Compression of Large Sets of Sequence Data Reveals Fine Diversification of Functional Profiles in Multigene Families of Proteins: A Study for Peptidyl-Prolyl cis/trans Isomerases (PPIase)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
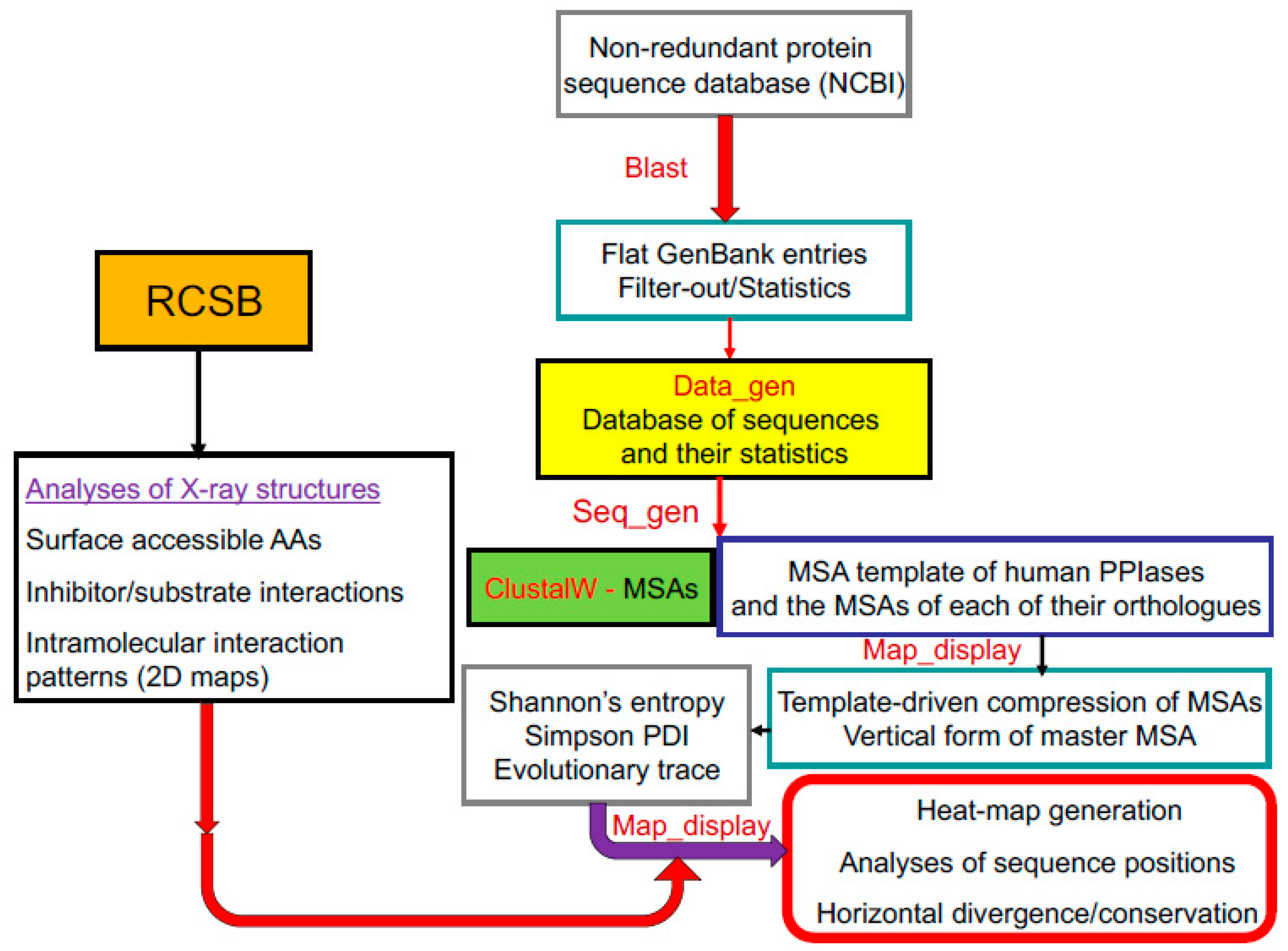
2. Materials and Methods
2.1. BLAST Searches
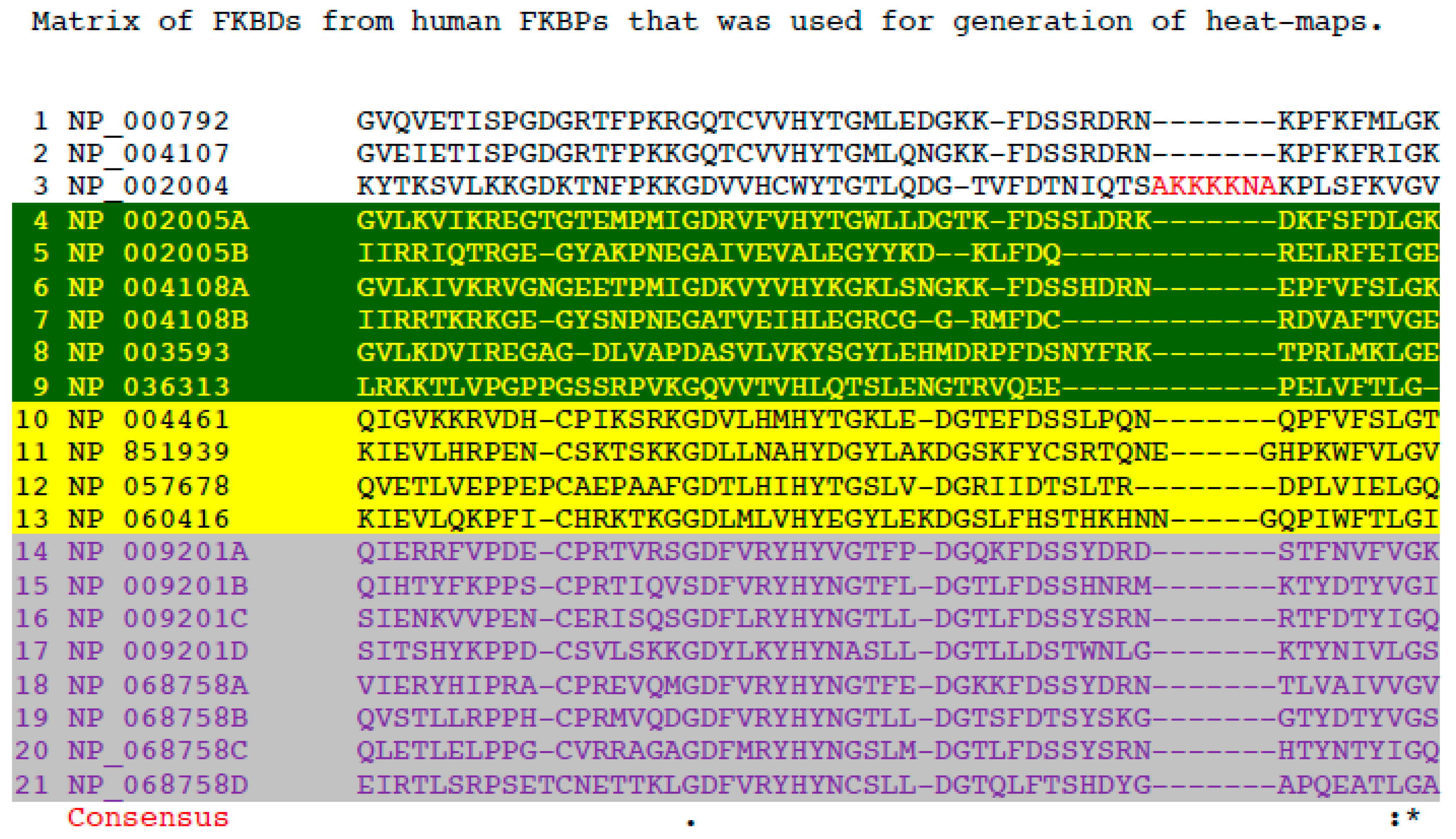
2.2. Multiple Sequence Alignment
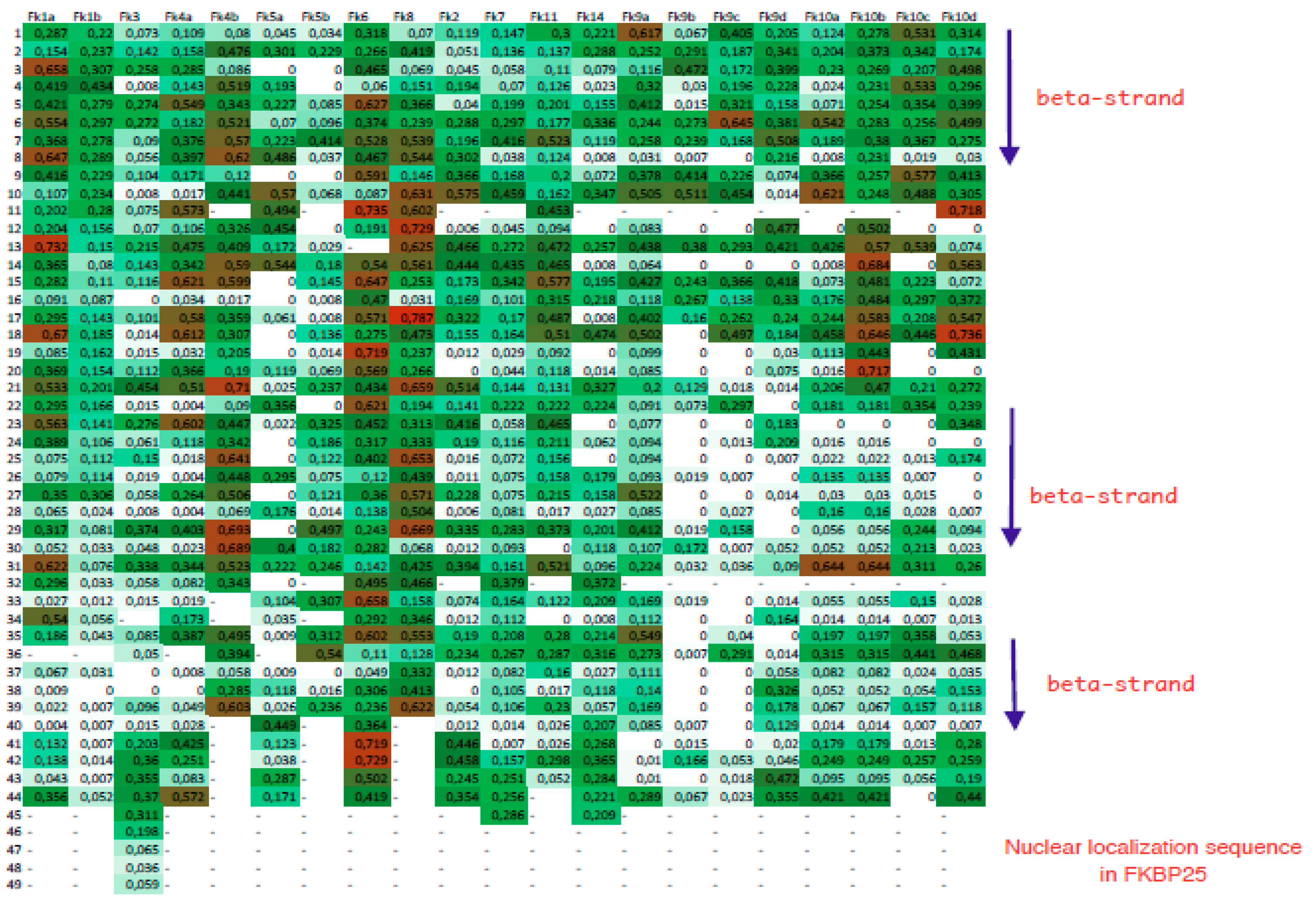
2.3. Calculation of Some Sequence Attributes and Their Statistics
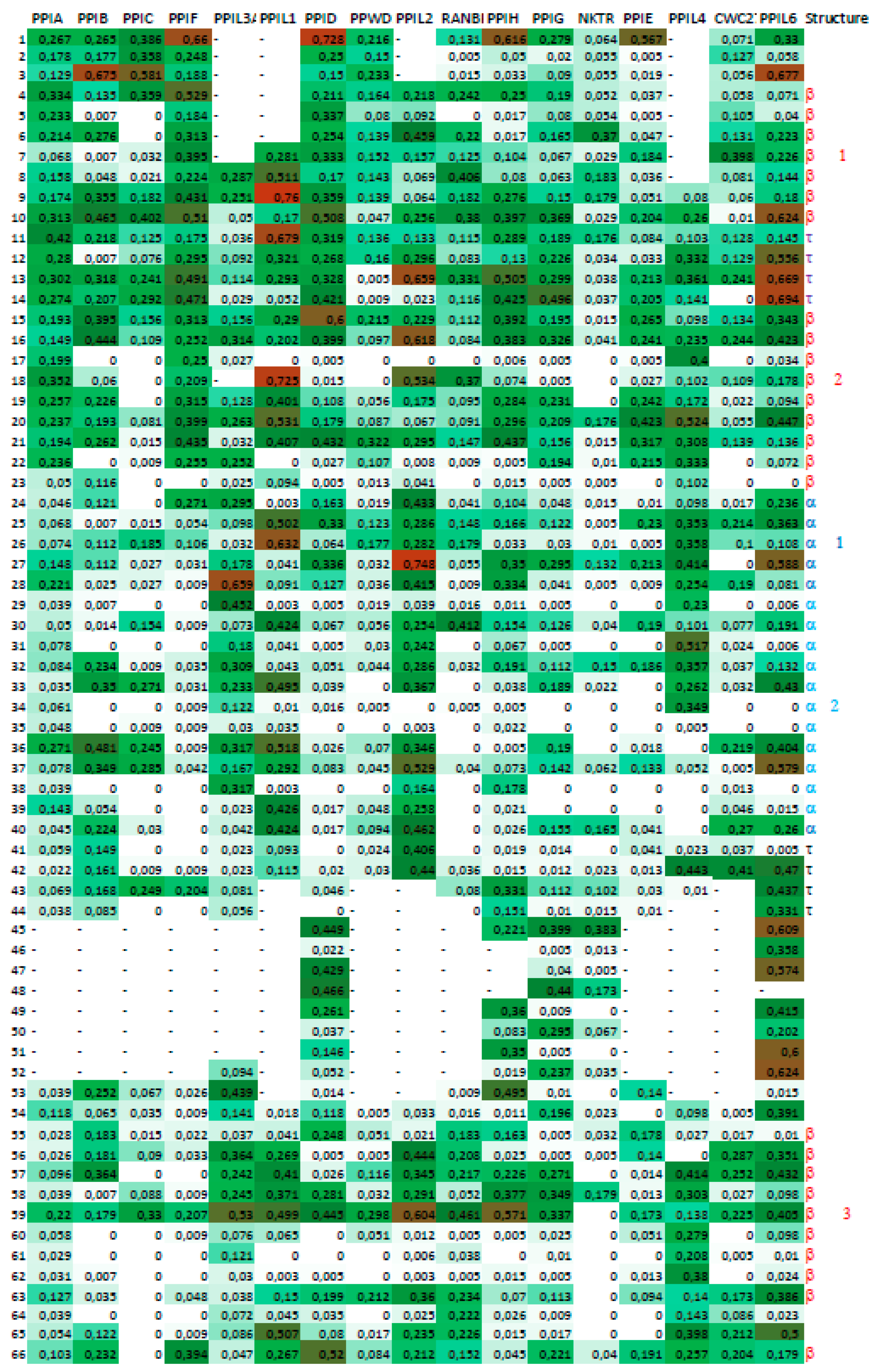
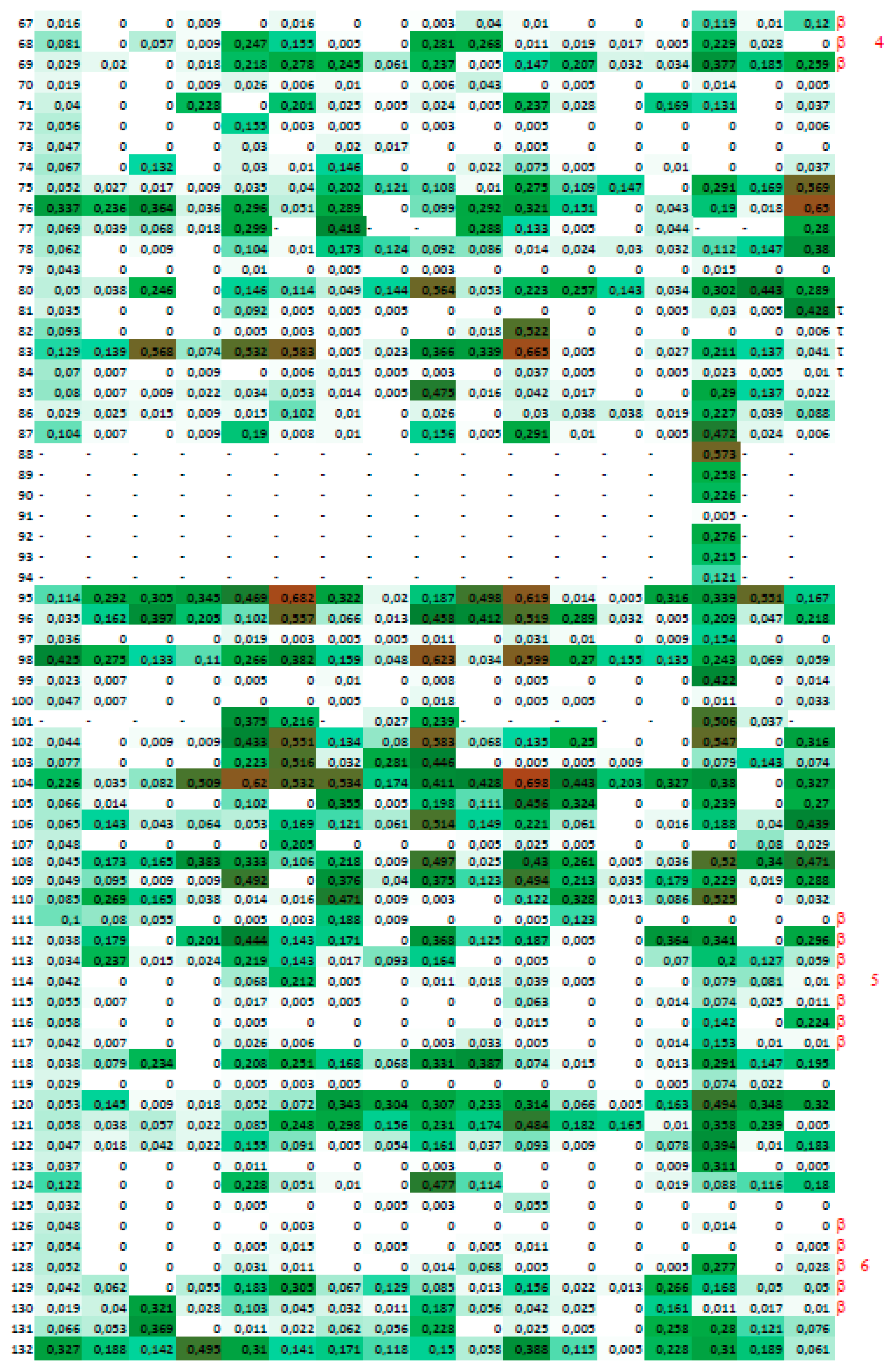
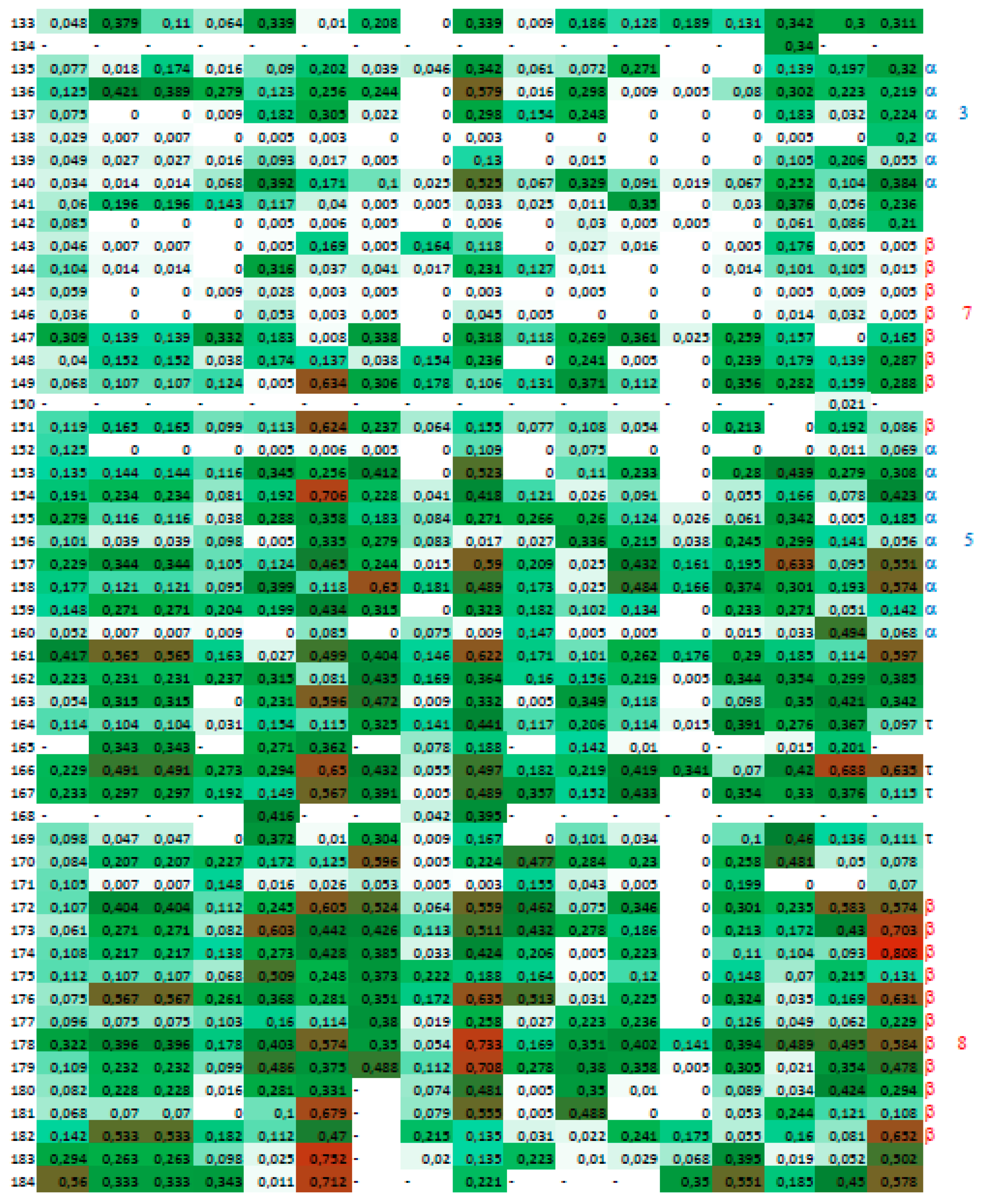
2.4. A Strategy for Analyses of Large-Scale Multiple Sequence Alignments
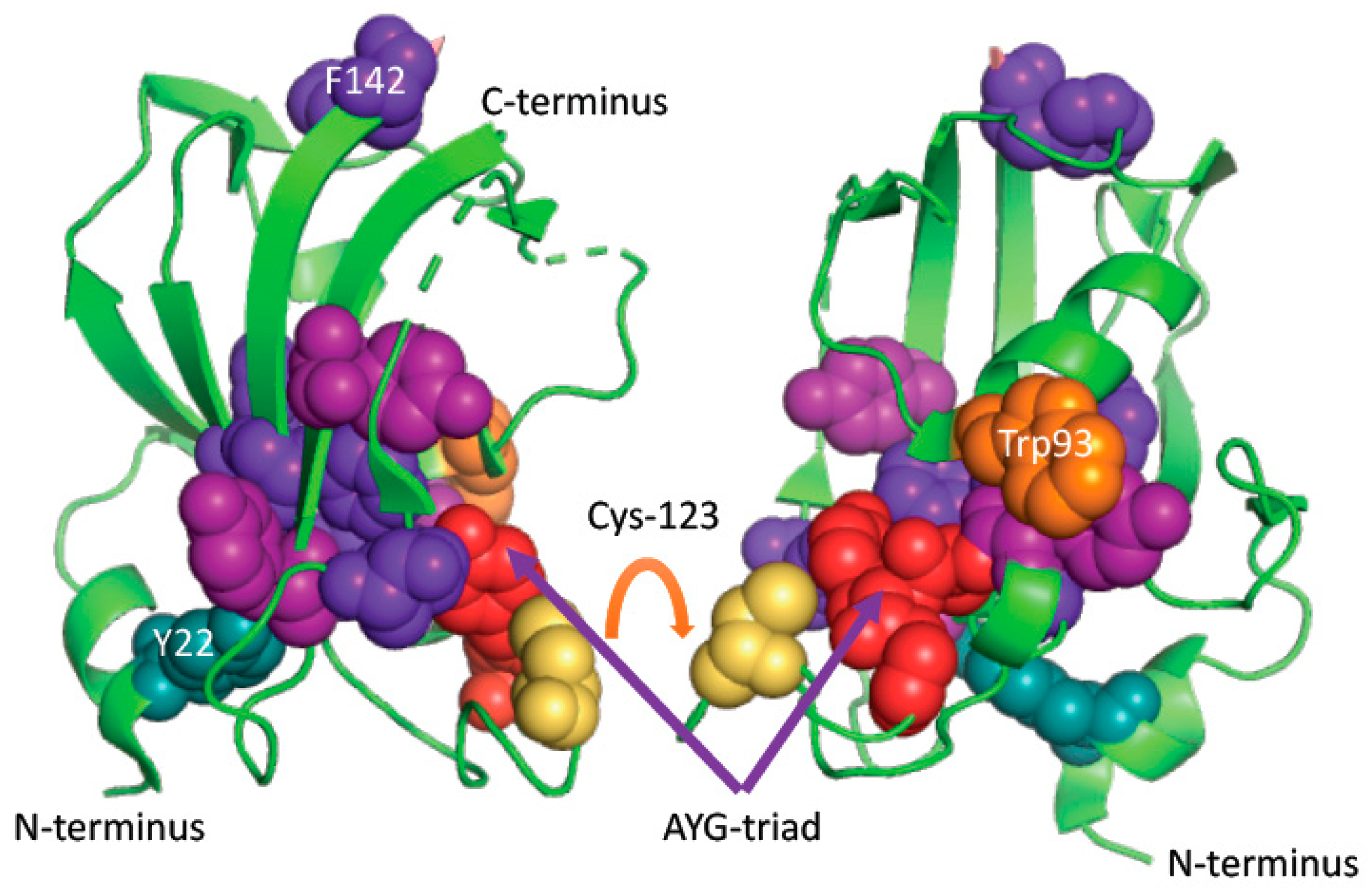
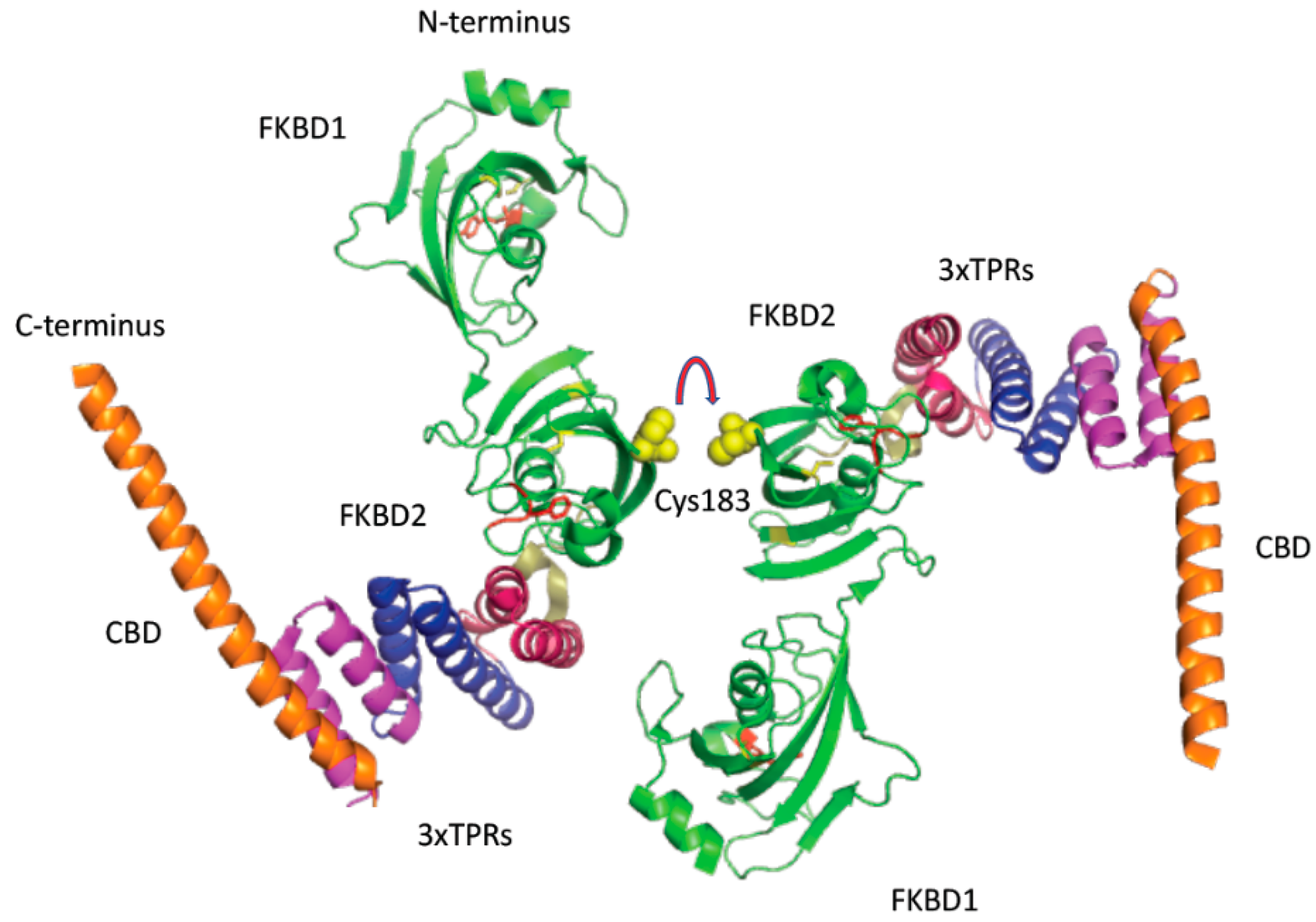
2.5. Analyses of Crystallographic Structures
2.6. In Home-Made Software
3. Results
3.1. FK506-Binding Proteins
3.2. Cyclophilins
4. Discussion
Supplementary Materials
Conflicts of Interest
Appendix A

References
- Schreiber, S.L. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 1991, 251, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Sigal, N.H.; Dumont, F.J. Cyclosporin A, FK-506, and rapamycin: Pharmacologic probes of lymphocyte signal transduction. Annu. Rev. Immunol. 1992, 10, 519–560. [Google Scholar] [CrossRef]
- Galat, A.; Riviere, S. Peptidyl-Prolyl Cis/Trans Isomerases: Immunophilins; Oxford Univeresity Press: Oxford, UK, 1998. [Google Scholar]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008, 36, D13–D21. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schäffer, A.A.; Yu, Y.K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Peptidylprolyl Isomerases as In Vivo Carriers for Drugs That Target Various Intracellular Entities. Biomolecules 2017, 7, 72. [Google Scholar] [CrossRef]
- Galat, A. On transversal hydrophobicity of some proteins and their modules. J. Chem. Inf. Model. 2009, 49, 1821–1830. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Edgar, R.C. Quality measures for protein alignment benchmarks. Nucleic Acids Res. 2010, 38, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Multidimensional drift of sequence attributes and functional profiles in the superfamily of the three-finger proteins and their structural homologues. J. Chem. Inf. Model. 2015, 55, 2026–2041. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. The PyMOL Molecular Graphics System. 2010. Available online: https://pymol.org/ (accessed on 21 December 2018).
- Berman, H.M.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [PubMed]
- Press, W.H.; Flannery, B.P.; Teukolsky, S.A.; Vettering, W.T. Numerical Recipes—The Art of Scientific Computing; Cambridge University Press: Cambridge, UK, 1986. [Google Scholar]
- Chapman, S.J. Fortran 90/95 for Scientists and Engineers; McGraw-Hill: Boston, MA, USA, 1998. [Google Scholar]
- Galat, A. Functional diversity and pharmacological profiles of the FKBPs and their complexes with small natural ligands. Cell. Mol. Life Sci. 2013, 70, 3243–3275. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Sequence diversification of the FK506-binding proteins in several different genomes. Eur. J. Biochem. 2000, 267, 4945–4959. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Davis, T.; Butler-Cole, B.; Paramanathan, R.; Weigelt, J.; Arrowsmith, J.C.; Edwards, A.M.; Bochkarev, A.; De-Paganon, S. Human FKBP 6 Protein. 3B7X.pdb in Brookhaven Structural Database; Structural Genomics Consortium (SGC): Oxford, UK; Toronto, ON, Canada, 2007. [Google Scholar]
- Kumar, R.; Moche, M.; Winblad, B.; Pavlov, P.F. Combined X-ray crystallography and computational modeling approach to investigate the Hsp90 C-terminal peptide binding to FKBP51. Sci. Rep. 2017, 7, 14288. [Google Scholar] [CrossRef] [PubMed]
- Munn, K.; Steward, R. The shut-down gene of Drosophila melanogaster encodes a novel FK506-binding protein essential for the formation of germline cysts during oogenesis. Genetics 2000, 156, 245–256. [Google Scholar] [PubMed]
- Galat, A.; Thai, R.; Stura, E.A. Diversified targets of FKBP25 and its complex with rapamycin. Int. J. Biol. Macromol. 2014, 69, 344–352. [Google Scholar] [CrossRef]
- Thapar, R. Roles of Prolyl Isomerases in RNA-Mediated Gene Expression. Biomolecules 2015, 5, 974–999. [Google Scholar] [CrossRef]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Ouyang, H.; Lee, W.H.; et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef]
- Wang, Z.; Song, J.; Milne, T.A.; Wang, G.G.; Li, H.; Allis, C.D.; Patel, D.J. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell 2010, 141, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Grow, E.J.; Wysocka, J. Flipping MLL1’s switch one proline at a time. Cell 2010, 141, 1108–1110. [Google Scholar] [CrossRef]
- Gallo, L.I.; Lagadari, M.; Piwien-Pilipuk, G.; Galigniana, M.D. The 90-kDa heat-shock protein (Hsp90)-binding immunophilin FKBP51 is a mitochondrial protein that translocates to the nucleus to protect cells against oxidative stress. J. Biol. Chem. 2011, 286, 30152–30160. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lu, X.; Morris, C.A.; Keating, M.T. A novel human gene FKBP6 is deleted in Williams syndrome. Genomics 1998, 52, 130–1337. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Kolas, N.K.; Noguchi, J.; Sarao, R.; Kikuchi, K.; Kaneko, H.; Kobayashi, E.; Kawai, Y.; Kozieradzki, I.; Landers, R.; et al. Essential role of FKBP6 in male fertility and homologous chromosome pairing in meiosis. Science 2003, 300, 1291–1295. [Google Scholar] [CrossRef]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, A.C.; Lindquist, S. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [PubMed]











© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galat, A. Compression of Large Sets of Sequence Data Reveals Fine Diversification of Functional Profiles in Multigene Families of Proteins: A Study for Peptidyl-Prolyl cis/trans Isomerases (PPIase). Biomolecules 2019, 9, 59. https://doi.org/10.3390/biom9020059
Galat A. Compression of Large Sets of Sequence Data Reveals Fine Diversification of Functional Profiles in Multigene Families of Proteins: A Study for Peptidyl-Prolyl cis/trans Isomerases (PPIase). Biomolecules. 2019; 9(2):59. https://doi.org/10.3390/biom9020059
Chicago/Turabian StyleGalat, Andrzej. 2019. "Compression of Large Sets of Sequence Data Reveals Fine Diversification of Functional Profiles in Multigene Families of Proteins: A Study for Peptidyl-Prolyl cis/trans Isomerases (PPIase)" Biomolecules 9, no. 2: 59. https://doi.org/10.3390/biom9020059