, 13, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the relb gene is regulated by NF-κB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Tchenio, T.; Piton, G.; Romeo, P.H.; Baud, V. Rela repression of relb activity induces selective gene activation downstream of tnf receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 14635–14640. [Google Scholar] [CrossRef] [PubMed]
- Madge, L.A.; May, M.J. Classical NF-κB activation negatively regulates noncanonical NF-κB-dependent cxcl12 expression. J. Biol. Chem. 2010, 285, 38069–38077. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Wang, W.; Wallach, D. Receptor-specific signaling for both the alternative and the canonical NF-κB activation pathways by NF-κB-inducing kinase. Immunity 2004, 21, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Davis, R.E.; Ngo, V.N.; Lenz, G.; Wright, G.; Xu, W.; Zhao, H.; Yu, X.; Dang, L.; Staudt, L.M. Compensatory IKKα activation of classical NF-κB signaling during IKKβ inhibition identified by an RNA interference sensitization screen. Proc. Natl. Acad. Sci. USA 2008, 105, 20798–20803. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Pujari, R.; Harhaj, N.S.; Abbott, D.W.; Harhaj, E.W. The kinase IKKα inhibits activation of the transcription factor NF-κB by phosphorylating the regulatory molecule tax1bp1. Nat. Immunol. 2011, 12, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, C.; Thome, M. IKKα takes control of canonical NF-κB activation. Nat. Immunol. 2011, 12, 815–816. [Google Scholar] [CrossRef] [PubMed]
- Gloire, G.; Horion, J.; El Mjiyad, N.; Bex, F.; Chariot, A.; Dejardin, E.; Piette, J. Promoter-dependent effect of IKKα on NF-κB/p65 DNA binding. J. Biol. Chem. 2007, 282, 21308–21318. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Kawauchi, K.; Tanaka, N. IKK/Nf-κB signaling pathway inhibits cell-cycle progression by a novel rb-independent suppression system for E2F transcription factors. Oncogene 2008, 27, 5696. [Google Scholar] [CrossRef] [PubMed]
- Sfikas, A.; Batsi, C.; Tselikou, E.; Vartholomatos, G.; Monokrousos, N.; Pappas, P.; Christoforidis, S.; Tzavaras, T.; Kanavaros, P.; Gorgoulis, V.G.; et al. The canonical NF-κB pathway differentially protects normal and human tumor cells from ros-induced DNA damage. Cell. Signal. 2012, 24, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Batsi, C.; Markopoulou, S.; Vartholomatos, G.; Georgiou, I.; Kanavaros, P.; Gorgoulis, V.G.; Marcu, K.B.; Kolettas, E. Chronic NF-κB activation delays rasv12-induced premature senescence of human fibroblasts by suppressing the DNA damage checkpoint response. Mech. Ageing Dev. 2009, 130, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin d1. Mol. Cell Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Krishnan, V.; O’Malley, B.W.; Yamamoto, Y.; Gaynor, R.B. Formation of an IKKα-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol. Cell 2005, 18, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IκB kinase promotes tumorigenesis through inhibition of forkhead foxo3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Chiu, C.F.; Chang, Y.W.; Kuo, K.T.; Shen, Y.S.; Liu, C.Y.; Yu, Y.H.; Cheng, C.C.; Lee, K.Y.; Chen, F.C.; Hsu, M.K.; et al. NF-κB-driven suppression of foxo3a contributes to egfr mutation-independent gefitinib resistance. Proc. Natl. Acad. Sci. USA 2016, 113, E2526–E2535. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. Gadd45β mediates the Nf-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wuerzberger-Davis, S.M.; Chang, P.Y.; Berchtold, C.; Miyamoto, S. Enhanced G2-M arrest by nuclear factor- κB-dependent p21waf1/cip1 induction. Mol. Cancer Res. 2005, 3, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.Y.; Miyamoto, S. Nuclear factor-κB dimer exchange promotes a p21waf1/cip1 superinduction response in human T leukemic cells. Mol. Cancer Res. 2006, 4, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Batsi, C.; Markopoulou, S.; Kontargiris, E.; Charalambous, C.; Thomas, C.; Christoforidis, S.; Kanavaros, P.; Constantinou, A.I.; Marcu, K.B.; Kolettas, E. Bcl-2 blocks 2-methoxyestradiol induced leukemia cell apoptosis by a p27(kip1)-dependent g1/s cell cycle arrest in conjunction with NF-κB activation. Biochem. Pharmacol. 2009, 78, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of NF-κB by akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Textor, B.; Pein, O.T.; Licht, A.H.; Andrecht, S.; Sator-Schmitt, M.; Fusenig, N.E.; Angel, P.; Schorpp-Kistner, M. Critical role for nf-κB-induced junb in vegf regulation and tumor angiogenesis. EMBO J. 2007, 26, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-κB and epithelial to mesenchymal transition of cancer. J. Cell Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.M.; Swingler, T.E.; Sampieri, C.L.; Edwards, D.R. The regulation of matrix metalloproteinases and their inhibitors. Int. J. Biochem. Cell Biol. 2008, 40, 1362–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanjul-Fernandez, M.; Folgueras, A.R.; Cabrera, S.; Lopez-Otin, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 2010, 1803, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Parks, W.C.; Wilson, C.L.; Lopez-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta 2017, 1864, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.M.; Kida, Y.; Zimmerman, P.V.; Smith, P.J. Timp1 and adverse prognosis in non-small cell lung cancer. Clin. Cancer Res. 1996, 2, 1369–1372. [Google Scholar] [PubMed]
- Aljada, I.S.; Ramnath, N.; Donohue, K.; Harvey, S.; Brooks, J.J.; Wiseman, S.M.; Khoury, T.; Loewen, G.; Slocum, H.K.; Anderson, T.M.; et al. Upregulation of the tissue inhibitor of metalloproteinase-1 protein is associated with progression of human non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 3218–3229. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of hif-1α. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and inflammation in cancer, focus on hif and NF-κB. Biomedicines 2017, 5, E21. [Google Scholar] [CrossRef] [PubMed]
- Bandarra, D.; Biddlestone, J.; Mudie, S.; Muller, H.A.; Rocha, S. Hif-1α restricts NF-κB-dependent gene expression to control innate immunity signals. Dis. Models Mech. 2015, 8, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and hif crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Xue, X.; Schwartz, A.J.; Jung, I.; Colacino, J.A.; Shah, Y.M. Epithelial hypoxia-inducible factor 2α facilitates the progression of colon tumors through recruiting neutrophils. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Butterworth, J.A.; Zhao, B.; Sellier, H.; Campbell, K.J.; Thomas, H.D.; Bacon, C.M.; Cockell, S.J.; Gewurz, B.E.; Perkins, N.D. The Nf-κB subunit c-rel regulates bach2 tumour suppressor expression in b-cell lymphoma. Oncogene 2015, 35, 3476. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Leslie, J.; Perkins, N.D. C-rel and its many roles in cancer: An old story with new twists. Br. J. Cancer 2016, 114, 1. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. P53-and mdm2-independent repression of Nf-κB transactivation by the arf tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef]
- Campbell, K.J.; Rocha, S.; Perkins, N.D. Active repression of antiapoptotic gene expression by RelA (p65) Nf-κB. Mol. Cell 2004, 13, 853–865. [Google Scholar] [CrossRef]
- Campbell, K.J.; Witty, J.M.; Rocha, S.; Perkins, N.D. Cisplatin mimics arf tumor suppressor regulation of RelA (p65) nuclear factor-κB transactivation. Cancer Res. 2006, 66, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Msaki, A.; Sanchez, A.M.; Koh, L.F.; Barre, B.; Rocha, S.; Perkins, N.D.; Johnson, R.F. The role of RelA (p65) threonine 505 phosphorylation in the regulation of cell growth, survival, and migration. Mol. Biol. Cell 2011, 22, 3032–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, N.D. The importance of the p50 Nf-κB subunit. Cell Cycle 2015, 14, 2877. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Butterworth, J.A.; Sanchez, A.; Hunter, J.E.; Leslie, J.; Sellier, H.; Tiniakos, D.; Cockell, S.J.; Mann, D.A.; Oakley, F.; et al. A RelA(p65) thr505 phospho-site mutation reveals an important mechanism regulating Nf-κB-dependent liver regeneration and cancer. Oncogene 2016, 35, 4623. [Google Scholar] [CrossRef] [PubMed]
- Klapproth, K.; Sander, S.; Marinkovic, D.; Baumann, B.; Wirth, T. The IKK2/Nf-κB pathway suppresses myc-induced lymphomagenesis. Blood 2009, 114, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhang, X.; Edwards, J.P.; Mosser, D.M. Nf-κB1 (p50) homodimers differentially regulate pro-and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 2006, 281, 26041–26050. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Paz-Priel, I.; Friedman, A.D. Nf-κB p50 regulates c/ebpα expression and inflammatory cytokine-induced neutrophil production. J. Immunol. 2009, 182, 5757–5762. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Oakley, F.; Lin, F.; Packham, G.; Mann, D.A.; Mann, J. The Nf-κB p50: P50: Hdac-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 2010, 53, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Voce, D.J.; Schmitt, A.M.; Uppal, A.; McNerney, M.E.; Bernal, G.M.; Cahill, K.E.; Wahlstrom, J.S.; Nassiri, A.; Yu, X.; Crawley, C.D. NFKB1 is a haploinsufficient DNA damage-specific tumor suppressor. Oncogene 2015, 34, 2807. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Jurk, D.; Fullard, N.; Banks, P.; Page, A.; Luli, S.; Elsharkawy, A.; Gieling, R.; Chakraborty, J.B.; Fox, C. NfκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015, 6, 6818. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I. Kpc1-mediated ubiquitination and proteasomal processing of Nf-κB1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.N.; Wang, C.; Willette-Brown, J.; Herjan, T.; Gulen, M.F.; Zhou, H.; Bulek, K.; Franchi, L.; Sato, T.; Alnemri, E.S. IKKα negatively regulates asc-dependent inflammasome activation. Nat. Commun. 2014, 5, 4977. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Jia, J.; Shi, Y.; Fu, C.; Chen, L.; Jiang, Y.; Zhou, L.; Liu, S.; Tao, Y. Opposed expression of IKKα: Loss in keratinizing carcinomas and gain in non-keratinizing carcinomas. Oncotarget 2015, 6, 25499. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, G.; Vlachos, I.S.; Zagganas, K.; Vergoulis, T.; Paraskevopoulou, M.D.; Kanellos, I.; Tsanakas, P.; Dellis, D.; Fevgas, A.; Dalamagas, T. Diana-mirgen v3. 0: Accurate characterization of microRNA promoters and their regulators. Nucleic Acids Res. 2015, 44, D190–D195. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Shi, Y.; Tan, G.; Yang, C.H.; Fan, M.; Pfeffer, L.M.; Wu, Z.-H. DNA damage induces Nf-κB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J. Biol. Chem. 2012, 287, 21783–21795. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, J.; Wang, Z.; Gu, X.; Fan, Y.; Zhang, W.; Xu, L.; Zhang, J.; Cai, D. NF-κB-dependent microRNA-425 upregulation promotes gastric cancer cell growth by targeting PTEN upon il-1β induction. Mol. Cancer 2014, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Takata, A.; Otsuka, M.; Yoshikawa, T.; Kishikawa, T.; Hikiba, Y.; Obi, S.; Goto, T.; Kang, Y.J.; Maeda, S.; Yoshida, H.; et al. MicroRNA-140 acts as a liver tumor suppressor by controlling Nf-κB activity by directly targeting DNA methyltransferase 1 (dnmt1) expression. Hepatology 2013, 57, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Takata, A.; Otsuka, M.; Kojima, K.; Yoshikawa, T.; Kishikawa, T.; Yoshida, H.; Koike, K. MicroRNA-22 and microRNA-140 suppress Nf-κB activity by regulating the expression of Nf-κB coactivators. Biochem. Biophys. Res. Commun. 2011, 411, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Huan, L.; Liang, L.-H.; He, X.-H. Role of microRNAs in inflammation-associated liver cancer. Cancer Biol Med. 2016, 13, 407. [Google Scholar] [PubMed]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27(kip1) tumor suppressor by mir-221 and mir-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorhoeve, P.M.; Agami, R. Classifying microRNAs in cancer: The good, the bad and the ugly. Biochim. Biophys. Acta 2007, 1775, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Kedde, M.; van Kouwenhove, M.; Zwart, W.; Oude Vrielink, J.A.; Elkon, R.; Agami, R. A pumilio-induced RNA structure switch in p27-3′ utr controls mir-221 and mir-222 accessibility. Nat. Cell Biol. 2010, 12, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2017, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ren, X.; Zhang, X.; Luo, Y.; Wang, G.; Huang, K.; Feng, S.; Bao, X.; He, X.; Liang, P. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting Nf-κB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene 2015, 34, 691. [Google Scholar] [CrossRef]
- Kong, X.-J.; Duan, L.-J.; Qian, X.-Q.; Xu, D.; Liu, H.-L.; Zhu, Y.-J.; Qi, J. Tumor-suppressive microRNA-497 targets IKKβ to regulate Nf-κB signaling pathway in human prostate cancer cells. Am. J. Cancer Res. 2015, 5, 1795–1804. [Google Scholar] [PubMed]
- Keklikoglou, I.; Koerner, C.; Schmidt, C.; Zhang, J.; Heckmann, D.; Shavinskaya, A.; Allgayer, H.; Gückel, B.; Fehm, T.; Schneeweiss, A. MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting Nf-κB and TGF-β signaling pathways. Oncogene 2012, 31, 4150. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving Nf-κB, lin28, let-7 microRNA, and il6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. Stat3 activation of mir-21 and mir-181b-1 via PTEN and cyld are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Olarerin-George, A.O.; Anton, L.; Hwang, Y.-C.; Elovitz, M.A.; Hogenesch, J.B. A functional genomics screen for microRNA regulators of NF-κB signaling. BMC Biol. 2013, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, H.; Ye, S.; Guan, J.; Tan, W.; Cheng, S.; Wei, G.; Wu, W.; Wu, F.; Zhou, Y. Up-regulation of microRNA-126 may contribute to pathogenesis of ulcerative colitis via regulating NF-κB inhibitor iκBα. PLoS ONE 2012, 7, e52782. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lin, J.; Zhang, H. Mir-126: A novel regulator in colon cancer. Biomed. Rep. 2016, 4, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Pal, A.S.; Hsu, A.Y.; Gurol, T.; Zhu, X.; Wirbisky-Hershberger, S.E.; Freeman, J.L.; Kasinski, A.L.; Deng, Q. MicroRNA-223 suppresses the canonical NF-κB pathway in basal keratinocytes to dampen neutrophilic inflammation. Cell Rep. 2018, 22, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; O’Neill, L.A.; Masters, S.L. Mir-223: Infection, inflammation and cancer. J. Intern. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, H.; Liu, Y.; Song, Y.; Lai, L.; Han, Q.; Cao, X.; Wang, Q. Inducible microRNA-223 down-regulation promotes tlr-triggered il-6 and il-1β production in macrophages by targeting stat3. PLoS ONE 2012, 7, e42971. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Morgan, M.J.; Choksi, S.; Zhang, Y.; Kim, Y.S.; Liu, Z.G. MicroRNAs modulate the noncanonical transcription factor NF-κB pathway by regulating expression of the kinase IKKα during macrophage differentiation. Nat. Immunol. 2010, 11, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Z.; Guo, F.; Qin, X.; Liu, B.; Lei, Z.; Song, Z.; Sun, L.; Zhang, H.T.; You, J.; et al. Mir-223 regulates migration and invasion by targeting artemin in human esophageal carcinoma. J. Biomed. Sci. 2011, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Zhang, H.; Liu, X.; Gong, T.; Li, M.; Sun, L.; Ji, G.; Shi, Y.; Han, Z.; et al. MiRNA-223 promotes gastric cancer invasion and metastasis by targeting tumor suppressor epb41l3. Mol. Cancer Res. 2011, 9, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, Y.; Chen, Q.; Qiu, N.; Zhao, Y.; You, X. Mir-223 inhibited cell metastasis of human cervical cancer by modulating epithelial-mesenchymal transition. Int. J. Clin. Exp. Pathol. 2015, 8, 11224–11229. [Google Scholar] [PubMed]
- Aqeilan, R.I.; Calin, G.A.; Croce, C.M. Mir-15a and mir-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2009, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Alvero, A.B.; Silasi, D.A.; Kelly, M.G.; Fest, S.; Visintin, I.; Leiser, A.; Schwartz, P.E.; Rutherford, T.; Mor, G. Regulation of IKKβ by mir-199a affects Nf-κB activity in ovarian cancer cells. Oncogene 2008, 27, 4712. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. Nf-κB dysregulation in microRNA-146a—Deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of mir-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.M.; Pu, Y.; Han, Z.; Liu, T.; Li, Y.X.; Liu, M.; Li, X.; Tang, H. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of Nf-κB1. FEBS J. 2009, 276, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, L.-C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.-Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFκB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Chen, Z.Q.; Cao, X.X.; Xu, J.D.; Xu, J.W.; Chen, Y.Y.; Wang, W.J.; Chen, Q.; Tang, F.; Liu, X.P.; et al. Involvement of Nf-κB/mir-448 regulatory feedback loop in chemotherapy-induced epithelial–mesenchymal transition of breast cancer cells. Cell Death Differ. 2010, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Erkeland, S.J.; Pester, R.E.; Chen, C.Y.; Ebert, M.S.; Sharp, P.A.; Jacks, T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl. Acad. Sci. USA 2008, 105, 3903–3908. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D. MicroRNA circuits regulate the cancer-inflammation link. Sci. Signal. 2014, 7, pe8. [Google Scholar] [CrossRef]
- Xiang, M.; Birkbak, N.J.; Vafaizadeh, V.; Walker, S.R.; Yeh, J.E.; Liu, S.; Kroll, Y.; Boldin, M.; Taganov, K.; Groner, B. Stat3 induction of mir-146b forms a feedback loop to inhibit the Nf-κB to il-6 signaling axis and stat3-driven cancer phenotypes. Sci. Signal. 2014, 7, ra11. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-H.; Park, S.-J.; Dickinson, S.I.; Luo, J.-L. A constitutive intrinsic inflammatory signaling circuit composed of mir-196b, Meis2, PPP3CC, and p65 drives prostate cancer castration resistance. Mol. Cell 2017, 65, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, G.L.; Alexiou, P.; Maragkakis, M.; Reczko, M.; Hatzigeorgiou, A.G. Diana-mirpath: Integrating human and mouse microRNAs in pathways. Bioinformatics 2009, 25, 1991–1993. [Google Scholar] [CrossRef] [PubMed]
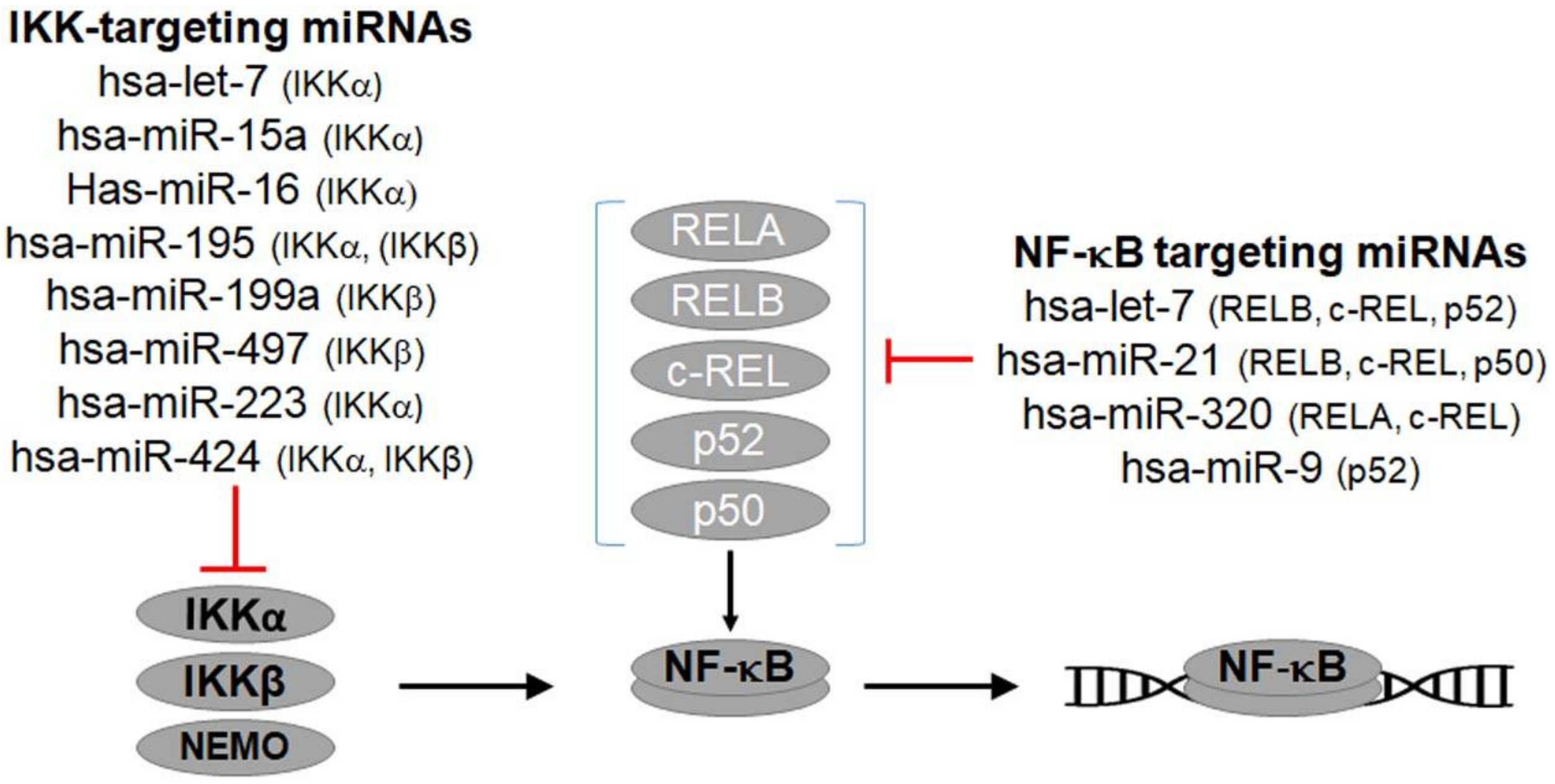

{kind=link}
{kind=link}
| miRNA Name | Chromosomal Location of Promoter (hg19) | Strand |
|---|---|---|
| hsa-let-7a-1 | chr9:96929483–96929484 | [+] |
| hsa-let-7d | chr9:96929483–96929484 | [+] |
| hsa-let-7f-1 | chr9:96929483–96929484 | [+] |
| hsa-let-7i | chr12:62997400–62997401 | [+] |
| hsa-mir-101-1 | chr1:65532138–65532139 | [−] |
| hsa-mir-1204 | chr8:128806768–128806769 | [+] |
| hsa-mir-1205 | chr8:128806768–128806769 | [+] |
| hsa-mir-1206 | chr8:128806768–128806769 | [+] |
| hsa-mir-1207 | chr8:128806759–128806760 | [+] |
| hsa-mir-1208 | chr8:128806759–128806760 | [+] |
| hsa-mir-124-1 | chr8:9763203–9763204 | [−] |
| hsa-mir-125b-1 | chr11:121971206–121971207 | [−] |
| hsa-mir-1289-1 | chr20:34042503–34042504 | [−] |
| hsa-mir-135b | chr1:205426509–205426510 | [−] |
| hsa-mir-137 | chr1:98520169–98520170 | [−] |
| hsa-mir-146a | chr5:159894835–159894836 | [+] |
| hsa-mir-148a | chr7:25990290–25990291 | [−] |
| hsa-mir-193a | chr17:29886484–29886485 | [+] |
| hsa-mir-22 | chr17:1618561–1618562 | [−] |
| hsa-mir-223 | chrX:65219544–65219545 | [+] |
| hsa-mir-23a | chr19:13953455–13953456 | [−] |
| hsa-mir-24-2 | chr19:13953455–13953456 | [−] |
| hsa-mir-2682 | chr1:98520169–98520170 | [−] |
| hsa-mir-27a | chr19:13953455–13953456 | [−] |
| hsa-mir-2861 | chr9:130548069–130548070 | [+] |
| hsa-mir-29a | chr7:130794752–130794753 | [−] |
| hsa-mir-29b-1 | chr7:130794752–130794753 | [−] |
| hsa-mir-30a | chr6:72130555–72130556 | [−] |
| hsa-mir-30c-2 | chr6:72130555–72130556 | [−] |
| hsa-mir-3142 | chr5:159894835–159894836 | [+] |
| hsa-mir-3199-2 | chr22:28315414–28315415 | [+] |
| hsa-mir-365b | chr17:29886484–29886485 | [+] |
| hsa-mir-3667 | chr22:50051180–50051181 | [−] |
| hsa-mir-3672 | chrX:120325891–120325892 | [+] |
| hsa-mir-3679 | chr2:134877461–134877462 | [+] |
| hsa-mir-3960 | chr9:130548069–130548070 | [+] |
| hsa-mir-4725 | chr17:29886484–29886485 | [+] |
| hsa-mir-505 | chrX:139015225–139015226 | [−] |
| hsa-mir-5194 | chr8:131028942–131028943 | [−] |
| hsa-mir-612 | chr11:65190256–65190257 | [+] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Alabasi, G.; Sandaltzopoulos, R.; Marcu, K.B.; Kolettas, E. Roles of NF-κB Signaling in the Regulation of miRNAs Impacting on Inflammation in Cancer. Biomedicines 2018, 6, 40. https://doi.org/10.3390/biomedicines6020040
Markopoulos GS, Roupakia E, Tokamani M, Alabasi G, Sandaltzopoulos R, Marcu KB, Kolettas E. Roles of NF-κB Signaling in the Regulation of miRNAs Impacting on Inflammation in Cancer. Biomedicines. 2018; 6(2):40. https://doi.org/10.3390/biomedicines6020040
Chicago/Turabian StyleMarkopoulos, Georgios S., Eugenia Roupakia, Maria Tokamani, Georgia Alabasi, Raphael Sandaltzopoulos, Kenneth B. Marcu, and Evangelos Kolettas. 2018. "Roles of NF-κB Signaling in the Regulation of miRNAs Impacting on Inflammation in Cancer" Biomedicines 6, no. 2: 40. https://doi.org/10.3390/biomedicines6020040