
Cell Fate Decisions During Breast Cancer Development

{kind=link}
Abstract
:1. Introduction
2. Breast Cancer and Its Complex Heterogeneity

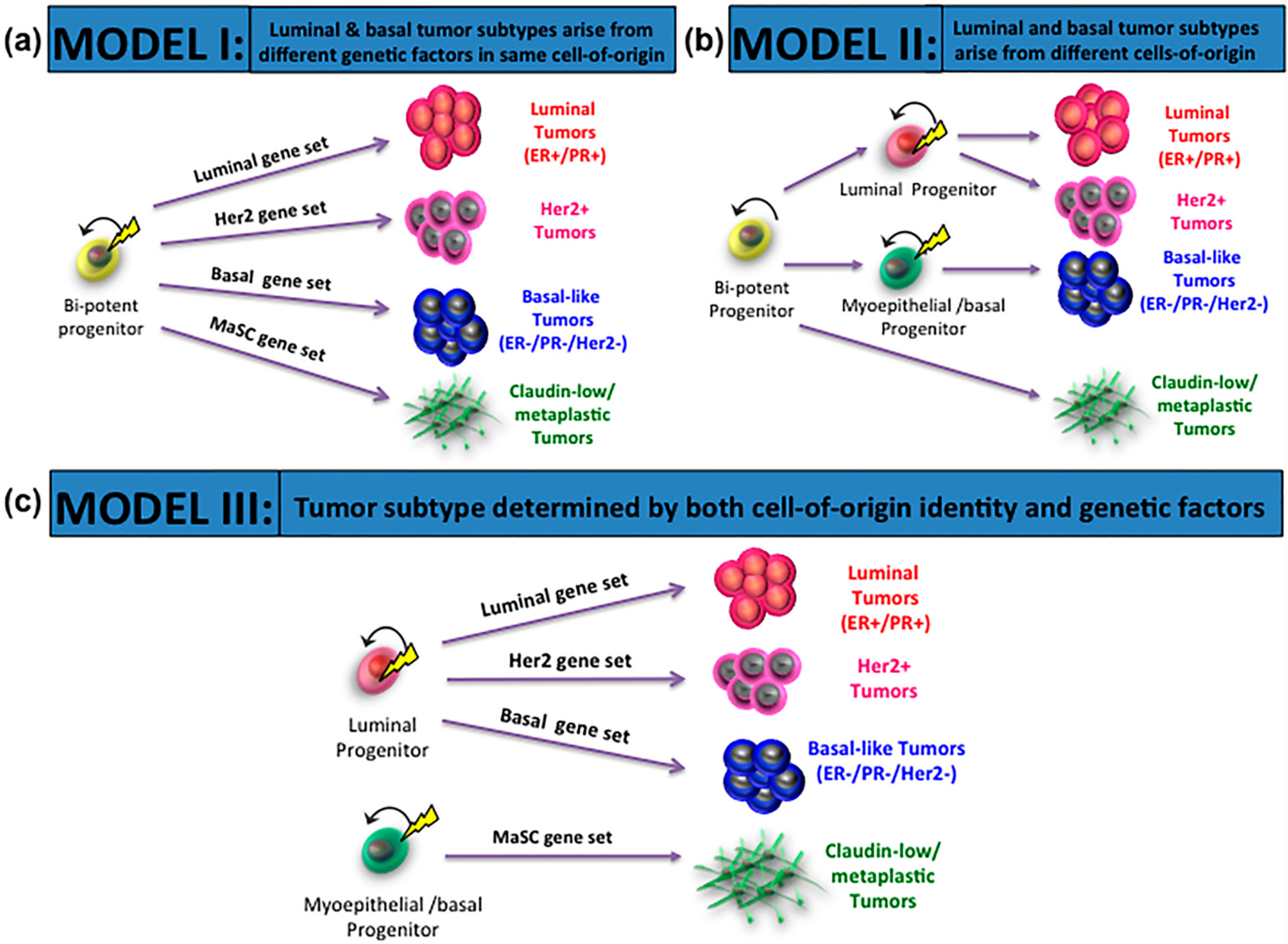
3. Cells-of-Origin and Impact on Breast Cancer Subtypes
4. Mutations-of-Origin and Impact on Breast Cancer Subtypes
5. Mechanisms Affecting Cell Fate Changes
6. Exploiting Cell Fate Changes During Development
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bertos, N.R.; Park, M. Breast cancer—One term, many entities? J. Clin. Investig. 2011, 121, 3789–3796. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009, 23, 2563–2577. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumors. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Kuperwasser, C.; Brunet, J.P.; Ramaswamy, S.; Kuo, W.L.; Gray, J.W.; Naber, S.P.; Weinberg, R.A. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat. Genet. 2005, 37, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Ince, T.A.; Richardson, A.L.; Bell, G.W.; Saitoh, M.; Godar, S.; Karnoub, A.E.; Iglehart, J.D.; Weinberg, R.A. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007, 12, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Raouf, A.; Zhao, Y.; To, K.; Stingl, J.; Delaney, A.; Barbara, M.; Iscove, N.; Jones, S.; McKinney, S.; Emerman, J.; et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell 2008, 3, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.J.; Lin, A.F.; Arendt, L.M.; Klebba, I.; Jones, A.D.; Rudnick, J.A.; DiMeo, T.A.; Gilmore, H.; Jefferson, D.M.; Graham, R.A.; et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res 2010, 12, R87. [Google Scholar] [CrossRef] [PubMed]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.J.; Arendt, L.M.; Skibinski, A.; Logvinenko, T.; Klebba, I.; Dong, S.; Smith, A.E.; Prat, A.; Perou, C.M.; Gilmore, H.; et al. Defining the cellular precursors to human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; van Bragt, M.P.A.; Li, Z. A long-lived luminal subpopulation enriched with alveolar progenitors serves as cellular origin of heterogeneous mammary tumors. Stem Cell Rep. 2015, 5, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; MacKay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Molyneux, G.; Mackay, A.; Magnay, F.A.; Atienza, M.; Kendrick, H.; Nava-Rodrigues, D.; López-García, M.Á.; Milanezi, F.; Greenow, K.; et al. Identification of cellular and genetic drivers of breast cancer heterogeneity in genetically engineered mouse tumour models: Breast cancer heterogeneity in mouse tumour models. J. Pathol. 2014, 233, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.S.; Brinkhaus, H.; Muller, U.; Muller, M.; Cardiff, R.D.; Bentires-Alj, M. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res. 2011, 71, 4344–4351. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Santiago, S.; Raeder, M.; Zhang, F.; Isabella, A.; Yang, J.; Semaan, D.J.; Chen, C.; Fox, E.A.; et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway–dependent and PI3K pathway–independent mechanisms. Nat. Med. 2011, 17, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.S.; Koren, S.; Leroy, C.; Brinkhaus, H.; Müller, U.; Klebba, I.; Müller, M.; Cardiff, R.D.; Bentires-Alj, M. Expression of PIK3CA mutant E545K in the mammary gland induces heterogeneous tumors but is less potent than mutant H1047R. Oncogenesis 2013, 2, e74. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Reavie, L.; do Couto, J.P.; de Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CAH1047R induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Hein, S.M.; Haricharan, S.; Johnston, A.N.; Toneff, M.J.; Reddy, J.P.; Dong, J.; Bu, W.; Li, Y. Luminal epithelial cells within the mammary gland can produce basal cells upon oncogenic stress. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.; Prat, A.; Sedic, M.; Proia, T.; Wronski, A.; Mazumdar, S.; Skibinski, A.; Shirley, S.H.; Perou, C.M.; Gill, G.; et al. Cell-state transitions regulated by SLUG are critical for tissue regeneration and tumor initiation. Stem Cell Rep. 2014, 2, 633–647. [Google Scholar] [CrossRef] [PubMed]
- De la O, J.P.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef] [PubMed]
- Habbe, N.; Shi, G.; Meguid, R.A.; Fendrich, V.; Esni, F.; Chen, H.; Feldmann, G.; Stoffers, D.A.; Konieczny, S.F.; Leach, S.D.; et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18913–18918. [Google Scholar] [CrossRef] [PubMed]
- Youssef, K.K.; Lapouge, G.; Bouvrée, K.; Rorive, S.; Brohée, S.; Appelstein, O.; Larsimont, J.C.; Sukumaran, V.; van de Sande, B.; Pucci, D.; et al. Adult interfollicular tumour-initiating cells are reprogrammed into an embryonic hair follicle progenitor-like fate during basal cell carcinoma initiation. Nat. Cell Biol. 2012, 14, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, K.E.; Kendrick, H.; Robertson, D.; Isacke, C.M.; Ashworth, A.; Smalley, M.J. Dissociation of estrogen receptor expression and in vivo stem cell activity in the mammary gland. J. Cell Biol. 2007, 176, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R.; Bowman, A.N.; Nusse, R. Developmental stage and time dictate the fate of Wnt/β-catenin-responsive stem cells in the mammary gland. Cell Stem Cell 2012, 11, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, A.; Breindel, J.L.; Prat, A.; Galván, P.; Smith, E.; Rolfs, A.; Gupta, P.B.; LaBaer, J.; Kuperwasser, C. The Hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell Rep. 2014, 6, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Lindley, L.E.; Curtis, K.M.; Sanchez-Mejias, A.; Rieger, M.E.; Robbins, D.J.; Briegel, K.J. The Wnt-controlled transcriptional regulator LBH is required for mammary stem cell expansion and maintenance of the basal lineage. Development 2015, 142, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.E.; Sims, A.H.; Coats, E.R.; Clarke, R.B.; Briegel, K.J. The Embryonic transcription cofactor LBH is a direct target of the Wnt signaling pathway in epithelial development and in aggressive basal subtype breast cancers. Mol. Cell. Biol. 2010, 30, 4267–4279. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Villarreal-Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Dev. Cell 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Bloushtain-Qimron, N.; Yao, J.; Snyder, E.L.; Shipitsin, M.; Campbell, L.L.; Mani, S.A.; Hu, M.; Chen, H.; Ustyansky, V.; Antosiewicz, J.E.; et al. Cell type-specific DNA methylation patterns in the human breast. Proc. Natl. Acad. Sci. USA 2008, 105, 14076–14081. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Bilenky, M.; Sigaroudinia, M.; Zhao, J.; Li, L.; Carles, A.; Delaney, A.; Tam, A.; Kamoh, B.; Cho, S.; et al. Epigenetic and transcriptional determinants of the human breast. Nat. Commun. 2015, 6, 6351. [Google Scholar] [CrossRef] [PubMed]
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 6910. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Bouras, T.; Shi, W.; Vaillant, F.; Sheridan, J.M.; Fu, N.; Breslin, K.; Jiang, K.; Ritchie, M.E.; Young, M.; et al. Global changes in the mammary epigenome are induced by hormonal cues and coordinated by Ezh2. Cell Rep. 2013, 3, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Watanabe, K.; Sun, P.; Fallahi, M.; Dai, X. Chromatin effector Pygo2 mediates Wnt-Notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. Cell Stem Cell 2013, 13, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Choudhury, S.; Kowalczyk, A.; Bessarabova, M.; Beresford-Smith, B.; Conway, T.; Kaspi, A.; Wu, Z.; Nikolskaya, T.; Merino, V.F.; et al. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet. 2011, 7, e1001369. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.U.; Boulanger, C.A.; Henry, M.D.; Sgagias, M.; Hennighausen, L.; Smith, G.H. An adjunct mammary epithelial cell population in parous females: Its role in functional adaptation and tissue renewal. Development 2002, 129, 1377–1386. [Google Scholar] [PubMed]
- Asselin-Labat, M.L.; Vaillant, F.; Sheridan, J.M.; Pal, B.; Wu, D.; Simpson, E.R.; Yasuda, H.; Smyth, G.K.; Martin, T.J.; Lindeman, G.J.; et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010, 465, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Kaanta, A.S.; Virtanen, C.; Selfors, L.M.; Brugge, J.S.; Neel, B.G. Evidence for a multipotent mammary progenitor with pregnancy-specific activity. Breast Cancer Res 2013, 15, R65. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.H.; Kunasegaran, K.; Tarulli, G.A.; de Silva, D.; Voorhoeve, P.M.; Pietersen, A.M. New insights into lineage restriction of mammary gland epithelium using parity-identified mammary epithelial cells. Breast Cancer Res. 2014, 16, R1. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Stingl, J. Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 2014, 28, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Lambe, M.; Hsieh, C.; Trichopoulos, D.; Ekbom, A.; Pavia, M.; Adami, H.O. Transient increase in the risk of breast cancer after giving birth. N. Engl. J. Med. 1994, 331, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, S.H.; Lee, Y.J.; Kang, E.S.; Lee, B.W.; Cha, B.S.; Kim, J.W.; Song, D.H.; Lee, H.C. Transcription factor Snail is a novel regulator of adipocyte differentiation via inhibiting the expression of peroxisome proliferator-activated receptor γ. Cell. Mol. Life Sci. 2013, 70, 3959–3971. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, S.; Honeth, G.; Ginestier, C.; Shinomiya, I.; Marlow, R.; Buchupalli, B.; Gazinska, P.; Brown, J.; Catchpole, S.; Liu, S.; et al. Growth hormone is secreted by normal breast epithelium upon progesterone stimulation and increases proliferation of stem/progenitor cells. Stem Cell Rep. 2014, 2, 780–793. [Google Scholar] [CrossRef]
- Rajaram, R.D.; Buric, D.; Caikovski, M.; Ayyanan, A.; Rougemont, J.; Shan, J.; Vainio, S.J.; Yalcin-Ozuysal, O.; Brisken, C. Progesterone and Wnt4 control mammary stem cells via myoepithelial crosstalk. EMBO J. 2015, 34, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Badders, N.M.; Goel, S.; Clark, R.J.; Klos, K.S.; Kim, S.; Bafico, A.; Lindvall, C.; Williams, B.O.; Alexander, C.M. The Wnt receptor, Lrp5, is expressed by mouse mammary stem cells and is required to maintain the basal lineage. PLoS ONE 2009, 4, e6594. [Google Scholar] [CrossRef] [PubMed]
- Shiah, Y.J.; Tharmapalan, P.; Casey, A.E.; Joshi, P.A.; McKee, T.D.; Jackson, H.W.; Beristain, A.G.; Chan-Seng-Yue, M.A.; Bader, G.D.; Lydon, J.P.; et al. A progesterone-CXCR4 axis controls mammary progenitor cell fate in the adult gland. Stem Cell Rep. 2015, 4, 313–322. [Google Scholar] [CrossRef]
- Anonymous. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet 1997, 350, 1047–1059. [Google Scholar]
- Chlebowski, R.T.; Hendrix, S.L.; Langer, R.D.; Stefanick, M.L.; Gass, M.; Lane, D.; Rodabough, R.J.; Gilligan, M.A.; Cyr, M.G.; Thomson, C.A.; et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: The women’s health initiative randomized trial. JAMA 2003, 289, 3243–3253. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.K.; Paganini-Hill, A.; Wan, P.C.; Pike, M.C. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J. Natl. Cancer Inst. 2000, 92, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Ao, X.; Grill, C.; Russo, I.H. Pattern of distribution of cells positive for estrogen receptor α and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res. Treat. 1999, 53, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.B.; Spence, K.; Anderson, E.; Howell, A.; Okano, H.; Potten, C.S. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev. Biol. 2005, 277, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Petz, L.N.; Nardulli, A.M. Estrogen receptor α and Sp1 regulate progesterone receptor gene expression. Mol. Cell. Endocrinol. 2003, 201, 165–175. [Google Scholar] [CrossRef]
- Santos, S.J.; Haslam, S.Z.; Conrad, S.E. Signal transducer and activator of transcription 5a mediates mammary ductal branching and proliferation in the nulliparous mouse. Endocrinology 2010, 151, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, E.O.; Deal, A.M.; Anders, C.K.; Prat, A.; Perou, C.M.; Carey, L.A.; Muss, H.B. Age-specific changes in intrinsic breast cancer subtypes: A focus on older women. Oncologist 2014, 19, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.J.; McClelland, R.A.; Candlish, W.; Blamey, R.W.; Nicholson, R.I. Hetergeneity of oestrogen receptor expression in normal and malignant breast tissue. Eur. J. Cancer 1992, 28, 34–37. [Google Scholar] [CrossRef]
- Shoker, V.S.; Jarvis, C.; Clarke, R.B.; Anderson, E.; Hewlett, J.; Davies, M.P.A.; Sibson, D.R.; Sloane, J.P. Estrogen receptor-positive proliferating cells in the normal and precancerous breast. Am. J. Pathol. 1999, 155, 1811–1815. [Google Scholar] [CrossRef]
- Shoker, B.S.; Jarvis, C.; Sibson, D.R.; Walker, C.; Sloane, J.P. Oestrogen receptor expression in the normal and pre-cancerous breast. J. Pathol. 1999, 188, 237–244. [Google Scholar] [CrossRef]
- Walker, R.; Martin, C. The aged breast. J. Pathol. 2007, 211, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Garbe, J.C.; Pepin, F.; Pelissier, F.A.; Sputova, K.; Fridriksdottir, A.J.; Guo, D.E.; Villadsen, R.; Park, M.; Petersen, O.W.; Borowsky, A.D.; et al. Accumulation of multipotent progenitors with a basal differentiation bias during aging of human mammary epithelia. Cancer Res. 2012, 72, 3687–3701. [Google Scholar] [CrossRef] [PubMed]
- Pelissier, F.A.; Garbe, J.C.; Ananthanarayanan, B.; Miyano, M.; Lin, C.; Jokela, T.; Kumar, S.; Stampfer, M.R.; Lorens, J.B.; LaBarge, M.A. Age-related dysfunction in mechanotransduction impairs differentiation of human mammary epithelial progenitors. Cell Rep. 2014, 7, 1926–1939. [Google Scholar] [CrossRef] [PubMed]
- Kannan, N.; Huda, N.; Tu, L.; Droumeva, R.; Aubert, G.; Chavez, E.; Brinkman, R.R.; Lansdorp, P.; Emerman, J.; Abe, S.; et al. The luminal progenitor compartment of the normal human mammary gland constitutes a unique site of telomere dysfunction. Stem Cell Rep. 2013, 1, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Sputova, K.; Garbe, J.C.; Pelissier, F.A.; Chang, E.; Stampfer, M.R.; LaBarge, M.A. Aging phenotypes in cultured normal human mammary epithelial cells are correlated with decreased telomerase activity independent of telomere length. Genome Integr. 2013, 4. [Google Scholar] [CrossRef]
- Pirone, J.R.; D’Arcy, M.; Stewart, D.A.; Hines, W.C.; Johnson, M.; Gould, M.N.; Yaswen, P.; Jerry, D.J.; Schneider, S.S.; Troester, M.A. Age-associated gene expression in normal breast tissue mirrors qualitative age-at-incidence patterns for breast cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Koestler, D.C.; Cheng, C.; Christensen, B.C. Age-related DNA methylation in normal breast tissue and its relationship with invasive breast tumor methylation. Epigenetics 2014, 9, 268–275. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, K.; Wronski, A.; Skibinski, A.; Phillips, S.; Kuperwasser, C. Cell Fate Decisions During Breast Cancer Development. J. Dev. Biol. 2016, 4, 4. https://doi.org/10.3390/jdb4010004
Gross K, Wronski A, Skibinski A, Phillips S, Kuperwasser C. Cell Fate Decisions During Breast Cancer Development. Journal of Developmental Biology. 2016; 4(1):4. https://doi.org/10.3390/jdb4010004
Chicago/Turabian StyleGross, Kayla, Ania Wronski, Adam Skibinski, Sarah Phillips, and Charlotte Kuperwasser. 2016. "Cell Fate Decisions During Breast Cancer Development" Journal of Developmental Biology 4, no. 1: 4. https://doi.org/10.3390/jdb4010004