The Direct and Indirect Roles of NF-κB in Cancer: Lessons from Oncogenic Fusion Proteins and Knock-in Mice

 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
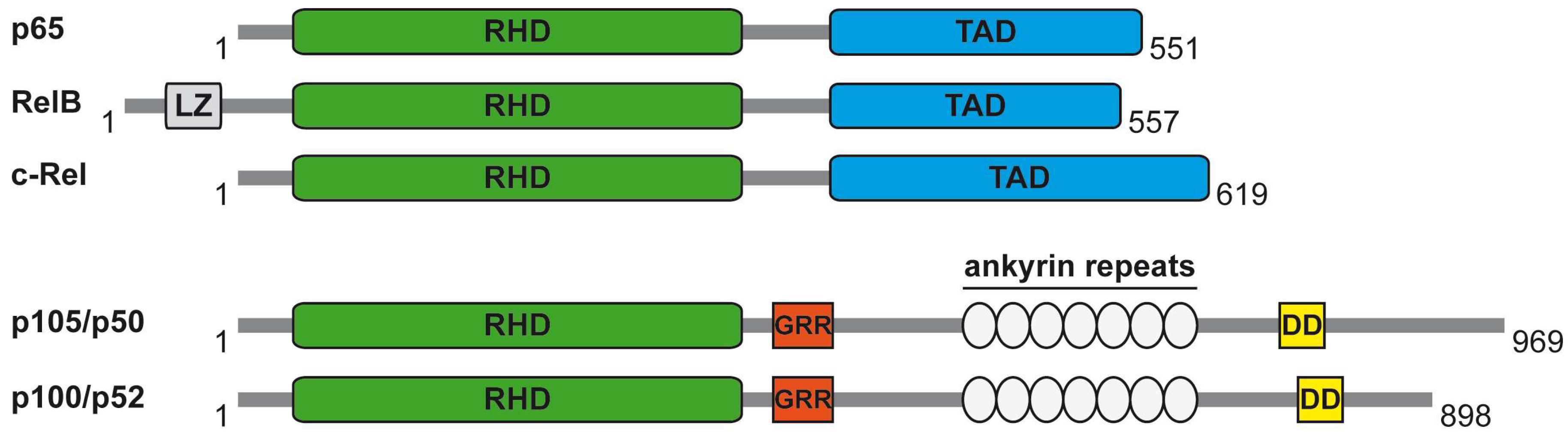
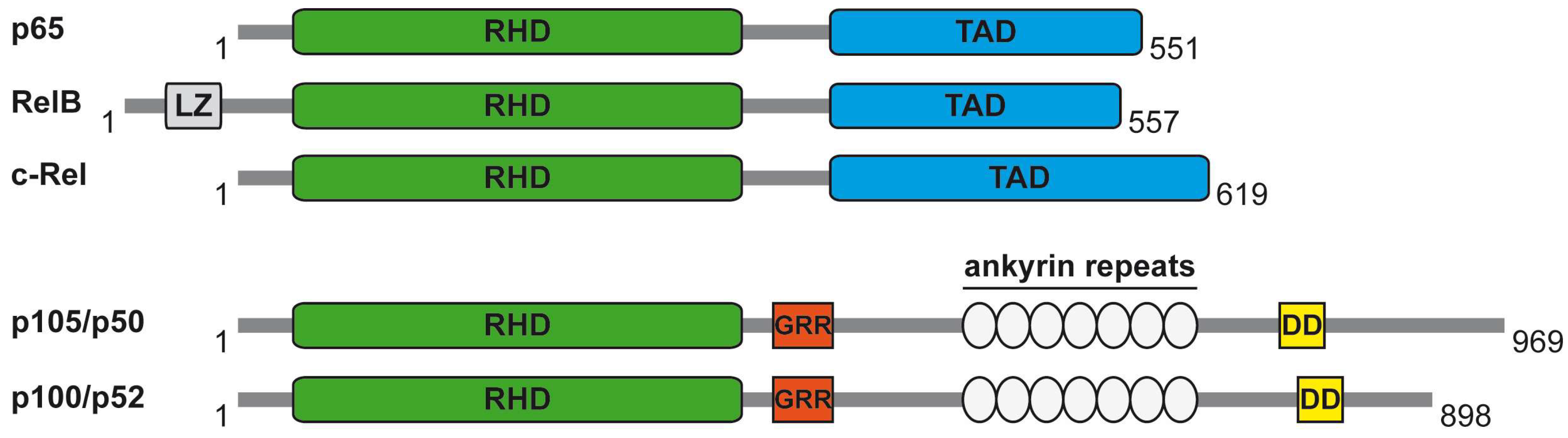
1. The NF-κB System
2. The Core NF-κB Activation Pathways
3. Auxiliary NF-κB Regulating Mechanisms
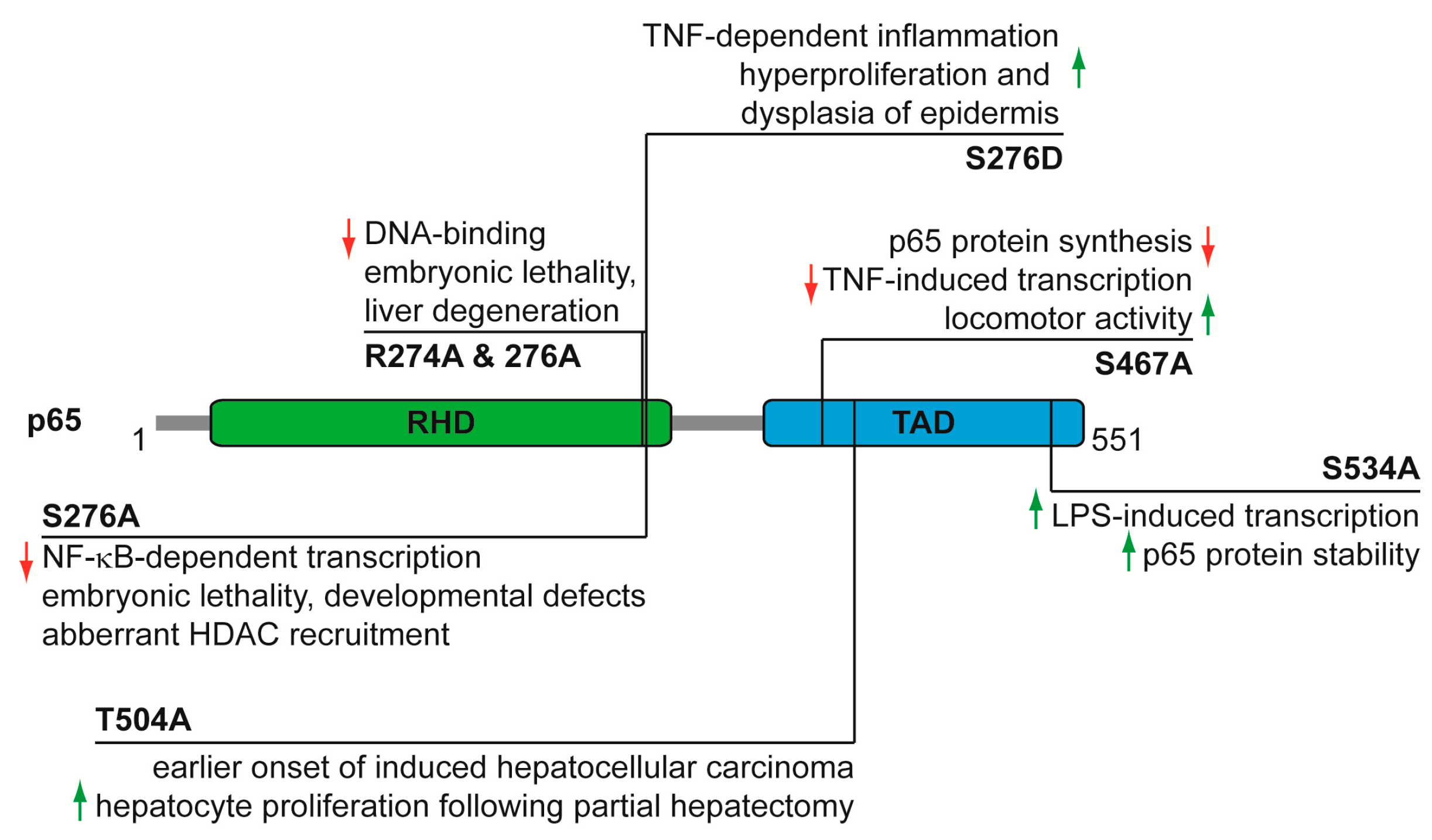
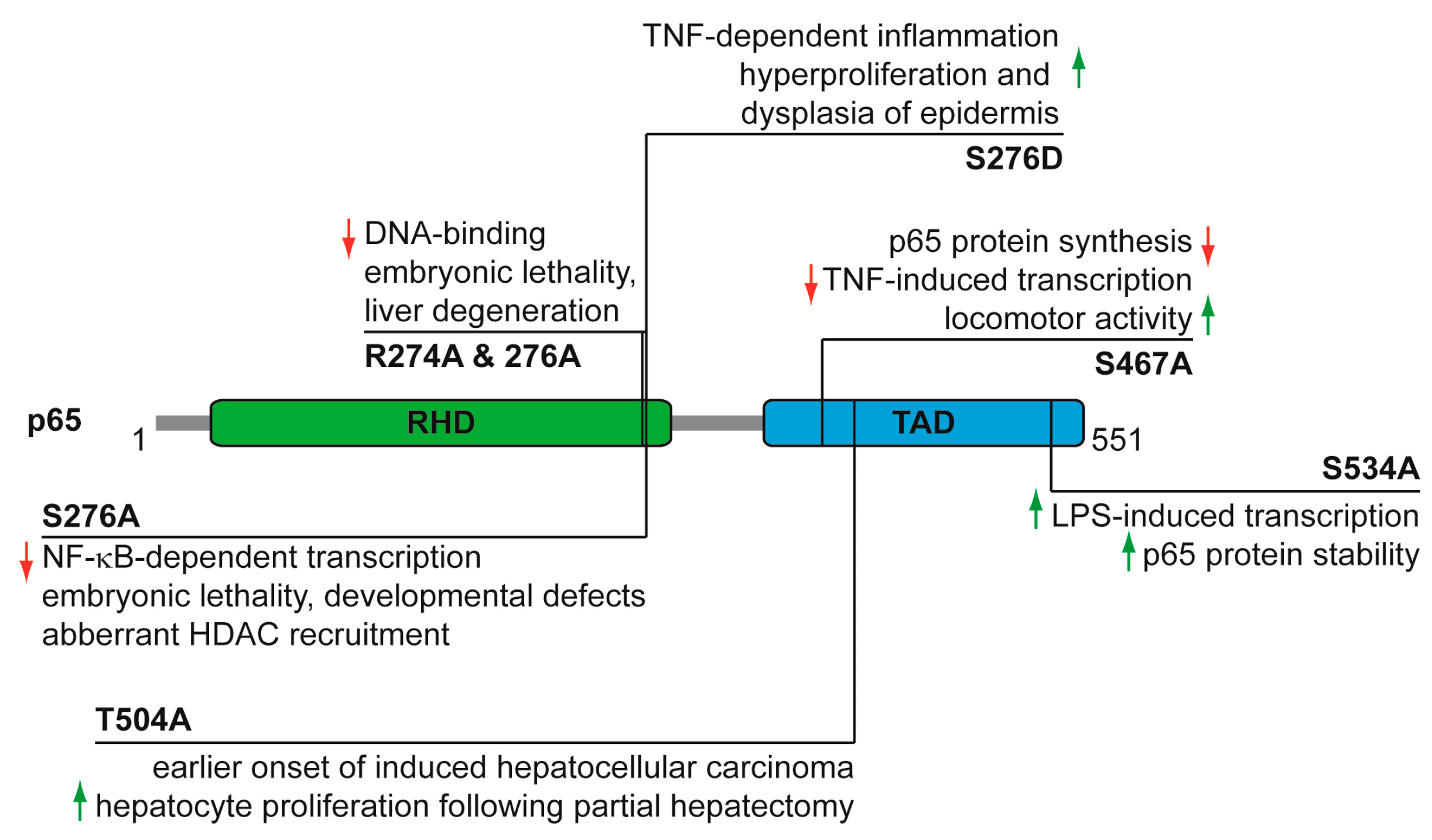
4. Functional Analysis of p65 Phosphorylations In Vivo
5. The Multiple Roles of NF-κB in Cancer
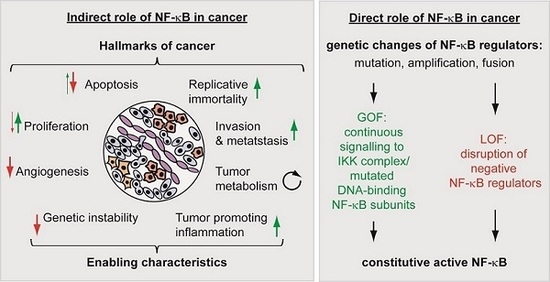
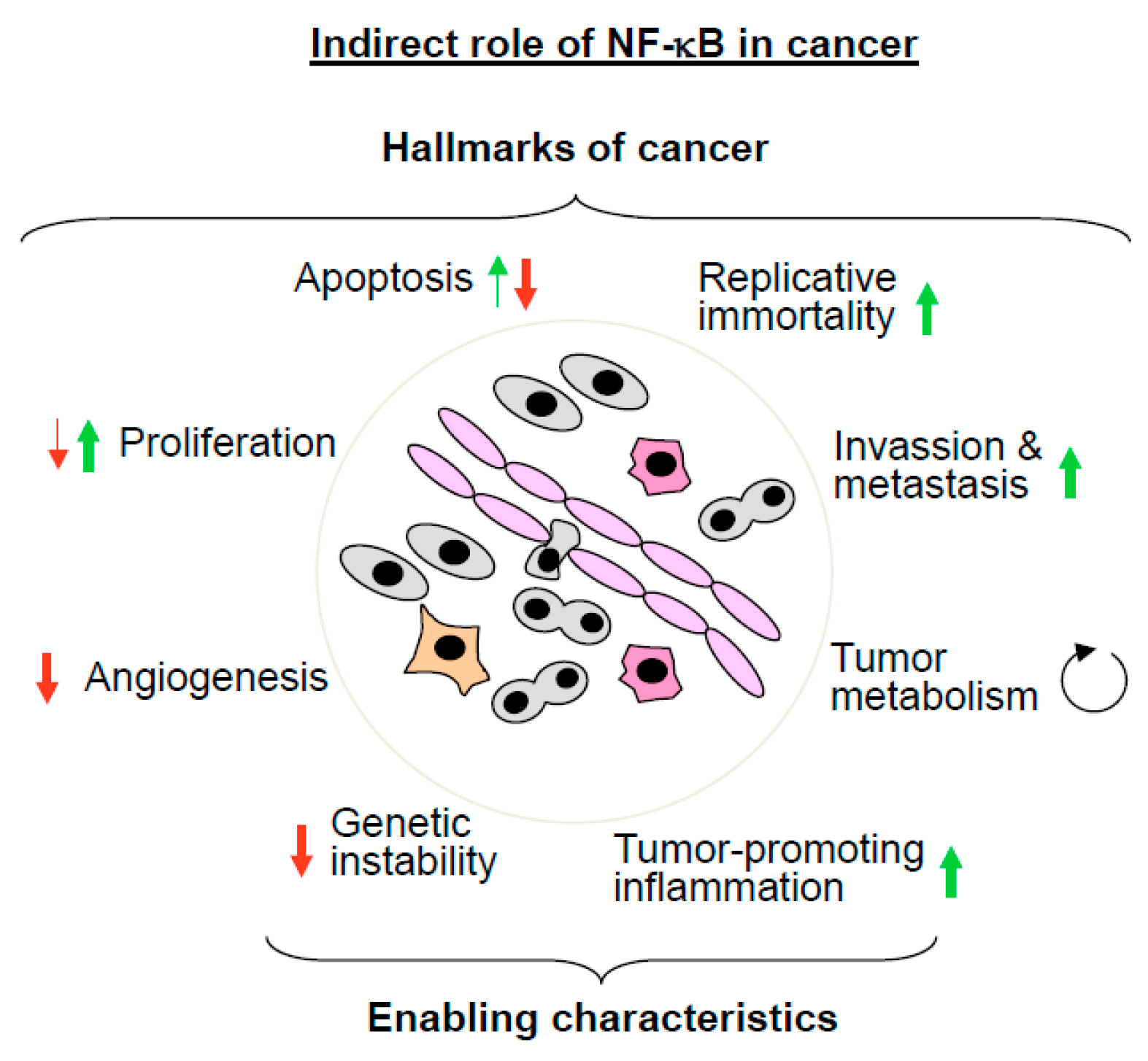
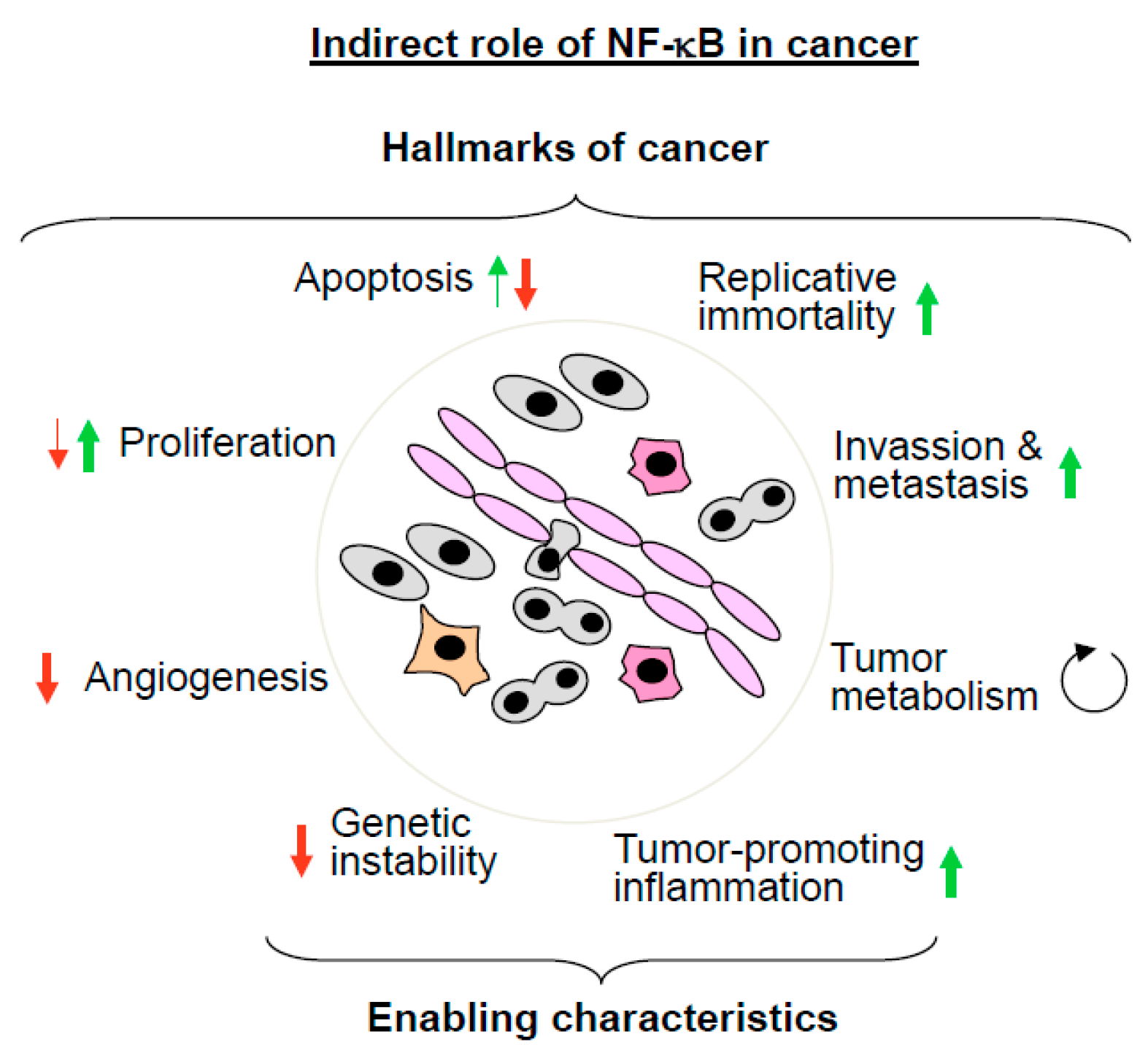
5.1. The Indirect Roles of NF-κB in Cancer
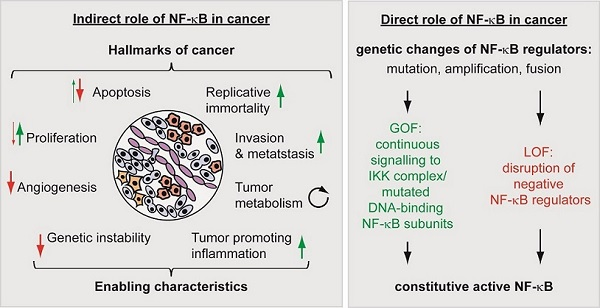
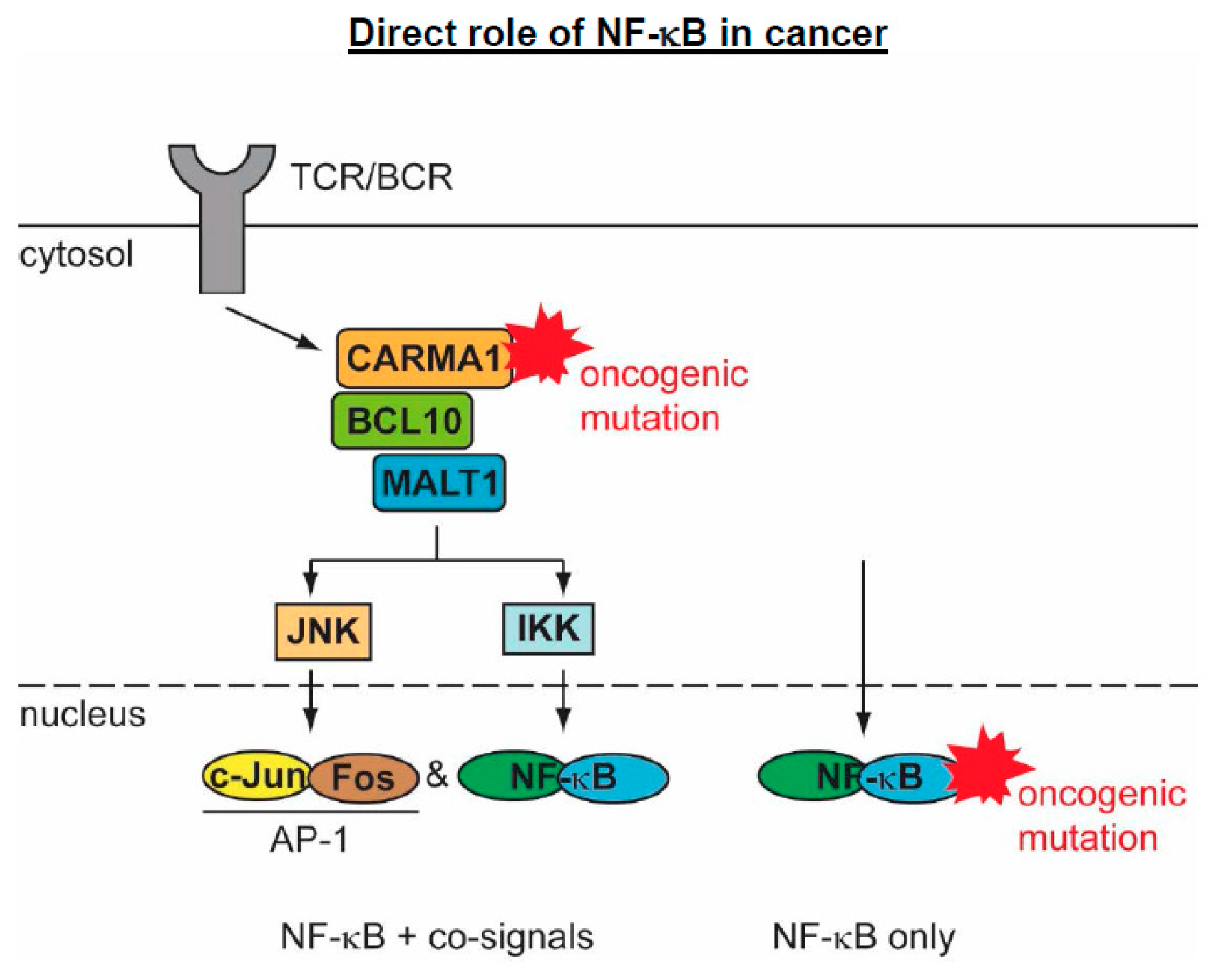
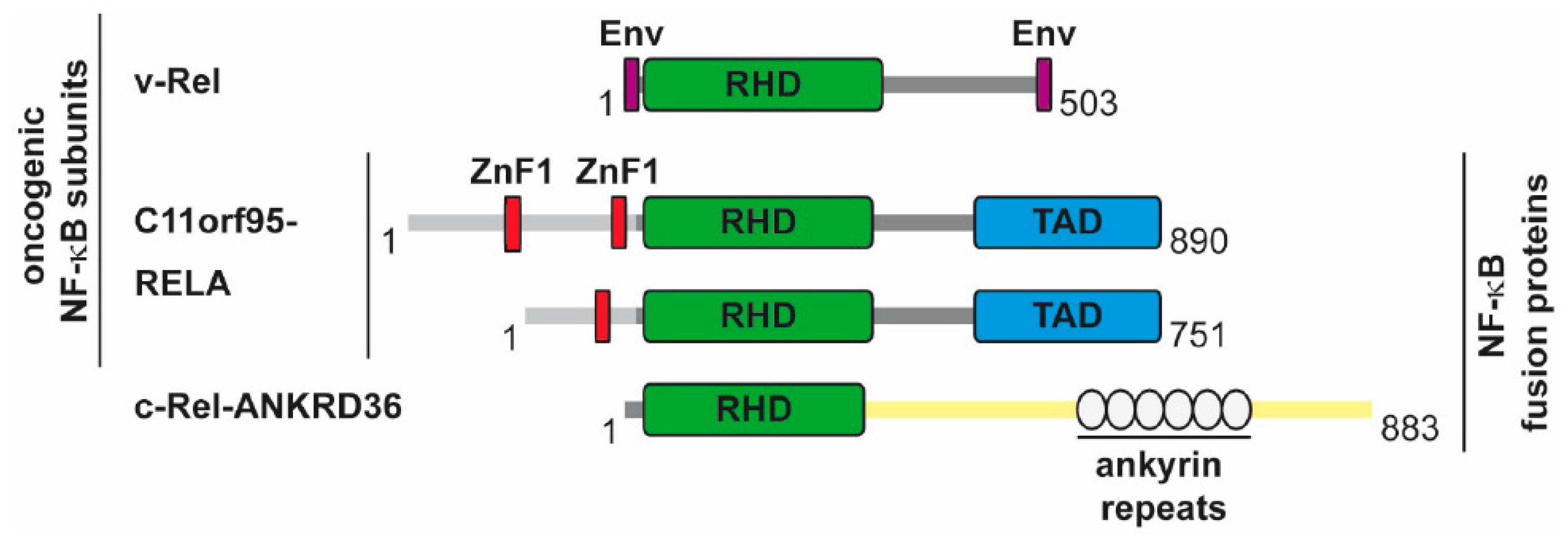
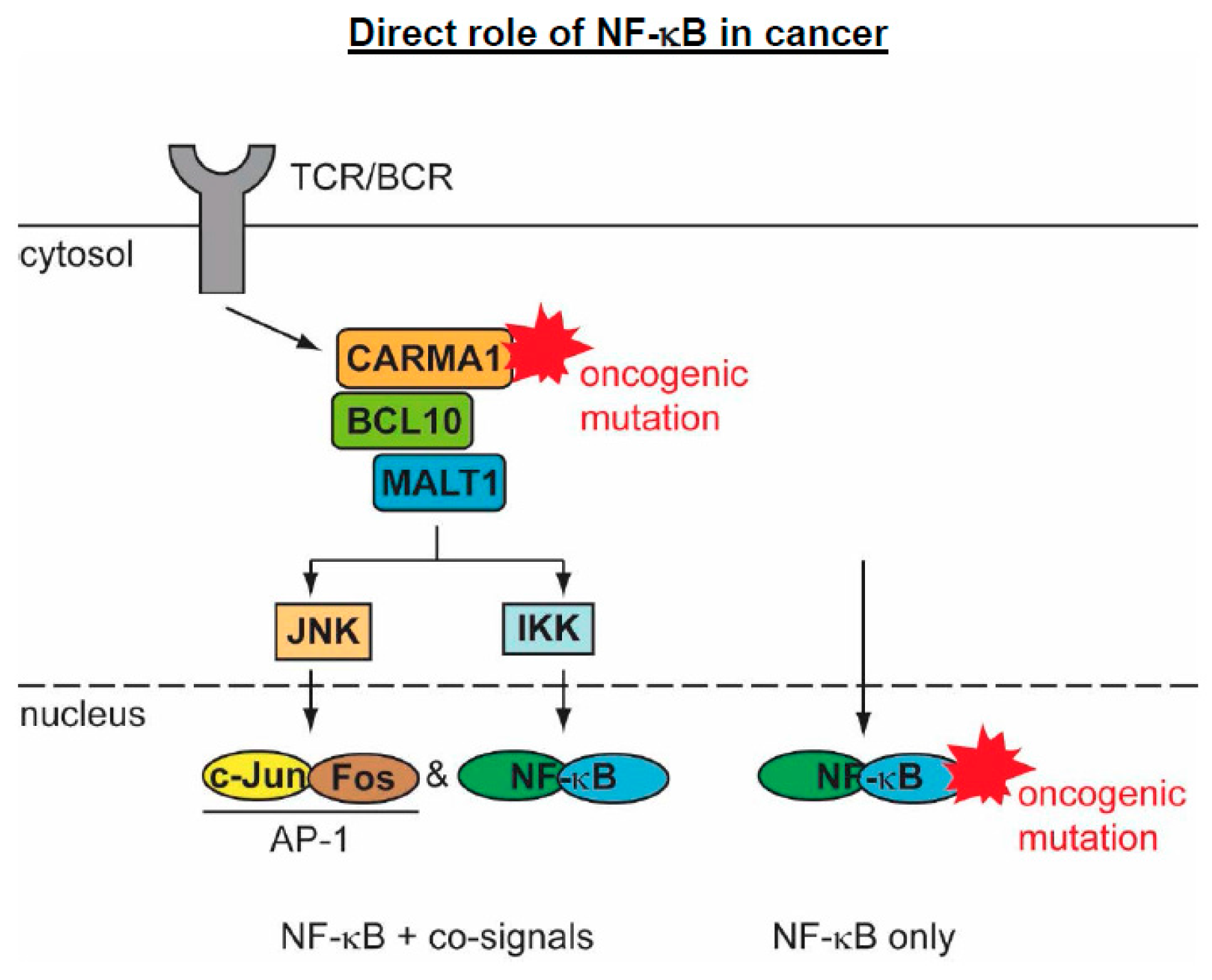
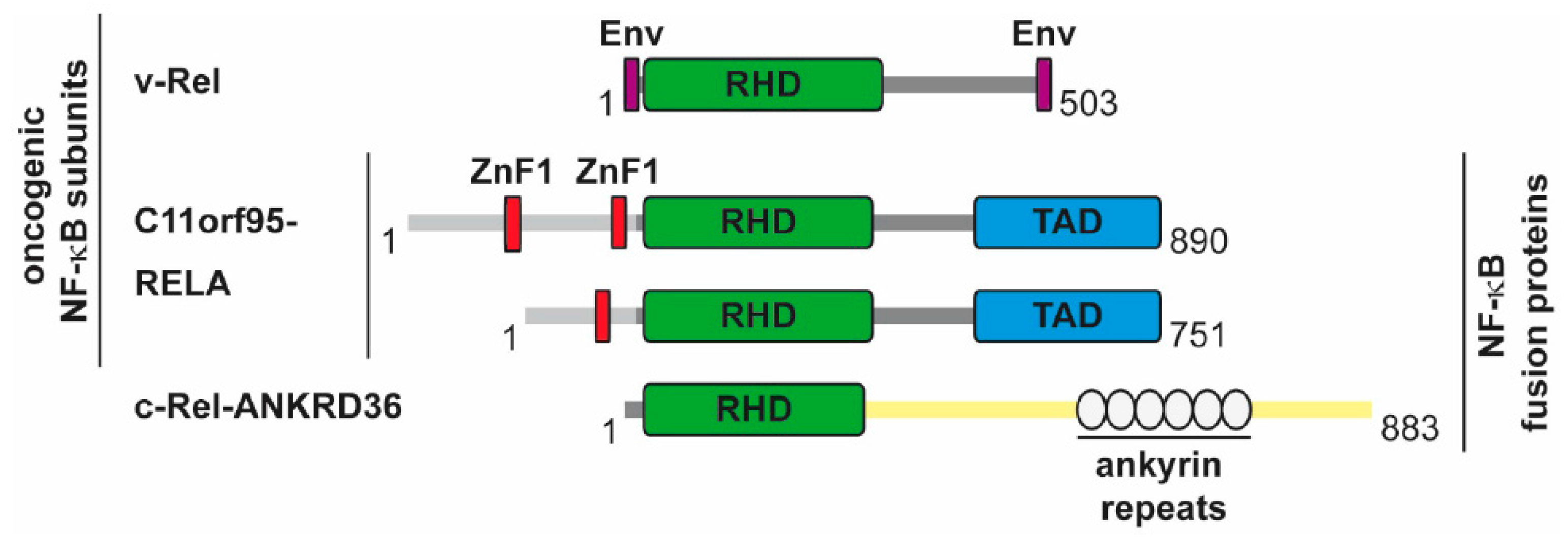
5.2. The Direct Roles of NF-κB in Cancer
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gilmore, T.D.; Wolenski, F.S. NF-κB: Where did it come from and why? Immunol. Rev. 2012, 246, 14–35. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Rocha, S. Hypoxia Induced NF-κB. Cells 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2011, 21, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NF-κB system. Wiley. Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Baeuerle, P.A. The inducible transcription factor NF-κB: Structure-function relationship of its protein subunits. Biochem. J. 1993, 290, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.B.; Kofahl, B.; Beaudette, P.; Baum, K.; Ipenberg, I.; Weih, F.; Wolf, J.; Dittmar, G.; Scheidereit, C. Quantitative dissection and modeling of the NF-κB p100-p105 module reveals interdependent precursor proteolysis. Cell Rep. 2014, 9, 1756–1769. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Baeuerle, P.A. The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO J. 1991, 10, 3805–3817. [Google Scholar] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Kracht, M.; Saul, V.V. The intricate interplay between RNA viruses and NF-κB. Biochim. Biophys. Acta 2014, 1843, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F. Linear ubiquitination signals in adaptive immune responses. Immunol. Rev. 2015, 266, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Rittinger, K.; Ikeda, F. Linear ubiquitin chains: Enzymes, mechanisms and biology. Open. Biol. 2017, 7, 170026. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Mabb, A.M.; Wuerzberger-Davis, S.M.; Miyamoto, S. PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat. Cell Biol. 2006, 8, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G. Tuning up inflammation: How DNA sequence and chromatin organization control the induction of inflammatory genes by NF-κB. FEBS Lett. 2006, 580, 2843–2849. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Teixeira, A.; Oikonomopoulos, S.; Humburg, P.; Lone, I.N.; Saliba, D.; Siggers, T.; Bulyk, M.; Angelov, D.; Dimitrov, S.; et al. Extensive characterization of NF-κB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011, 12, R70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, B.; Baldwin, A.S., Jr.; Ballard, D.W.; Greene, W.C.; Angel, P.; Herrlich, P. Cross-coupling of the NF-κB p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 1993, 12, 3879–3891. [Google Scholar] [PubMed]
- Dryden, N.H.; Sperone, A.; Martin-Almedina, S.; Hannah, R.L.; Birdsey, G.M.; Khan, S.T.; Layhadi, J.A.; Mason, J.C.; Haskard, D.O.; Gottgens, B.; et al. The transcription factor Erg controls endothelial cell quiescence by repressing activity of nuclear factor (NF)-κB p65. J. Biol. Chem. 2012, 287, 12331–12342. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.A.; Yao, F.; Wong, J.J.; George, J.; Xu, H.; Chiu, K.P.; Sung, W.K.; Lipovich, L.; Vega, V.B.; Chen, J.; et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-κB upon TLR4 activation. Mol. Cell 2007, 27, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, R.; Wong, K.A.; Getman, C.; Stein, N.; Teitell, M.A.; Cheng, G.; Wu, H.; Shuai, K. Negative regulation of NF-κB signaling by PIAS1. Mol. Cell Biol. 2005, 25, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Huq, M.D.; Hu, X.; Wei, L.N. Tyrosine nitration on p65: A novel mechanism to rapidly inactivate nuclear factor-κB. Mol. Cell Proteom. 2005, 4, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Reintjes, A.; Fuchs, J.E.; Kremser, L.; Lindner, H.H.; Liedl, K.R.; Huber, L.A.; Valovka, T. Asymmetric arginine dimethylation of RelA provides a repressive mark to modulate TNFα/NF-κB response. Proc. Natl. Acad. Sci. USA 2016, 113, 4326–4331. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jimi, E.; Zhong, H.; Hayden, M.S.; Ghosh, S. Repression of gene expression by unphosphorylated NF-κB p65 through epigenetic mechanisms. Genes Dev. 2008, 22, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jimi, E.; Zeiss, C.; Hayden, M.S.; Ghosh, S. Constitutively active NF-κB triggers systemic TNFα-dependent inflammation and localized TNFα-independent inflammatory disease. Genes Dev. 2010, 24, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.E.; Prichard, L.; Shiojiri, N.; Fausto, N. Prevention of hepatic apoptosis and embryonic lethality in RelA/TNFR-1 double knockout mice. Am. J. Pathol. 2000, 156, 997–1007. [Google Scholar] [CrossRef]
- Poligone, B.; Hayden, M.S.; Chen, L.; Pentland, A.P.; Jimi, E.; Ghosh, S. A role for NF-κB activity in skin hyperplasia and the development of keratoacanthomata in mice. PLoS ONE 2013, 8, e71887. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Butterworth, J.A.; Sanchez, A.; Hunter, J.E.; Leslie, J.; Sellier, H.; Tiniakos, D.; Cockell, S.J.; Mann, D.A.; Oakley, F.; et al. A RelA(p65) Thr505 phospho-site mutation reveals an important mechanism regulating NF-κB-dependent liver regeneration and cancer. Oncogene 2016, 35, 4623. [Google Scholar] [CrossRef] [PubMed]
- Msaki, A.; Sanchez, A.M.; Koh, L.F.; Barre, B.; Rocha, S.; Perkins, N.D.; Johnson, R.F. The role of RelA (p65) threonine 505 phosphorylation in the regulation of cell growth, survival, and migration. Mol. Biol. Cell 2011, 22, 3032–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradere, J.P.; Hernandez, C.; Koppe, C.; Friedman, R.A.; Luedde, T.; Schwabe, R.F. Negative regulation of NF-κB p65 activity by serine 536 phosphorylation. Sci. Signal 2016, 9, ra85. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Wittwer, T.; Dittrich-Breiholz, O.; Kracht, M.; Schmitz, M.L. Phosphorylation of NF-κB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009, 10, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. IKKα limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Gluck, N.; Li, D.; Maine, G.N.; Li, H.; Zaidi, I.W.; Repaka, A.; Mayo, M.W.; Burstein, E. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-κB/RelA. Genes Dev. 2009, 23, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, T.; Dommerholt, M.B.; Wijshake, T.; Kruit, J.K.; Huijkman, N.; Dekker, D.; Koster, M.; Kloosterhuis, N.; Koonen, D.P.Y.; de Bruin, A.; et al. NF-κB p65 serine 467 phosphorylation sensitizes mice to weight gain and TNFα-or diet-induced inflammation. Biochim. Biophys. Acta 2017, 1864, 1785–1798. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C. NF-κB in Long-Term Memory and Structural Plasticity in the Adult Mammalian Brain. Front. Mol. Neurosci. 2015, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Gilmore, T.D. Mutations in the NF-κB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baltimore, D. An essential role for NF-κB in preventing TNF-alpha-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-κB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Baldwin, A.S., Jr. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science 1996, 274, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.Q.; Huang, X.B.; Ke, S.Z.; Jiang, Y.N.; Zhang, Y.H.; Wang, Y.N.; Li, J.; Gao, F.G. Interleukin 6 augments lung cancer chemotherapeutic resistance via ataxia-telangiectasia mutated/NF-κB pathway activation. Cancer Sci. 2014, 105, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Tusquets, I.; Ferrer, B.; Corominas, J.M.; Bellosillo, B.; Campas, C.; Suarez, M.; Fabregat, X.; Campo, E.; Gascon, P.; et al. Activation of nuclear factor-κB is linked to resistance to neoadjuvant chemotherapy in breast cancer patients. Endocr. Relat Cancer 2006, 13, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-κB to overcome resistance to chemotherapy. Front Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohma, I.; Fujiwara, Y.; Sugita, Y.; Yoshioka, A.; Shirakawa, M.; Moon, J.H.; Takiguchi, S.; Miyata, H.; Yamasaki, M.; Mori, M.; et al. Parthenolide, an NF-κB inhibitor, suppresses tumor growth and enhances response to chemotherapy in gastric cancer. Cancer Genom. Proteom. 2011, 8, 39–47. [Google Scholar]
- Enzler, T.; Sano, Y.; Choo, M.K.; Cottam, H.B.; Karin, M.; Tsao, H.; Park, J.M. Cell-selective inhibition of NF-κB signaling improves therapeutic index in a melanoma chemotherapy model. Cancer Discov. 2011, 1, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C.; Hofmann, T.G.; Hehner, S.P.; Droge, W.; Schmitz, M.L. The pro- or anti-apoptotic function of NF-κB is determined by the nature of the apoptotic stimulus. Eur. J. Biochem. 2000, 267, 3828–3835. [Google Scholar] [CrossRef] [PubMed]
- Jennewein, C.; Karl, S.; Baumann, B.; Micheau, O.; Debatin, K.M.; Fulda, S. Identification of a novel pro-apoptotic role of NF-κB in the regulation of T. Oncogene 2012, 31, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Hideshima, T.; Hayashi, T.; Tai, Y.T.; Mitsiades, C.S.; Mitsiades, N.; Chauhan, D.; Richardson, P.; Munshi, N.C.; Anderson, K.C. Nuclear factor-κB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res. 2003, 63, 18–21. [Google Scholar] [PubMed]
- Yin, L.; Hubbard, A.K.; Giardina, C. NF-κB regulates transcription of the mouse telomerase catalytic subunit. J. Biol. Chem. 2000, 275, 36671–36675. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-κB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Diaz, J.A.; Maia, A.M.; Correa, S.; Abdelhay, E.S. NF-κB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hikiba, Y.; Sakamoto, K.; Nakagawa, H.; Hirata, Y.; Hayakawa, Y.; Yanai, A.; Ogura, K.; Karin, M.; Omata, M. Ikappa B kinasebeta/nuclear factor-κB activation controls the development of liver metastasis by way of interleukin-6 expression. Hepatology 2009, 50, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De, S.E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Witzel, I.I.; Perkins, N.D. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-κB. Cancer Res. 2011, 71, 5588–5597. [Google Scholar] [CrossRef] [PubMed]
- Köntgen, F.; Grumont, R.J.; Strasser, A.; Metcalf, D.; Li, R.; Tarlinton, D.; Gerondakis, S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995, 9, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Tumang, J.R.; Owyang, A.; Andjelic, S.; Jin, Z.; Hardy, R.R.; Liou, M.L.; Liou, H.C. c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur. J. Immunol. 1998, 28, 4299–4312. [Google Scholar] [CrossRef]
- Snapper, C.M.; Rosas, F.R.; Zelazowski, P.; Moorman, M.A.; Kehry, M.R.; Bravo, R.; Weih, F. B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J. Exp. Med. 1996, 184, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Fullard, N.; Moles, A.; O’Reilly, S.; van Laar, J.M.; Faini, D.; Diboll, J.; Reynolds, N.J.; Mann, D.A.; Reichelt, J.; Oakley, F. The c-Rel subunit of NF-κB regulates epidermal homeostasis and promotes skin fibrosis in mice. Am. J. Pathol. 2013, 182, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Seitz, C.S.; Lin, Q.; Deng, H.; Khavari, P.A. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proc. Natl. Acad. Sci. USA 1998, 95, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.L.; Liu, S.H.; Ai, Z.H.; Tao, M.F.; Ma, L.; Wen, S.Y.; Dai, M.; Liu, F.; Liu, H.S.; Jiang, R.Z.; et al. RelB/NF-κB links cell cycle transition and apoptosis to endometrioid adenocarcinoma tumorigenesis. Cell Death. Dis. 2016, 7, e2402. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C.; Hehner, S.P.; Droge, W.; Schmitz, M.L. Repression of NF-κB impairs HeLa cell proliferation by functional interference with cell cycle checkpoint regulators. Oncogene 1999, 18, 3213–3225. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Song, L.; Vorontchikhina, M.; Feirt, N.; Kitajewski, J.; Schindler, C. NF-κB regulation of endothelial cell function during LPS-induced toxemia and cancer. J. Clin. Investig. 2006, 116, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmuller, L. NF-κB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res. 2012, 40, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jacob, N.K.; Ladner, K.J.; Beg, A.; Perko, J.D.; Tanner, S.M.; Liyanarachchi, S.; Fishel, R.; Guttridge, D.C. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep. 2009, 10, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Elton, L.; Carpentier, I.; Beyaert, R. MALT1--a universal soldier: Multiple strategies to ensure NF-κB activation and target gene expression. FEBS J. 2015, 282, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nat. Immunol. 2008, 9, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Rebeaud, F.; Hailfinger, S.; Posevitz-Fejfar, A.; Tapernoux, M.; Moser, R.; Rueda, D.; Gaide, O.; Guzzardi, M.; Iancu, E.M.; Rufer, N.; et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat. Immunol. 2008, 9, 272–281. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Li, Y.; Ma, J.; Garner, E.; Zhang, X.; Wu, H. Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc. Natl. Acad. Sci. USA 2018, 115, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Juilland, M.; Thome, M. Role of the CARMA1/BCL10/MALT1 complex in lymphoid malignancies. Curr. Opin. Hematol. 2016, 23, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Meininger, I.; Krappmann, D. Lymphocyte signaling and activation by the CARMA1-BCL10-MALT1 signalosome. Biol. Chem. 2016, 397, 1315–1333. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Jeong, Y.; Lee, S.; Im, H.; Tae, H.S.; Kim, B.G.; Park, H.D.; Park, J.; Hong, S. Identification of beta-Lapachone Analogs as Novel MALT1 Inhibitors To Treat an Aggressive Subtype of Diffuse Large B-Cell Lymphoma. J. Med. Chem. 2015, 58, 8491–8502. [Google Scholar] [CrossRef] [PubMed]
- Nagel, D.; Spranger, S.; Vincendeau, M.; Grau, M.; Raffegerst, S.; Kloo, B.; Hlahla, D.; Neuenschwander, M.; Peter von Kries, J.; Hadian, K.; et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 2012, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Saba, N.S.; Wong, D.H.; Tanios, G.; Iyer, J.R.; Lobelle-Rich, P.; Dadashian, E.L.; Liu, D.; Fontan, L.; Flemington, E.K.; Nichols, C.M.; et al. MALT1 Inhibition Is Efficacious in Both Naive and Ibrutinib-Resistant Chronic Lymphocytic Leukemia. Cancer Res. 2017, 77, 7038–7048. [Google Scholar] [CrossRef] [PubMed]
- Knies, N.; Alankus, B.; Weilemann, A.; Tzankov, A.; Brunner, K.; Ruff, T.; Kremer, M.; Keller, U.B.; Lenz, G.; Ruland, J. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-κB and JNK activation. Proc. Natl. Acad. Sci. USA 2015, 112, E7230–E7238. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Temin, H.M. Different localization of the product of the v-rel oncogene in chicken fibroblasts and spleen cells correlates with transformation by REV-T. Cell 1986, 44, 791–800. [Google Scholar] [CrossRef]
- Stephens, R.M.; Rice, N.R.; Hiebsch, R.R.; Bose, H.R., Jr.; Gilden, R.V. Nucleotide sequence of v-rel: The oncogene of reticuloendotheliosis virus. Proc. Natl. Acad. Sci. USA 1983, 80, 6229–6233. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.; Rizzo, C.A.; Dorfman, K.; Bravo, R. The v-rel oncogene promotes malignant T-cell leukemia/lymphoma in transgenic mice. EMBO J. 1996, 15, 3640–3650. [Google Scholar] [PubMed]
- Walker, W.H.; Stein, B.; Ganchi, P.A.; Hoffman, J.A.; Kaufman, P.A.; Ballard, D.W.; Hannink, M.; Greene, W.C. The v-rel oncogene: Insights into the mechanism of transcriptional activation, repression, and transformation. J. Virol. 1992, 66, 5018–5029. [Google Scholar] [PubMed]
- Ballard, D.W.; Dixon, E.P.; Peffer, N.J.; Bogerd, H.; Doerre, S.; Stein, B.; Greene, W.C. The 65-kDa subunit of human NF-κB functions as a potent transcriptional activator and a target for v-Rel-mediated repression. Proc. Natl. Acad. Sci. USA 1992, 89, 1875–1879. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Temin, H.M. v-rel oncoproteins in the nucleus and in the cytoplasm transform chicken spleen cells. J. Virol. 1988, 62, 703–714. [Google Scholar] [PubMed]
- Sachdev, S.; Diehl, J.A.; McKinsey, T.A.; Hans, A.; Hannink, M. A threshold nuclear level of the v-Rel oncoprotein is required for transformation of avian lymphocytes. Oncogene 1997, 14, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Nehyba, J.; Bose, H.R., Jr. Interferon regulatory factor 4 contributes to transformation of v-Rel-expressing fibroblasts. Mol. Cell Biol. 2001, 21, 6369–6386. [Google Scholar] [PubMed]
- Liss, A.S.; Tiwari, R.; Kralova, J.; Bose, H.R., Jr. Cell transformation by v-Rel reveals distinct roles of AP-1 family members in Rel/NF-κB oncogenesis. Oncogene 2010, 29, 4925–4937. [Google Scholar] [CrossRef] [PubMed]
- Sif, S.; Gilmore, T.D. Interaction of the v-Rel oncoprotein with cellular transcription factor Sp1. J. Virol. 1994, 68, 7131–7138. [Google Scholar] [PubMed]
- Fan, Y.; Gelinas, C. An optimal range of transcription potency is necessary for efficient cell transformation by c-Rel to ensure optimal nuclear localization and gene-specific activation. Oncogene 2007, 26, 4038–4043. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Reynolds, J.G.; Gilmore, T.D. Deletion of either C-terminal transactivation subdomain enhances the in vitro transforming activity of human transcription factor REL in chicken spleen cells. Oncogene 2003, 22, 6928–6936. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Gerondakis, S. The c-Rel Transcription Factor in Development and Disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S. Genetic alterations in primary cutaneous CD30+ anaplastic large cell lymphoma. Genes Chromosomes. Cancer 2003, 37, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Klapper, W.; Sotnikova, A.; Callet-Bauchu, E.; Harder, L.; Bastard, C.; Schmitz, R.; Grohmann, S.; Hoppner, J.; Riemke, J.; et al. Chromosomal breakpoints affecting immunoglobulin loci are recurrent in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Cancer Res. 2006, 66, 10332–10338. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.J.; Takakuwa, T.; Ham, M.F.; Wada, N.; Liu, A.; Fujita, S.; Sakane-Ishikawa, E.; Aozasa, K. Epstein-Barr virus is integrated between REL and BCL-11A in American Burkitt lymphoma cell line (NAB-2). Lab Investig. 2004, 84, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Belguise, K.; Sonenshein, G.E. PKCtheta promotes c-Rel-driven mammary tumorigenesis in mice and humans by repressing estrogen receptor alpha synthesis. J. Clin. Investig. 2007, 117, 4009–4021. [Google Scholar] [PubMed]
- Kalaitzidis, D.; Gilmore, T.D. Genomic organization and expression of the rearranged REL proto-oncogene in the human B-cell lymphoma cell line RC-K8. Genes Chromosomes Cancer 2002, 34, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riedlinger, T.; Haas, J.; Busch, J.; Van de Sluis, B.; Kracht, M.; Schmitz, M.L. The Direct and Indirect Roles of NF-κB in Cancer: Lessons from Oncogenic Fusion Proteins and Knock-in Mice. Biomedicines 2018, 6, 36. https://doi.org/10.3390/biomedicines6010036
Riedlinger T, Haas J, Busch J, Van de Sluis B, Kracht M, Schmitz ML. The Direct and Indirect Roles of NF-κB in Cancer: Lessons from Oncogenic Fusion Proteins and Knock-in Mice. Biomedicines. 2018; 6(1):36. https://doi.org/10.3390/biomedicines6010036
Chicago/Turabian StyleRiedlinger, Tabea, Jana Haas, Julia Busch, Bart Van de Sluis, Michael Kracht, and M. Lienhard Schmitz. 2018. "The Direct and Indirect Roles of NF-κB in Cancer: Lessons from Oncogenic Fusion Proteins and Knock-in Mice" Biomedicines 6, no. 1: 36. https://doi.org/10.3390/biomedicines6010036