
Complementary PTM Profiling of Drug Response in Human Gastric Carcinoma by Immunoaffinity and IMAC Methods with Total Proteome Analysis

Abstract
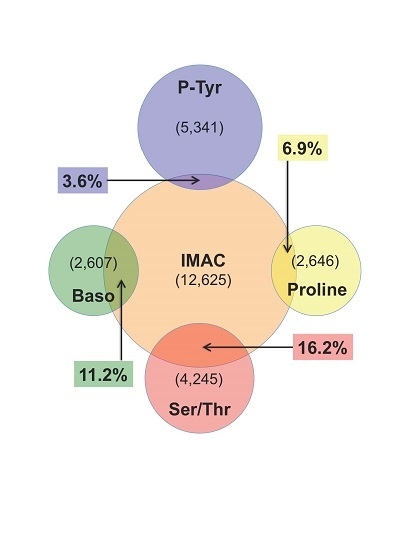
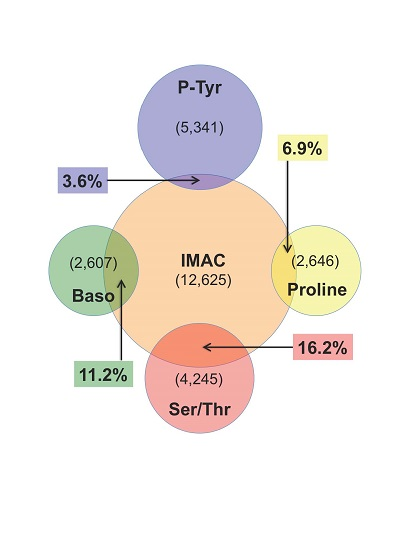
:
{kind=link}
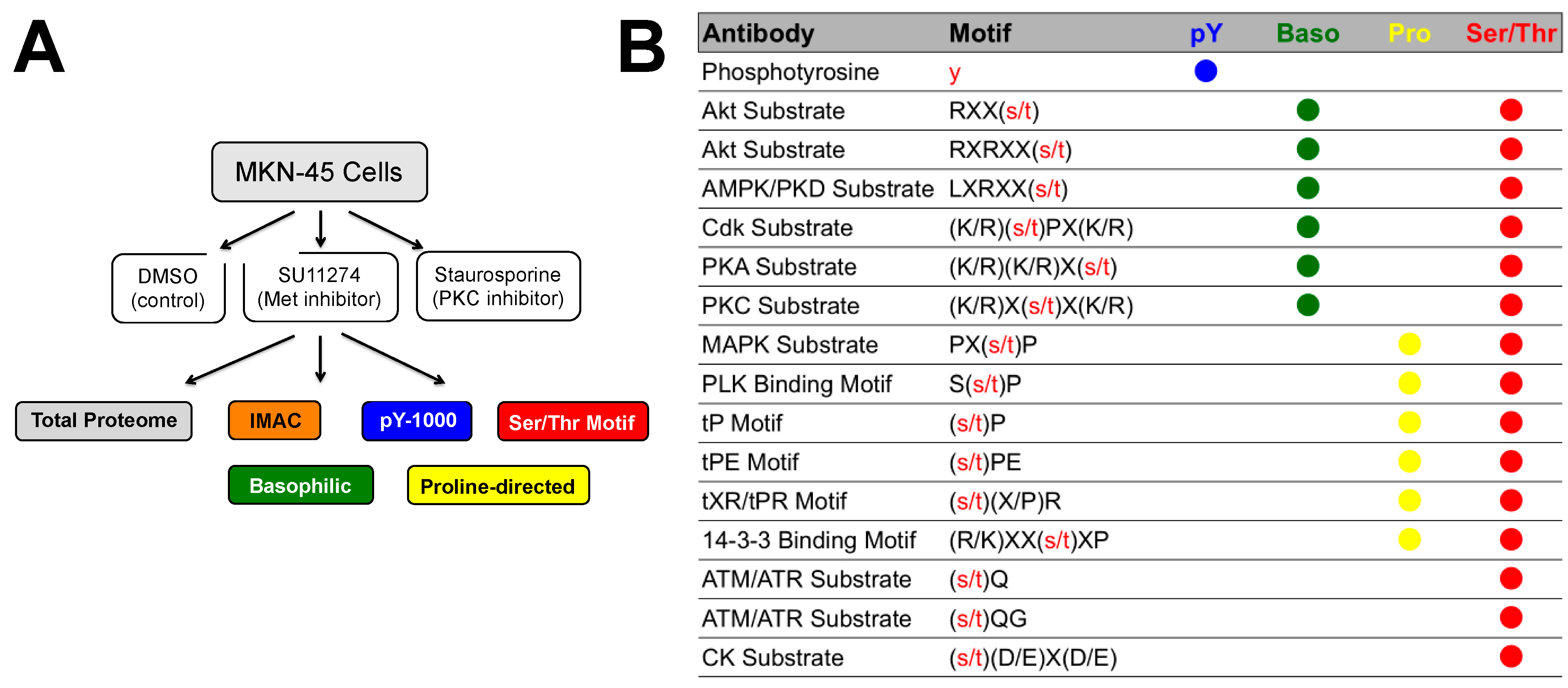{kind=link}
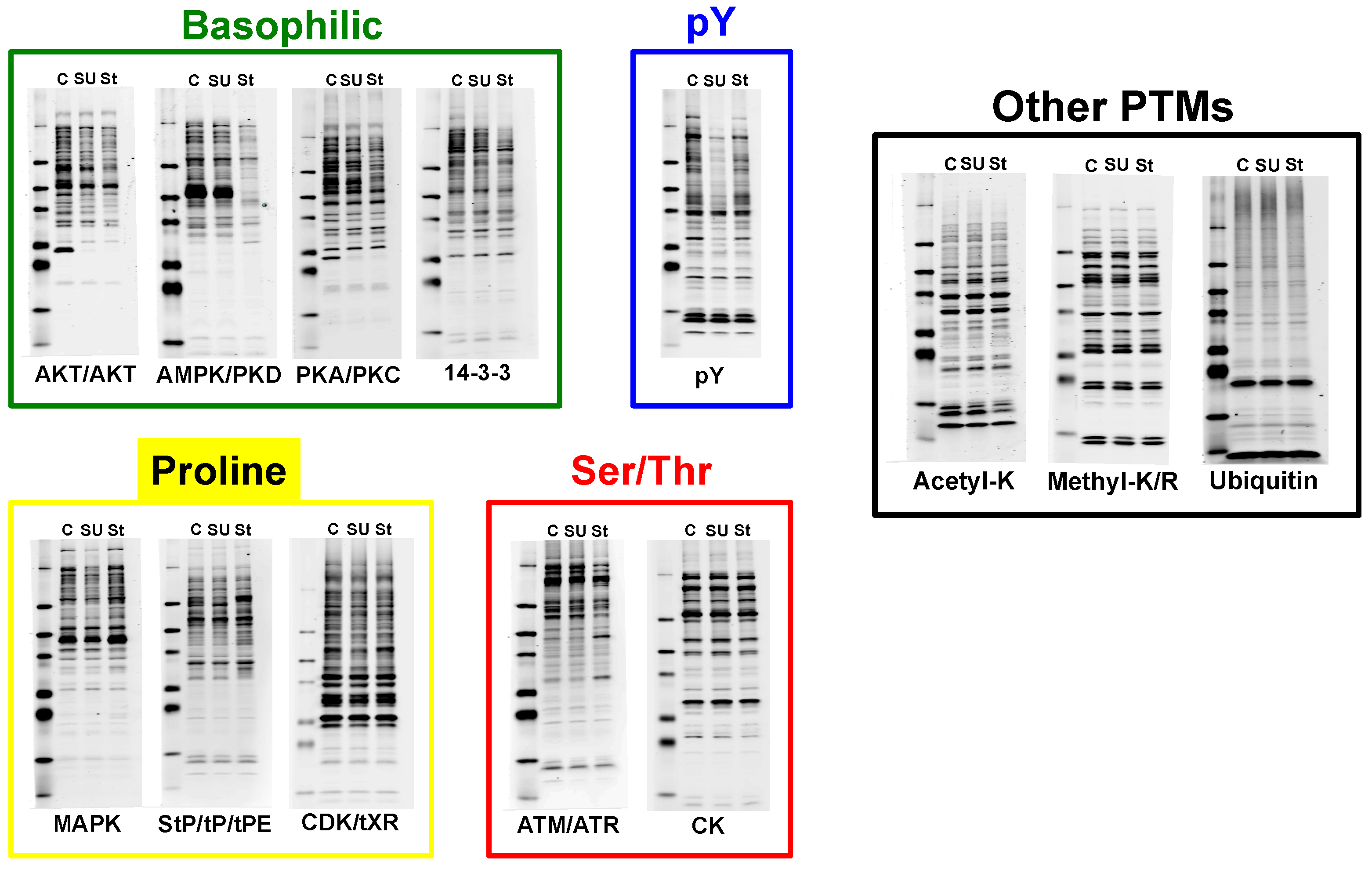{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
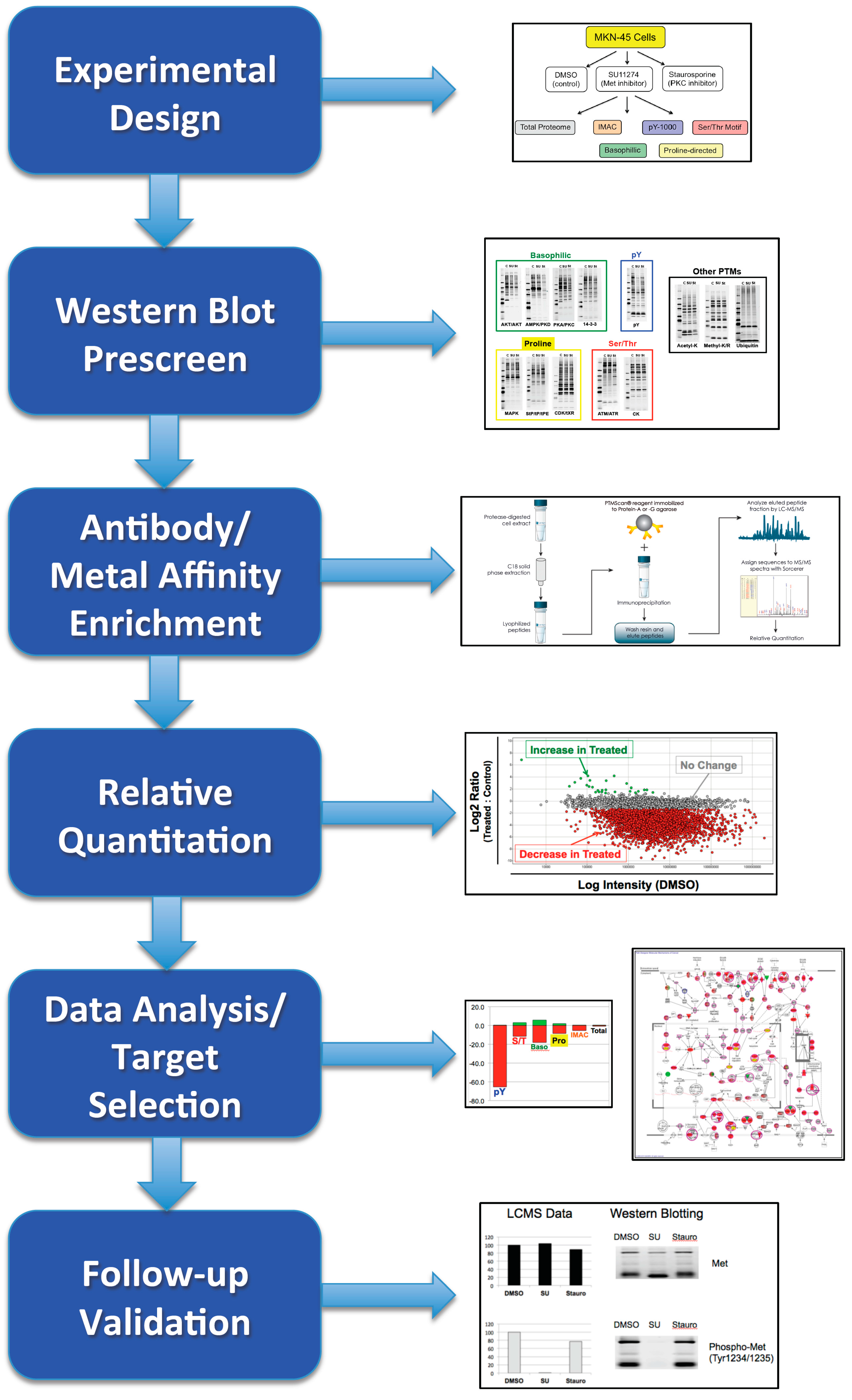
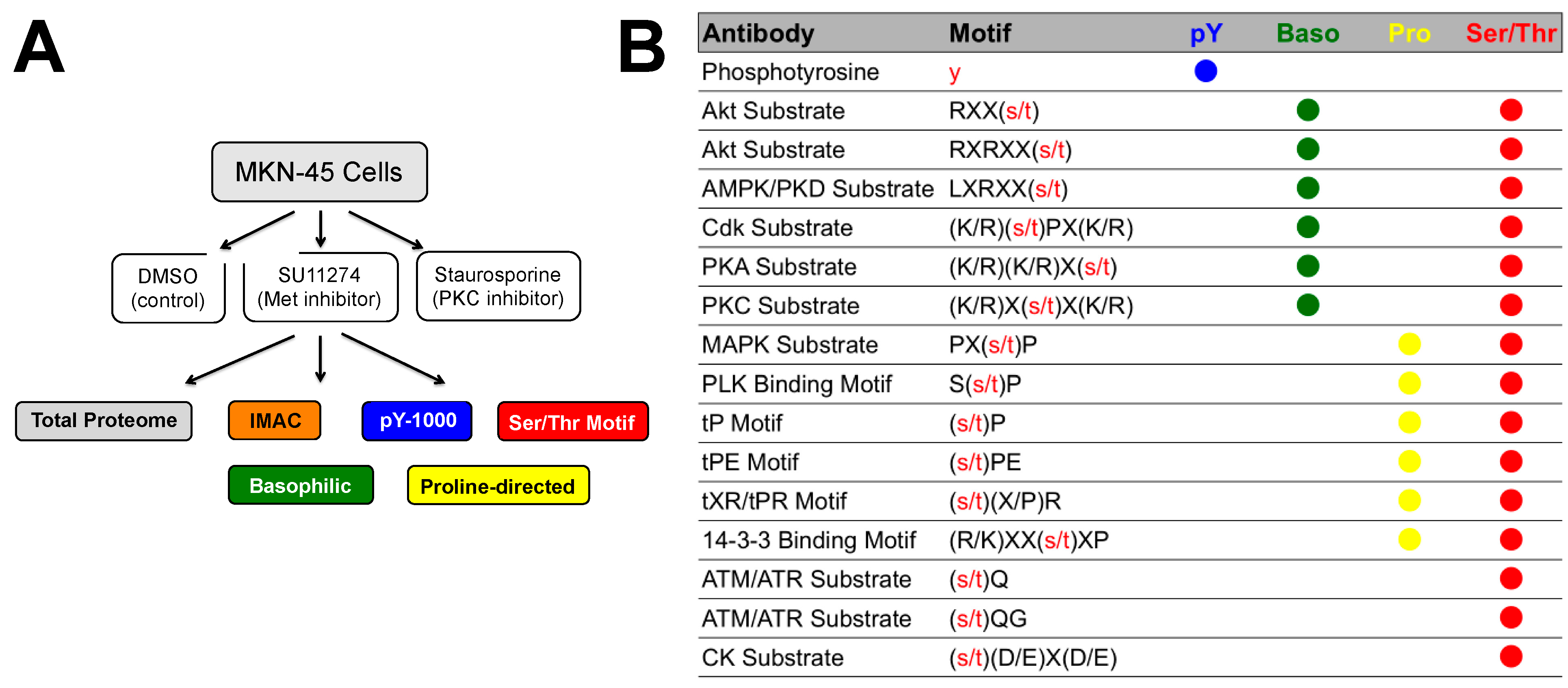
2.1. Overview
2.2. Cell Lines and Tissues
2.3. Cell Lysate Preparation
2.4. Immunoprecipitation
2.5. IMAC
2.6. LC-MS/MS Analysis
2.7. Data Analysis
2.8. Western Blotting
3. Results
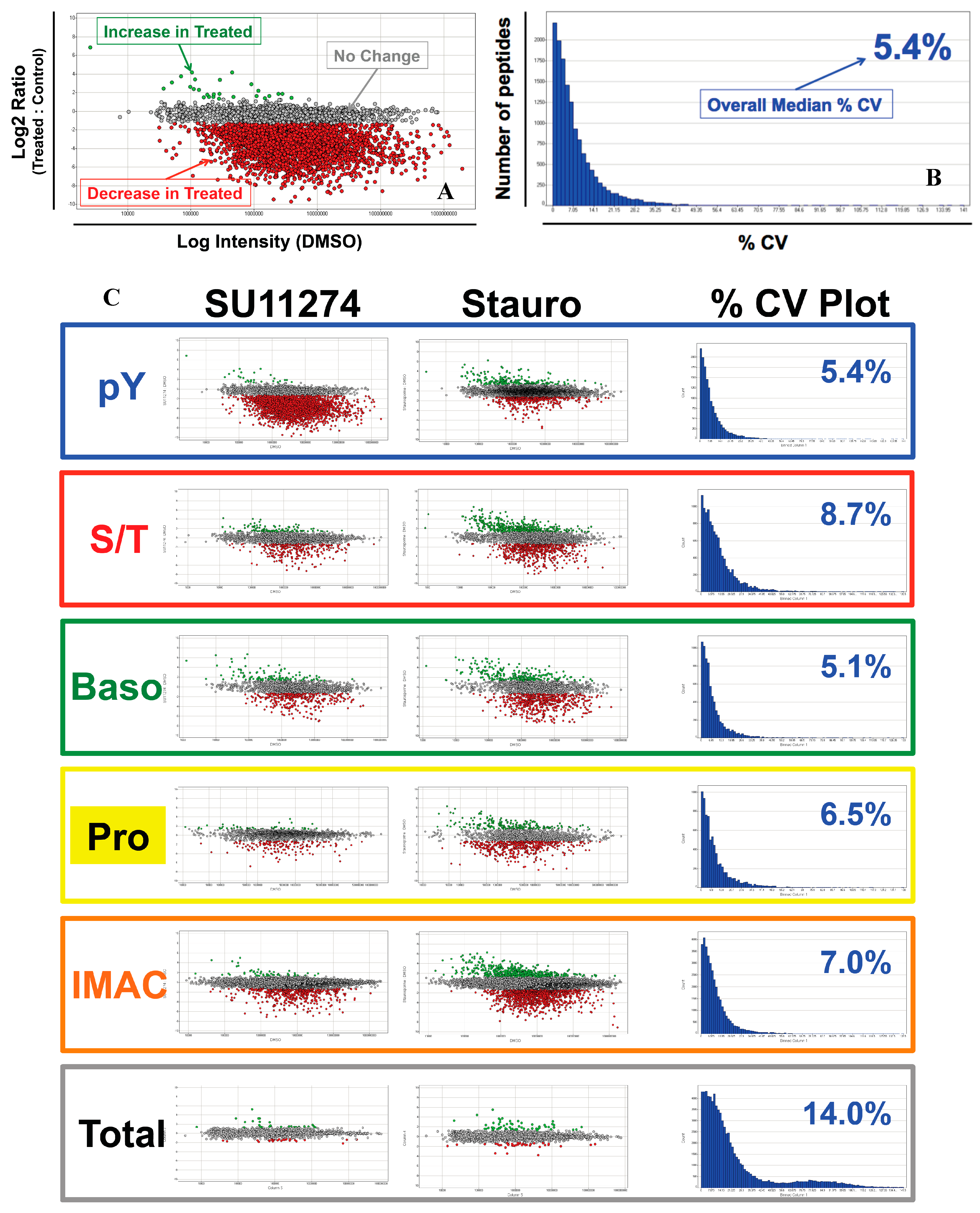
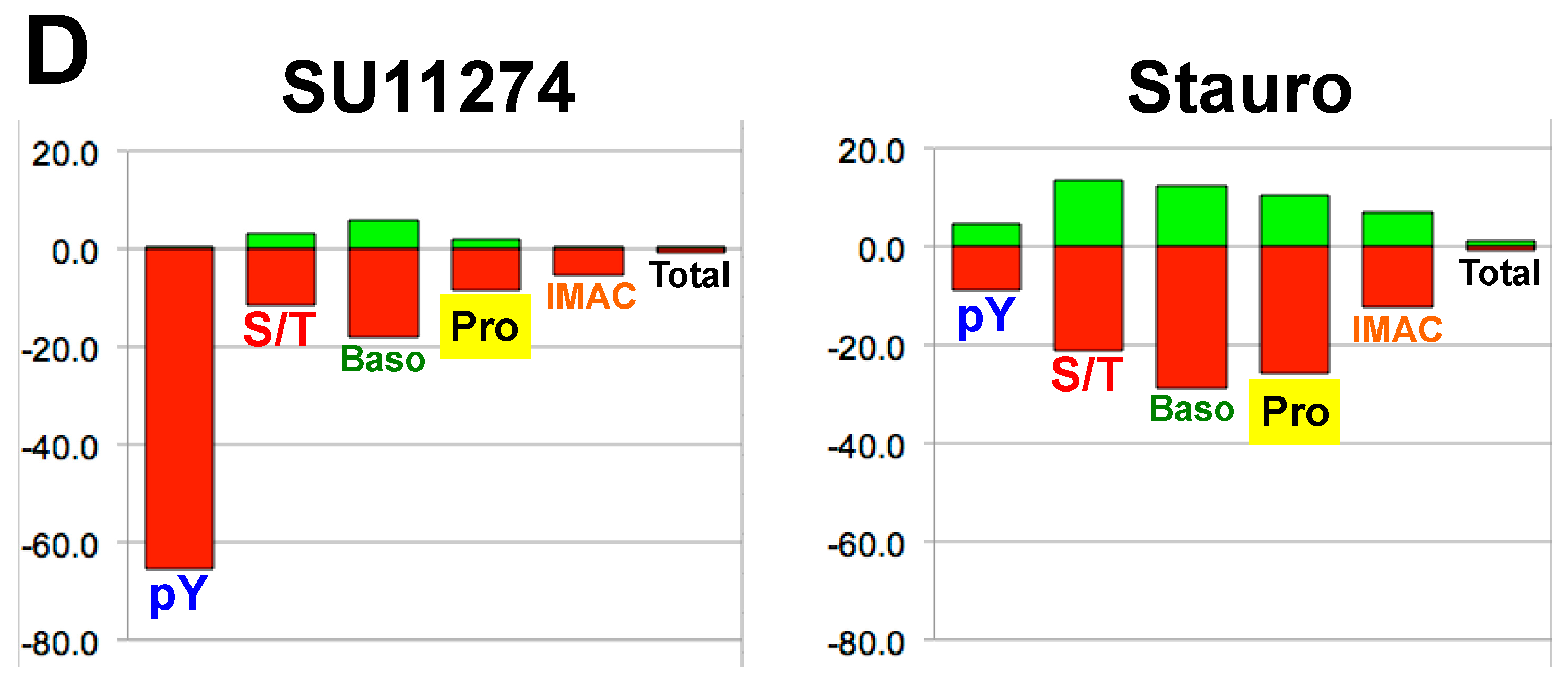
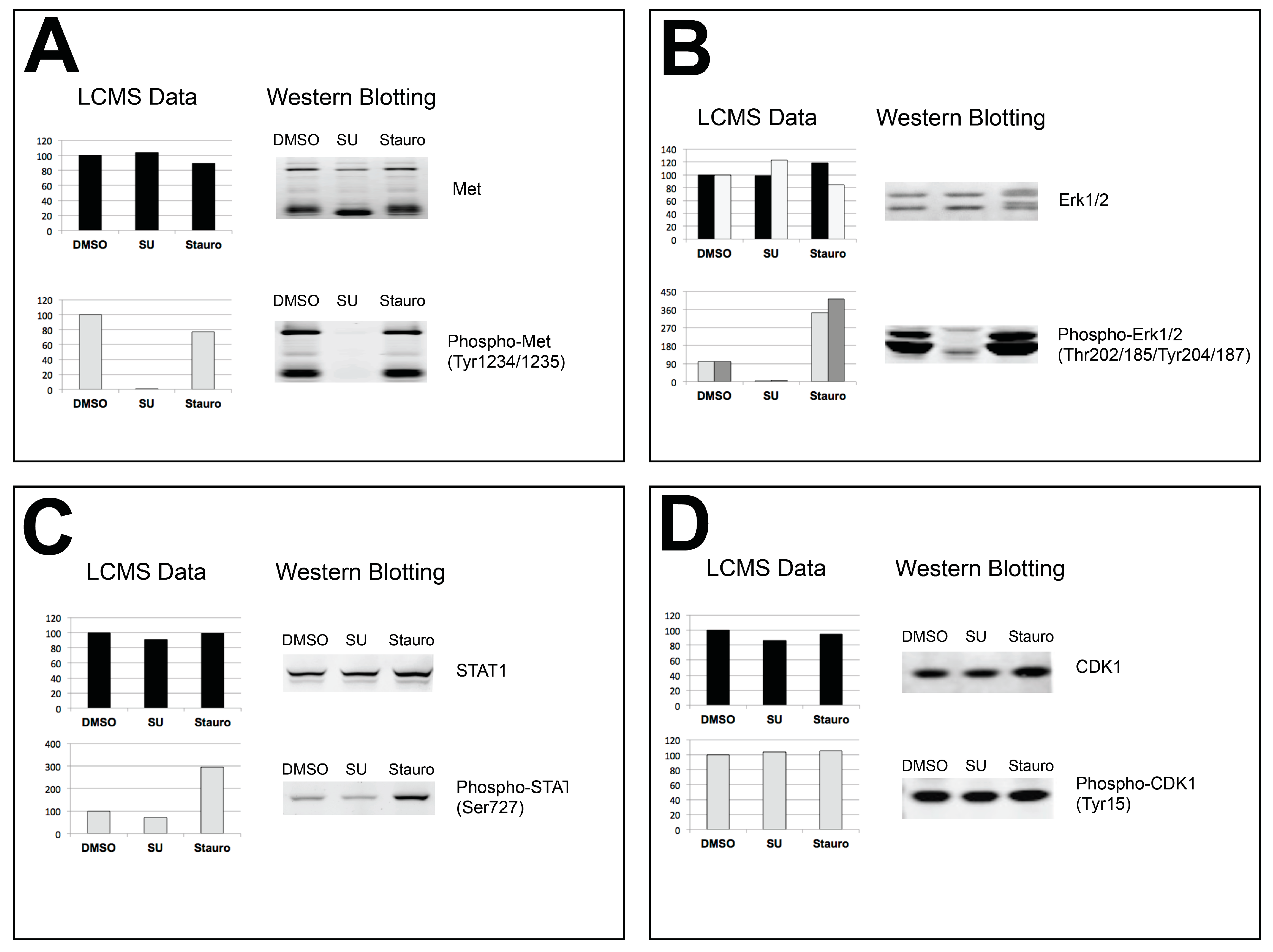
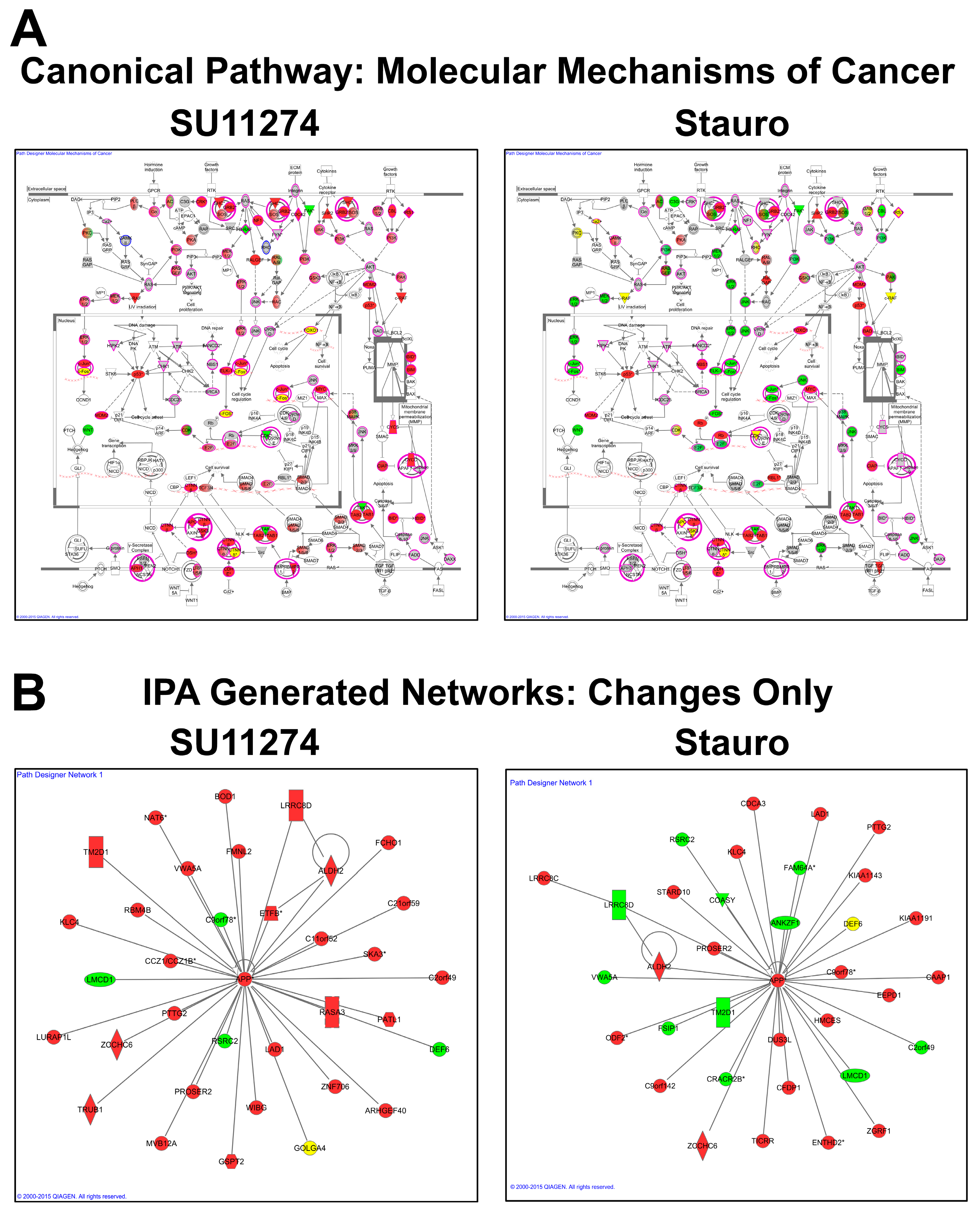
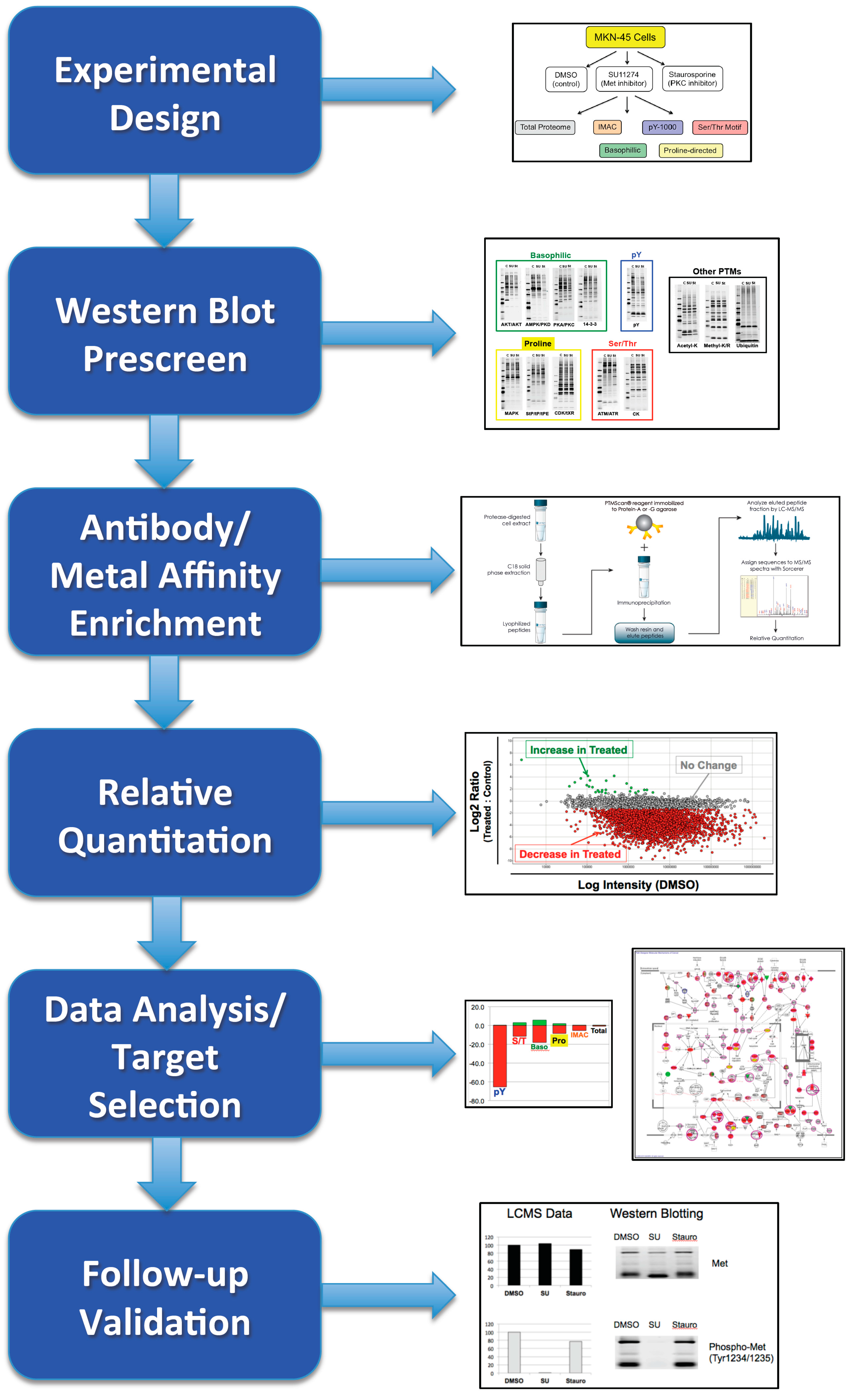
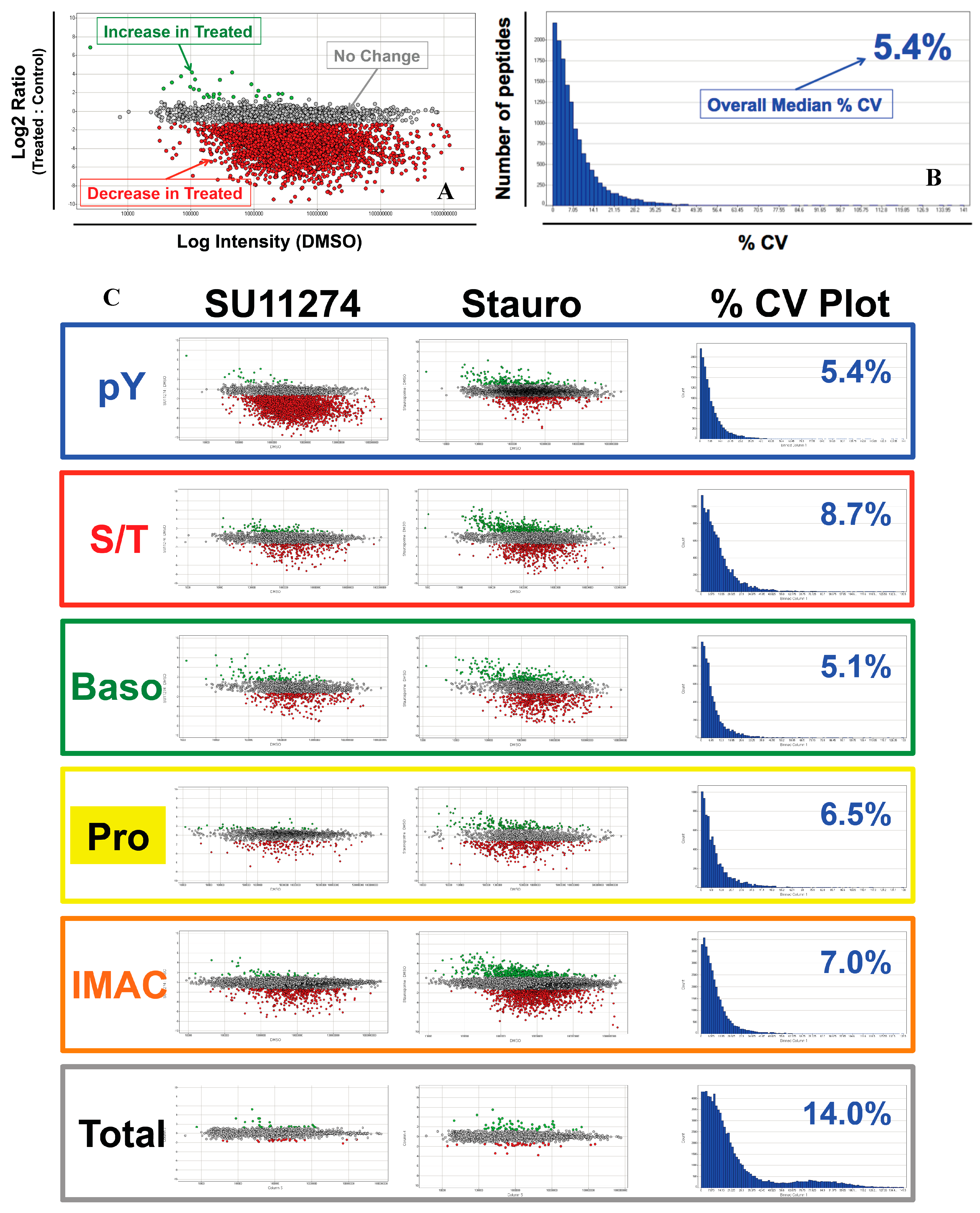
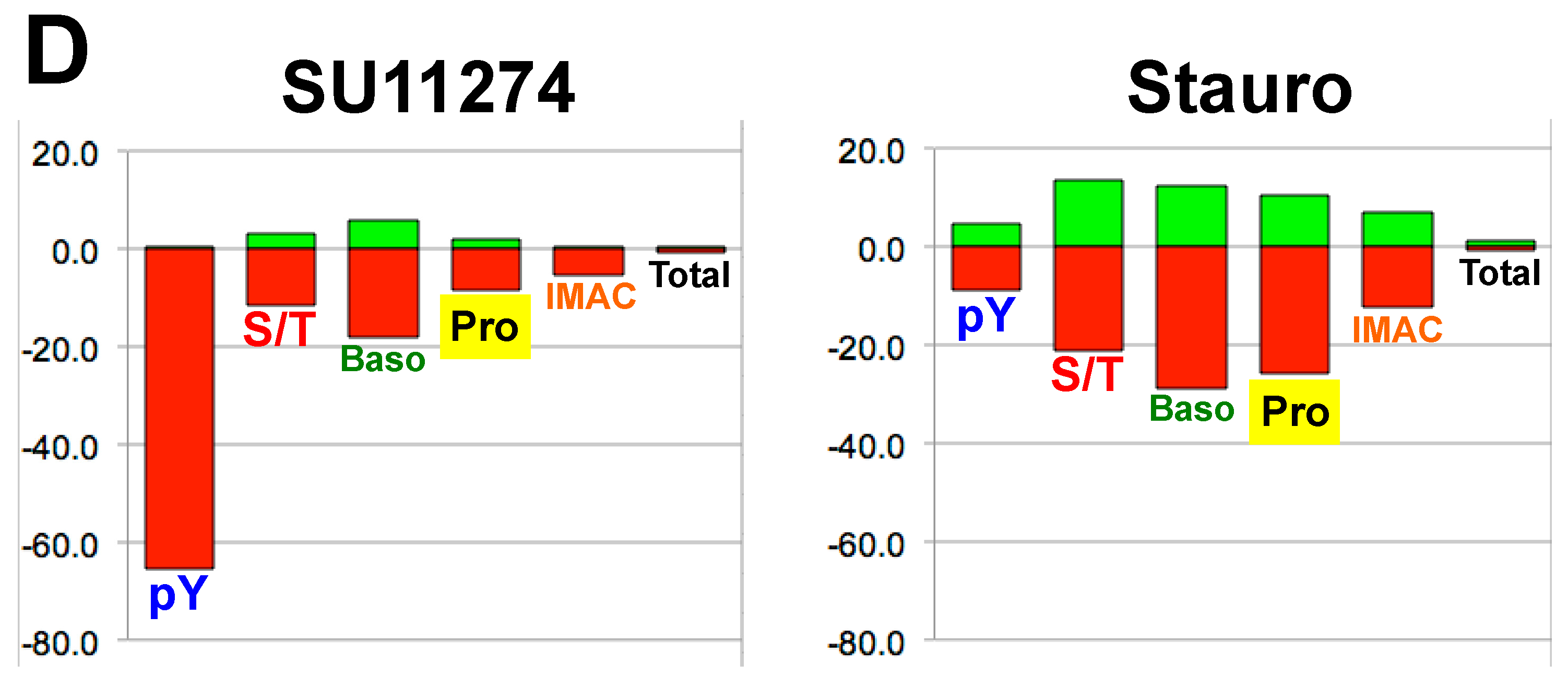
4. Discussion
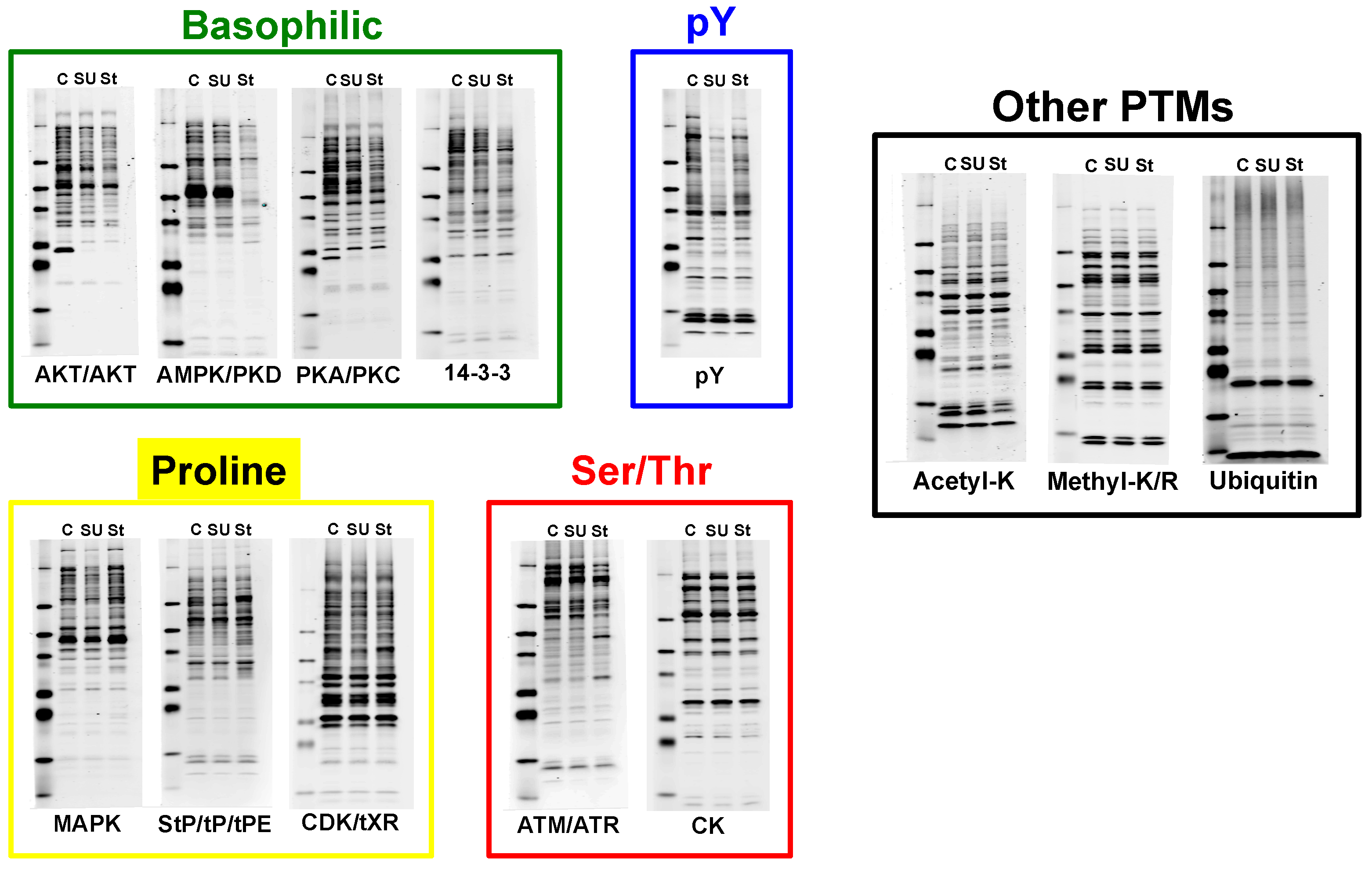
4.1. Experimental Design/Western Blot Prescreen
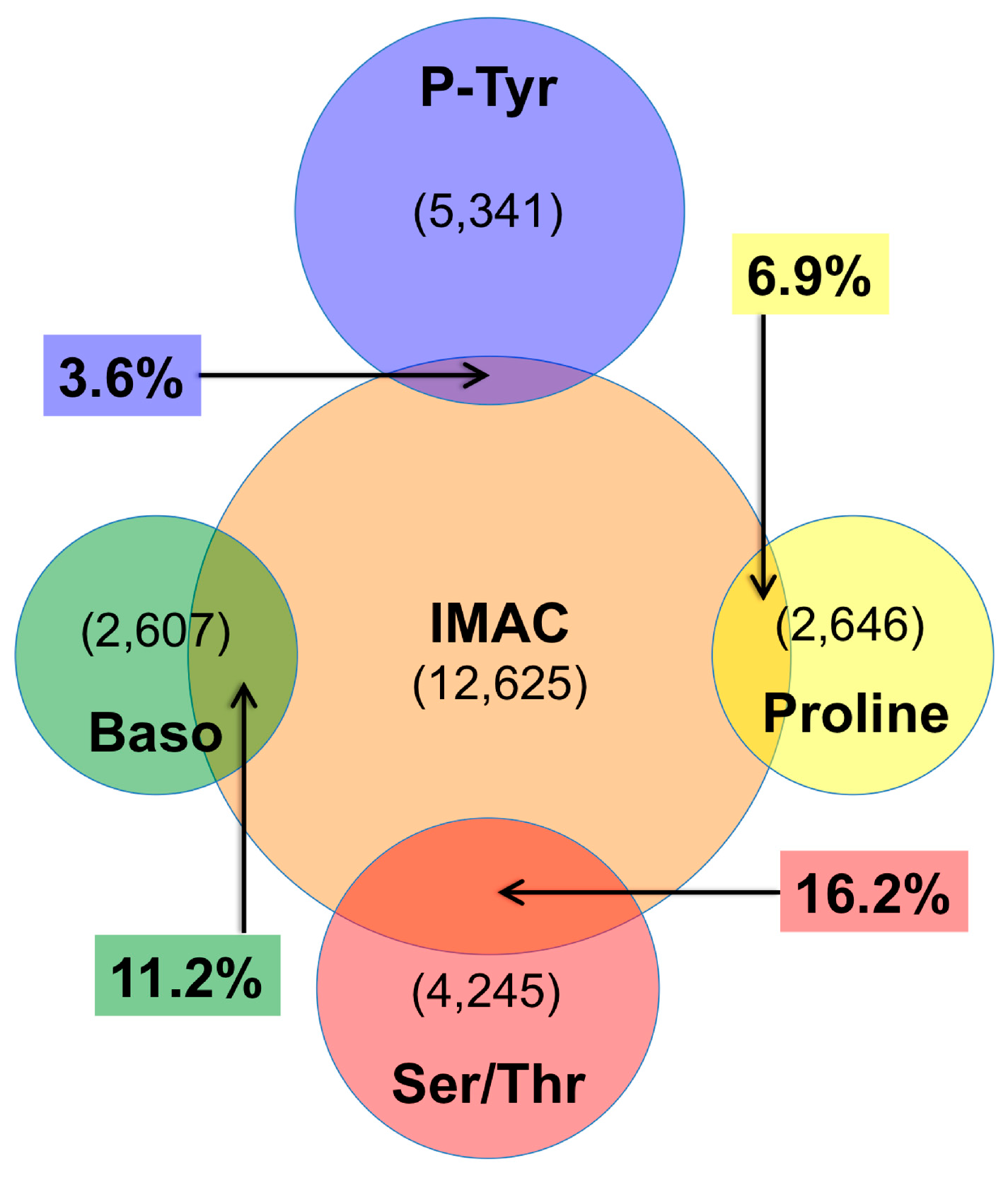
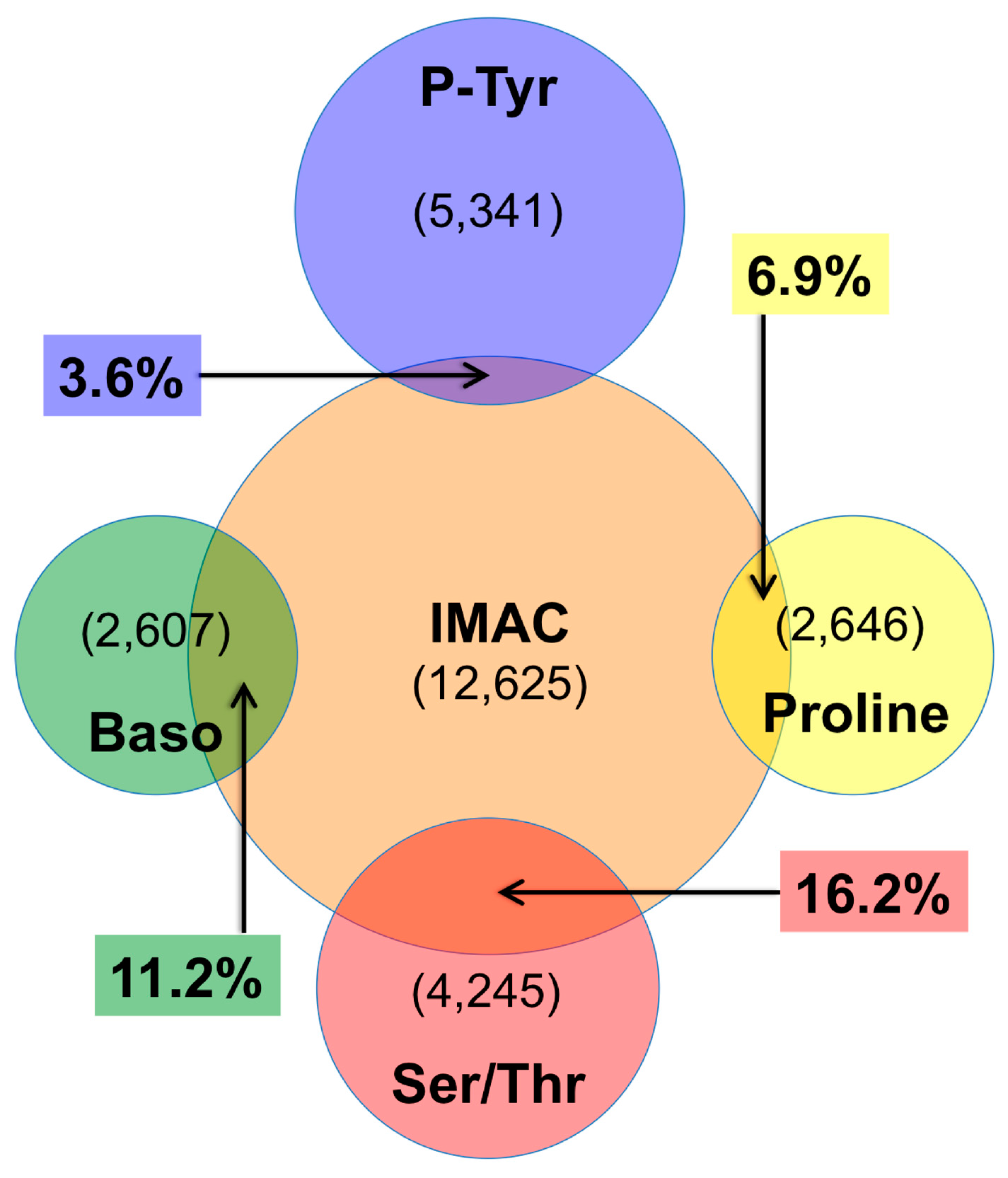
4.2. IMAC versus Antibody Enrichment
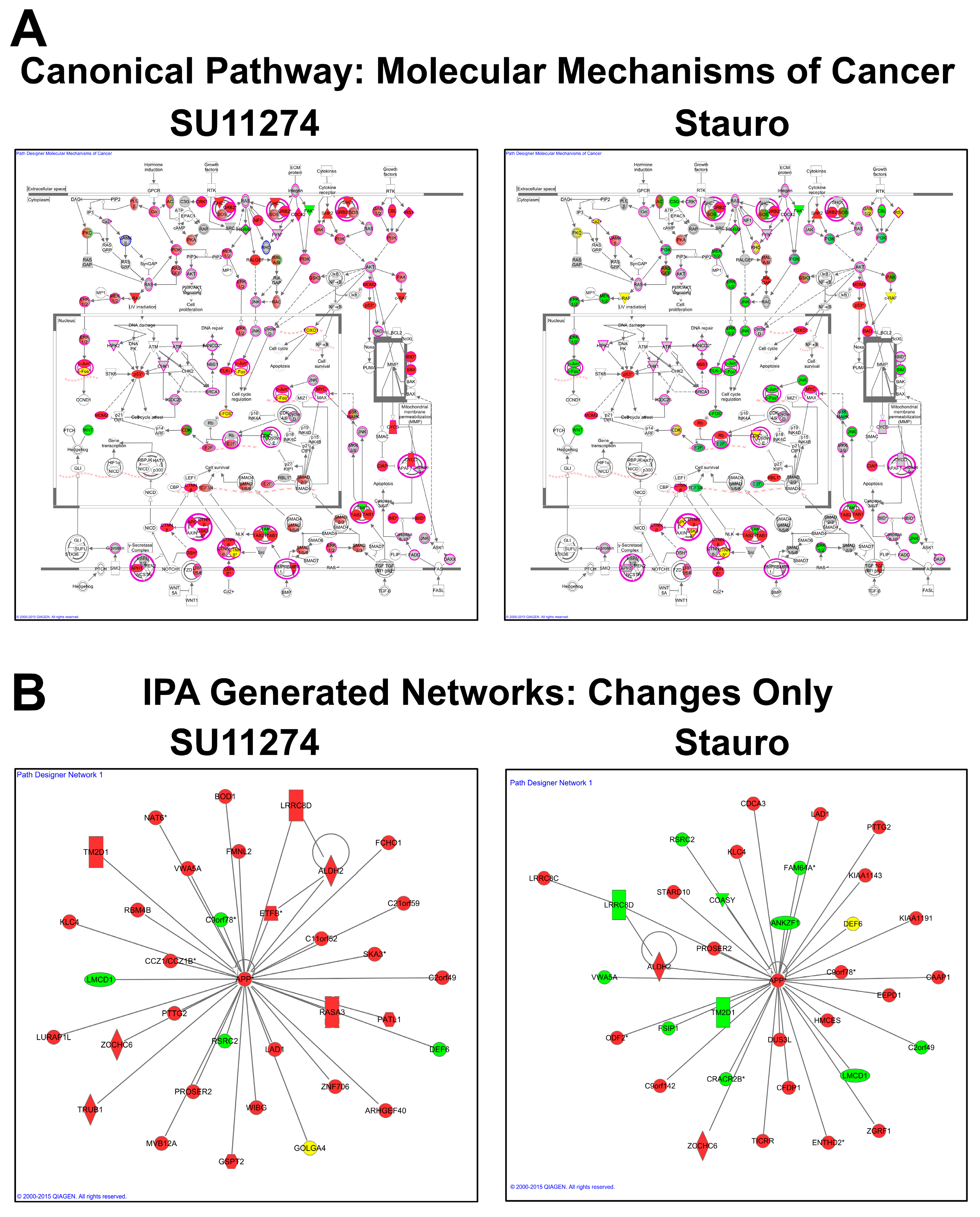
4.3. Data and Network Analysis
4.4. Validation of Results
5. Summary
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Andersen, J.N.; Sathyanarayanan, S.; Di Bacco, A.; Chi, A.; Zhang, T.; Chen, A.H.; Dolinski, B.; Kraus, M.; Roberts, B.; Arthur, W.; et al. Pathway-based identification of biomarkers for targeted therapeutics: Personalized oncology with PI3K pathway inhibitors. Sci. Transl. Med. 2010, 2, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Hunter, C.L. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell Proteomics 2006, 5, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G.; Pearson, T.W.; Borchers, C.H.; Paulovich, A.G.; Patterson, S.D.; Gillette, M.; Aebersold, R.; Carr, S.A. A human proteome detection and quantitation project. Mol. Cell Proteomics 2009, 8, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, D.; Villard, C.; Lafitte, D. Advances in top-down proteomics for disease biomarker discovery. J. Proteomics 2011, 74, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Cantin, G.T.; Yi, W.; Lu, B.; Park, S.K.; Xu, T.; Lee, J.D.; Yates, J.R., III. Combining protein-based IMAC, peptide-based IMAC, and MudPIT for efficient phosphoproteomic analysis. J. Proteome Res. 2008, 7, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Stults, J.T. Mapping the phosphorylation sites of proteins using on-line immobilized metal affinity chromatography/capillary electrophoresis/electrospray ionization multiple stage tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 1600–1606. [Google Scholar] [CrossRef]
- Choudhary, C.; Mann, M. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 2010, 11, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Dephe, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Gillette, M.; Carr, S.A.; Paulovich, A.G.; Smith, R.D.; Rodland, K.K.; Townsend, R.R.; Kinsinger, C.; Mesri, M.; Rodriguez, H.; et al. Connecting genomic alterations to cancer biology with proteomics: The NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013, 3, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Eyrich, B.; Sickmann, A.; Zahedi, R.P. Catch me if you can: Mass spectrometry-based phosphoproteomics and quantification strategies. Proteomics 2011, 11, 554–570. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Ye, M.; Zhou, H.; Jiang, X.; Zou, H.; Gong, B. Immobilized zirconium ion affinity chromatography for specific enrichment of phosphopeptides in phosphoproteome analysis. Mol. Cell Proteomics 2007, 6, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Cox, J.; Mann, M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell Proteomics 2010, 9, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Halvey, P.J.; Wang, X.; Wang, J.; Bhat, A.A.; Dhawan, P.; Li, M.; Zhang, B.; Liebler, D.C.; Slebos, R.J. Proteogenomic analysis reveals unanticipated adaptations of colorectal tumor cells to deficiencies in DNA mismatch repair. Cancer Res. 2014, 74, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, V.C.; Eagle, G.L.; Drew, P.J.; Lind, M.J.; Cawkwell, L. Biomarkers of chemotherapy resistance in breast cancer identified by proteomics: Current status. Cancer Lett. 2010, 294, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Burgess, M.W.; Gillette, M.A.; Mertins, P.; Clauser, K.R.; Mani, D.R.; Kuhn, E.W.; Farrell, L.A.; Gerszten, R.E.; Carr, S.A. Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol. Cell Proteomics 2015. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.R.; Thingholm, T.E.; Jensen, O.N.; Roepstorff, P.; Jorgensen, T.J. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell Proteomics 2005, 4, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Tabb, D.L.; Yates III, J.R. Large-scale protein identification using mass spectrometry. Biochim. Biophys. Acta 2003, 1646, 1–10. [Google Scholar] [CrossRef]
- Mann, M.; Ong, S.E.; Gronborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002, 20, 261–268. [Google Scholar] [CrossRef]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Damoc, E.; Hauschild, J.P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Monetti, M.; Nagaraj, N.; Sharma, K.; Mann, M. Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nat. Methods 2011, 8, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.L.; Williamson, J.C.; van Elburg, K.A.; Snijders, A.P.; Wright, P.C.; Dickman, M.J. Shotgun proteome analysis utilising mixed mode (reversed phase-anion exchange chromatography) in conjunction with reversed phase liquid chromatography mass spectrometry analysis. Proteom. 2010, 10, 2950–2960. [Google Scholar] [CrossRef] [PubMed]
- Rigbolt, K.T.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Stensballe, A.; Andersen, S.; Jensen, O.N. Characterization of phosphoproteins from electrophoretic gels by nanoscale Fe(III) affinity chromatography with off-line mass spectrometry analysis. Proteomics 2001, 1, 207–222. [Google Scholar] [CrossRef]
- Tan, F.; Zhang, Y.; Mi, W.; Wang, J.; Wei, J.; Cai, Y.; Qian, X. Enrichment of phosphopeptides by Fe3+-immobilized magnetic nanoparticles for phosphoproteome analysis of the plasma membrane of mouse liver. J. Proteome. Res. 2008, 7, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.S.; Geiger, T.; Chatterjee, B.; Bandilla, P.; Frohlich, F.; Cox, J.; Mann, M. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol. Cell Proteomics 2011, 10, M110-003699. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, T.D. Global and targeted quantitative proteomics for biomarker discovery. J. Chromatogr. B 2007, 847, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Villen, J.; Beausoleil, S.A.; Gerber, S.A.; Gygi, S.P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, F.; Gritsenko, M.A.; Wang, Y.; Clauss, T.; Liu, T.; Shen, Y.; Monroe, M.E.; Lopez-Ferrer, D.; Reno, T.; et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, J.R.; Zhao, L.; Abbatiello, S.E.; Burgess, M.; Kuhn, E.; Lin, C.; Pope, M.E.; Razavi, M.; Anderson, N.L.; Pearson, T.W.; et al. Evaluation of large scale quantitative proteomic assay development using peptide affinity-based mass spectrometry. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Doondeea, J.B.; Moghaddas Gholami, A.; Janning, M.C.; Lemeer, S.; Kramer, K.; Eccles, S.A.; Gollin, S.M.; Grenman, R.; Walch, A.; Feller, S.M.; Kuster, B. Quantitative chemical proteomics reveals new potential drug targets in head and neck cancer. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Shen, Y.; Camp, D.G.; Smith, R.D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 2012, 9, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Tian, R.; Ye, M.; Xu, S.; Feng, S.; Pan, C.; Jiang, X.; Li, X.; Zou, H. Highly specific enrichment of phosphopeptides by zirconium dioxide nanoparticles for phosphoproteome analysis. Electrophoresis 2007, 28, 2201–2215. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Gu, H.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.; et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteomics 2014, 13, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Villen, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Hammerle, L.P.; Andrews, P.S.; Stokes, M.P.; Mustelin, T.; Silva, J.C.; Black, R.A.; Doedens, J.R. Ubiquitin ligase substrate identification through quantitative proteomics at both the protein and peptide levels. J. Biol. Chem. 2011, 286, 41530–41538. [Google Scholar] [CrossRef] [PubMed]
- Moritz, A.; Li, Y.; Guo, A.; Villen, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal 2010, 3, ra64. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Rush, J.; Moritz, A.; Lee, K.A.; Guo, A.; Goss, V.L.; Spek, E.J.; Zhang, H.; Zha, X.M.; Polakiewicz, R.D.; Comb, M.J. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005, 23, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; Zhang, H.; Polakiewicz, R.D.; Comb, M.J. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 19855–19860. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.D.; Mertins, P.; Svinkina, T.; Carr, S.A. Large-scale identification of ubiquitination sites by mass spectrometry. Nat. Protoc. 2013, 8, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell Proteomics 2013, 12, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 2010, 28, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zha, X.; Tan, Y.; Hornbeck, P.V.; Mastrangelo, A.J.; Alessi, D.R.; Polakiewicz, R.D.; Comb, M.J. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem. 2002, 277, 39379–39387. [Google Scholar] [CrossRef] [PubMed]
- Bonnette, P.C.; Robinson, B.S.; Silva, J.C.; Stokes, M.P.; Brosius, A.D.; Baumann, A.; Buckbinder, L. Phosphoproteomic characterization of PYK2 signaling pathways involved in osteogenesis. J. Proteomics 2010, 73, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Brave, S.R.; Ratcliffe, K.; Wilson, Z.; James, N.H.; Ashton, S.; Wainwright, A.; Kendrew, J.; Dudley, P.; Broadbent, N.; Sproat, G.; et al. Assessing the activity of cediranib, a VEGFR-2/3 tyrosine kinase inhibitor, against VEGFR-1 and members of the structurally related PDGFR family. Mol. Cancer Ther. 2011, 10, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Paardekooper Overman, J.; Yi, J.S.; Bonetti, M.; Soulsby, M.; Preisinger, C.; Stokes, M.P.; Hui, L.; Silva, J.C.; Overvoorde, J.; Giansanti, P.; et al. PZR coordinates Shp2 Noonan and LEOPARD syndrome signaling in zebrafish and mice. Mol. Cell Biol. 2014, 34, 2874–2889. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, P.; Stokes, M.P.; Silva, J.C.; Scholten, A.; Heck, A.J. Interrogating cAMP-dependent kinase signaling in Jurkat T cells via a protein kinase A targeted immune-precipitation phosphoproteomics approach. Mol. Cell Proteomics 2013, 12, 3350–3359. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Comb, M.J. A wide-ranging cellular response to UV damage of DNA. Cell Cycle 2008, 7, 2097–2099. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Farnsworth, C.L.; Moritz, A.; Silva, J.C.; Jia, X.; Lee, K.A.; Guo, A.; Polakiewicz, R.D.; Comb, M.J. PTMScan Direct: Identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol. Cell Proteomics 2012, 11, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Silva, J.C.; Jia, X.; Lee, K.A.; Polakiewicz, R.D.; Comb, M.J. Quantitative Profiling of DNA Damage and Apoptotic Pathways in UV Damaged Cells Using PTMScan Direct. Int. J. Mol. Sci. 2012, 14, 286–307. [Google Scholar] [CrossRef] [PubMed]
- Soderblom, E.J.; Thompson, J.W.; Farnsworth, C.L.; Richardson, B.; Silva, J.C.; Moseley, M.A. Expanding the Depth of Coverage in Quantitative Phosphoproteomics through the Combination of Metal Oxide and Motif-Specific Antibody Enrichment Strategies. In Proceedings of American Society for Mass Spectrometry Annual Meeting, Minneapolis, MN, USA, 9–13 June 2013.
- Thompson, J.W.; Richardson, B.; Soderblom, E.J.; Farnsworth, C.L.; Silva, J.C.; Moseley, M.A. Quantitative and Qualitative Evaluation of Various Enrichment Methodologies for Posttranslational Modification Analysis by Mass Spectrometry. In Proceedings of American Society of Mass Spectrometry Annual Conference, Vancouver, BC, Canada, 20–24 May 2012.
- Berthou, S.; Aebersold, D.M.; Schmidt, L.S.; Stroka, D.; Heigl, C.; Streit, B.; Stalder, D.; Gruber, G.; Liang, C.; Howlett, A.R.; et al. The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene 2004, 23, 5387–5393. [Google Scholar] [CrossRef] [PubMed]
- Couldwell, W.T.; Hinton, D.R.; He, S.; Chen, T.C.; Sebat, I.; Weiss, M.H.; Law, R.E. Protein kinase C inhibitors induce apoptosis in human malignant glioma cell lines. FEBS lett. 1994, 345, 43–46. [Google Scholar] [CrossRef]
- Ruegg, U.T.; Burgess, G.M. Staurosporine, K-252 and UCN-01: Potent but nonspecific inhibitors of protein kinases. Trends Pharmacol. Sci. 1989, 10, 218–220. [Google Scholar] [CrossRef]
- Sattler, M.; Pride, Y.B.; Ma, P.; Gramlich, J.L.; Chu, S.C.; Quinnan, L.A.; Shirazian, S.; Liang, C.; Podar, K.; Christensen, J.G.; Salgia, R. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003, 63, 5462–5469. [Google Scholar] [PubMed]
- Fahrmann, M. Targeting protein kinase C (PKC) in physiology and cancer of the gastric cell system. Curr. Med. Chem. 2008, 15, 1175–1191. [Google Scholar] [CrossRef] [PubMed]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2316–2321. [Google Scholar] [CrossRef] [PubMed]
- Ficarro, S.B.; Zhang, Y.; Carrasco-Alfonso, M.J.; Garg, B.; Adelmant, G.; Webber, J.T.; Luckey, C.J.; Marto, J.A. Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; de Godoy, L.M.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell Proteomics 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Young, A.; Zhou, W.; Lyle, K.; Ong, C.C.; Stokes, M.P.; Silva, J.C.; Belvin, M.; Friedman, L.S.; Koeppen, H.; et al. Systems-wide analysis of K-Ras, Cdc42, and PAK4 signaling by quantitative phosphoproteomics. Mol. Cell Proteomics 2013, 12, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Di Palma, S.; Zoumaro-Djayoon, A.; Peng, M.; Post, H.; Preisinger, C.; Munoz, J.; Heck, A.J. Finding the same needles in the haystack? A comparison of phosphotyrosine peptides enriched by immuno-affinity precipitation and metal-based affinity chromatography. J. Proteomics 2013, 91, 331–337. [Google Scholar]
- Possemato, A.; Beausoleil, S.A. Complementarity between antibody-based and IMAC enrichment of phosphopeptides using TMT labeling and bRP fractionation. Cell Signaling Technology, Inc: Danvers, MA, USA, 2015. [Google Scholar]
- Hsu, J.L.; Huang, S.Y.; Chow, N.H.; Chen, S.H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75, 6843–6852. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.C.; Lahert, E.; Pike, I.; Ward, M. Quantitation of protein post-translational modifications using isobaric tandem mass tags. Bioanalysis 2015, 7, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Paliouras, G.N.; Abella, J.V.; Dube, N.; Monast, A.; Tremblay, M.L.; Park, M. Regulation of the Met receptor-tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J. Biol. Chem. 2008, 283, 34374–34383. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Bardelli, A.; Ponzetto, C.; Naldini, L.; Comoglio, P.M. Tyrosines1234-1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994, 9, 49–57. [Google Scholar] [PubMed]
- Cho, J.Y.; Katz, D.R.; Chain, B.M. Staurosporine induces rapid homotypic intercellular adhesion of U937 cells via multiple kinase activation. Br. J. Pharmacol. 2003, 140, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Shen, H.; Dai, Q.; Burzynski, F.J.; Minuk, G.Y.; Gong, Y. Extent of extracellular signal-regulated kinases phosphorylation determines the sensitivity of hepatic stellate cells to staurosporine-induced apoptosis. J. Cent. South Univ. 2012, 37, 11–16. [Google Scholar]
- Pettersson, F.; Couture, M.C.; Hanna, N.; Miller, W.H. Enhanced retinoid-induced apoptosis of MDA-MB-231 breast cancer cells by PKC inhibitors involves activation of ERK. Oncogene 2004, 23, 7053–7066. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, K.; Yonezawa, T.; Ohuchi, K. Signal transduction cascade in staurosporine-induced prostaglandin E2 production by rat peritoneal macrophages. J. Pharmacol. Exp. Ther. 2000, 293, 206–213. [Google Scholar] [PubMed]
- Uddin, S.; Sassano, A.; Deb, D.K.; Verma, A.; Majchrzak, B.; Rahman, A.; Malik, A.B.; Fish, E.N.; Platanias, L.C. Protein kinase C-δ (PKC-δ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 2002, 277, 14408–14416. [Google Scholar] [CrossRef] [PubMed]
- Vanhatupa, S.; Ungureanu, D.; Paakkunainen, M.; Silvennoinen, O. MAPK-induced Ser727 phosphorylation promotes SUMOylation of STAT1. Biochem. J. 2008, 409, 179–185. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stokes, M.P.; Farnsworth, C.L.; Gu, H.; Jia, X.; Worsfold, C.R.; Yang, V.; Ren, J.M.; Lee, K.A.; Silva, J.C. Complementary PTM Profiling of Drug Response in Human Gastric Carcinoma by Immunoaffinity and IMAC Methods with Total Proteome Analysis. Proteomes 2015, 3, 160-183. https://doi.org/10.3390/proteomes3030160
Stokes MP, Farnsworth CL, Gu H, Jia X, Worsfold CR, Yang V, Ren JM, Lee KA, Silva JC. Complementary PTM Profiling of Drug Response in Human Gastric Carcinoma by Immunoaffinity and IMAC Methods with Total Proteome Analysis. Proteomes. 2015; 3(3):160-183. https://doi.org/10.3390/proteomes3030160
Chicago/Turabian StyleStokes, Matthew P., Charles L. Farnsworth, Hongbo Gu, Xiaoying Jia, Camilla R. Worsfold, Vicky Yang, Jian Min Ren, Kimberly A. Lee, and Jeffrey C. Silva. 2015. "Complementary PTM Profiling of Drug Response in Human Gastric Carcinoma by Immunoaffinity and IMAC Methods with Total Proteome Analysis" Proteomes 3, no. 3: 160-183. https://doi.org/10.3390/proteomes3030160