1. Introduction
Iron is a trace element and is essential in terms of oxygen transport, oxygen utilization, and mitochondrial function [
1]. Iron metabolism and respective disorders are a major issue in cardiovascular medicine: patients with heart failure (HF) or acute myocardial infarction (MI) and concomitant iron deficiency are at increased risk of future cardiovascular events and see improvement with intravenous iron supplementation, which has emerged as a guideline-endorsed therapy [
2,
3,
4,
5].
Hepcidin, which was discovered in the last decade, is a peptide hormone synthesized mainly in the liver [
6]. It is now recognized that hepcidin is the key regulator of iron homeostasis in humans, who by nature have no capacity of active iron excretion [
6]. The hepcidin production is feedback-regulated by iron levels: in case of iron deficiency or increased iron needs, the resulting suppression of hepcidin levels increases serum iron by elevated iron absorption through enterocytes in the duodenum, and by forced release of iron from macrophages [
7].
Since the established factors of iron metabolism—ferritin and transferrin saturation—have already proven predictive value in inflammatory responses, cardiovascular disease, and cardiovascular mortality, the question raised is whether circulating levels of hepcidin might be an even better predictive parameter [
2,
8,
9,
10]. Various experimental studies reported strongly raised hepcidin levels during hypoxia, myocarditis, and myocardial ischemia [
11,
12,
13]. Interestingly, while hypoxia appears to induce hepcidin in heart tissue, the opposite is the case in the liver, where hepcidin is suppressed during hypoxia [
11]. Likewise, in epidemiological studies, altered hepcidin levels have been linked with metabolic syndrome, arterial hypertension, aortic stiffness, pulmonary arterial hypertension, atherosclerosis, severity of systolic HF, and with poor outcome in patients with stable and acutely decompensated HF [
8,
14,
15,
16,
17,
18,
19]. Although results from the aforementioned studies seem to be fairly consistent, discussion on hepcidin and its role in cardiovascular disease remains controversial, since all performed studies had methodological limitations—either due to small sample size, to a cross-sectional design, to testing in primary-prevention settings and interpretation of results in manifest disease settings, or due to missing adjustment for potential confounders.
We sought to elucidate the role of hepcidin in the prediction of cardiovascular death and non-fatal MI, beyond established and emerging cardiovascular risk factors, in a large prospective cohort of patients with coronary heart disease (CHD).
2. Materials and Methods
2.1. Study Population
A total of 3800 patients, who underwent coronary angiography at the Department of Medicine II of the Johannes Gutenberg University Mainz or the Bundeswehr-Zentralkrankenhaus Koblenz, were recruited in the AtheroGene Study between June 1999 and March 2004 [
20]. The exclusion criteria were evidence of haemodynamically significant valvular heart disease, surgery or trauma within the previous month, known cardiomyopathy, known cancer, febrile conditions, or use of oral anticoagulant therapy within the previous 4 weeks. Subjects with missing information on the clinical presentation, or missing information on the cause of death were additionally ruled out, resulting in 3423 patients with CHD. After inobservance of subjects with missing samples or low sample volume, measurement of hepcidin was performed in 2198 patients. There were no relevant differences in baseline characteristics between the subcohort and the overall CHD cohort (data not shown).
All subjects gave written informed consent. The study was performed in accordance with the Declaration of Helsinki and approved by the Ethics Board of the Johannes Gutenberg University Mainz and of the Physicians’ chamber of the State Rhineland-Palatinate (Mainz, Germany) under the ethical number 837.057.99.
2.2. Data Collection
At baseline, all participants were subjected to a standardized questionnaire containing socio-demographic information and medical history. In addition, information was taken from the patients’ hospital charts. Coronary artery disease (CAD) was diagnosed if the coronary angiogram showed at least one stenosis >30% in a major coronary artery. Acute coronary syndrome (ACS) comprised unstable angina and acute myocardial infarction (AMI). Unstable angina was diagnosed according to Braunwald criteria [
21]. AMI was either ST-segment elevation with significant elevation (STEMI) in at least two contiguous leads or non-ST-elevation myocardial infarction (NSTEMI) based on clinic and positive in-house troponin concentrations. Status of haemochromatosis was not assessed.
In all patients, active follow-up was performed until a median of 4.1 years after discharge. Information was obtained from the patients using a mailed standardized questionnaire. Information regarding adverse cardiovascular disease (CVD) events and treatment since discharge from the in-hospital rehabilitation clinic was obtained from the primary care physicians also by means of a standardized questionnaire. If a subject had died during follow-up, the death certificate was obtained from the local Public Health Department and the main cause of death was coded according to the International Classification of Diseases (ICD-9 pos. 390–459: ICD-10 pos. I0-I99 and R57.0). Adverse CVD events were defined as CVD as the main cause of death (as stated in the death certificate).
2.3. Laboratory Methods
At baseline, blood was drawn before angiography in a fasting state under standardized conditions and stored at −80 °C until analysis. Serum hepcidin was measured using the Hepcidin 25 (bioactive) HS ELISA from DRG [
13]. The inter- and intra-assay coefficient of variation was 9.7% and 8.7%, respectively. These measurements were performed from prior unthawed aliquots. C-reactive protein (CRP), N-terminal pro brain natriuretic peptide (NT-proBNP), troponin I, total cholesterol, high-density lipoprotein-cholesterol (HDL), and low-density lipoprotein-cholesterol (LDL) cholesterol measurements were done by routine methods in the participating hospitals. All biomarkers were measured in a blinded fashion.
2.4. Statistical Methods
The study population was described with respect to various sociodemographic and medical characteristics.
Cardiovascular death and non-fatal MI were defined as the outcome measures. The relation of hepcidin with (i) cardiovascular mortality and/or non-fatal MI, and (ii) cardiovascular morality only, during follow-up was assessed by Cox regression analyses adjusted for age, sex in model 1, and additionally for hypertension, smoking status, diabetes, hyperlipidemia, body mass index (BMI), hemoglobin, log (NT-proBNP), and log (troponin I) in model 2.
All statistical procedures were carried out using R 3.2.4 (
http://www.r-project.org/). A
p-value < 0.05 was considered as statistically significant.
3. Results
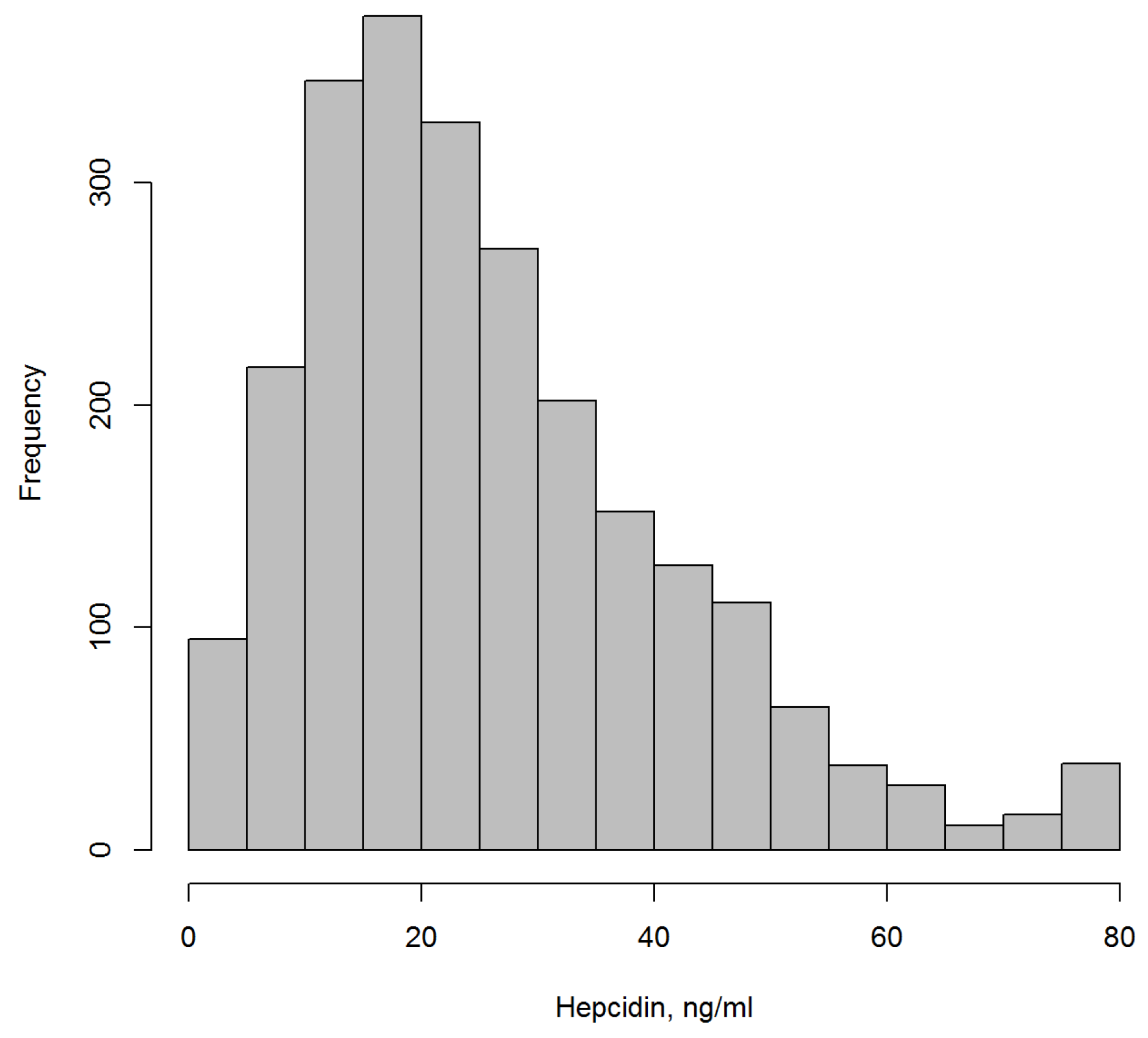
Table 1 shows the main sociodemographic and laboratory characteristics at baseline. Mean age was 63.0 years, and participants were predominantly men (75.2%). Median hepcidin was 22.8 ng/mL (25/75 percentiles: 14.2; 34.5).
Figure 1 shows the distribution of hepcidin levels.
During a median follow-up of 4.1 years, 10.3% of patients experienced cardiovascular death and/or non-fatal MI. In Cox regression analyses the hazard ratio (HR) for future cardiovascular death or MI per one SD increase of hepcidin was 1.03 after adjustment for sex and age ((95% CI 0.91–1.18),
p = 0.63) (
Table 2). This association virtually did not change after additional adjustment for BMI, smoking status, hypertension, diabetes, dyslipidemia, and surrogates of cardiac function (N-terminal pro B-type natriuretic peptide), size of myocardial necrosis (troponin I), and anemia (hemoglobin) (HR 0.95 (95% CI 0.79–1.14),
p = 0.57).
Cox regression analyses regarding cardiovascular mortality only (without non-fatal MI) during 4.1-years follow-up also did not yield significant association (
Table 3): the HR for future cardiovascular death per one SD increase of hepcidin was 1.04 after adjustment for sex and age ((95% CI 0.87–1.25),
p = 0.65), and 0.88 ((95% CI 0.69–1.12),
p = 0.29) in a fully adjusted model.
4. Discussion
Iron metabolism and respective disorders are of utmost importance in cardiovascular medicine. It is now recognized that the liver-derived peptide hepcidin is the key regulator of iron homeostasis in humans and mammals, and recent studies, mostly performed in primary-prevention settings, have highlighted hepcidin as most auspicious new marker of iron metabolism and its dysfunction in cardiovascular disease.
We sought to elucidate the role of hepcidin in the prediction of cardiovascular death and non-fatal MI, beyond established and emerging cardiovascular risk factors, in a large prospective cohort of patients with CHD. Of note, the diagnosis of CHD was exclusively based on coronary angiography.
We could not find any independent association between hepcidin levels and future risk of MI or cardiovascular death—neither in a basic model adjusted for age and sex, nor in a more comprehensively adjusted model. This is in contrast to previous, much smaller reports, and implicates a limited, if any, role for hepcidin in secondary cardiovascular risk prediction. Despite pathophysiological implications, we could not demonstrate any additional value for hepcidin in individuals with manifest CHD.
Experimental studies have suggested that hepcidin might mirror hypoxia and ischemia: in rat heart, hypoxia results in a strong upregulation of hepcidin expression on mRNA and protein level, accompanied by an increased immunoreactivity of hepcidin pronounced at the myocardial intercalated disc area [
11]. These findings were confirmed in a rat model of acute MI and extended to patients with acute myocarditis: hepcidin showed an abrupt increase of up to 100 times in human cardiomyocytes within one day after myocardial infarction and acute myocarditis [
12,
13]. Moreover, the receptor for hepcidin, ferroportin, is not just highly expressed in duodenal enterocytes, but also in reticuloendothelial macrophages, which themselves are involved in all stages of atherogenesis [
7,
22].
The discrepancy between our results and previous reports might arise from the fact that we studied the prognostic value of hepcidin in a cohort with existing cardiovascular disease, while experimental evidence for hepcidin mirroring hypoxia and ischemia comes from primary disease settings. This is in line with epidemiological studies, which were mostly performed in primary- prevention or in community-based settings, and which reported hepcidin to be associated with predictors of cardiovascular disease and very early stage of cardiovascular disease, like arterial hypertension, metabolic syndrome, aortic stiffness, or other measures of subclinical atherosclerosis [
17,
18,
19]. Additionally, it might be that circulating hepcidin levels measured in the present study do not mirror changes in cardiac hepcidin levels associated with CHD. As hepcidin is primarily expressed in the liver most of the circulating hepcidin in blood is derived from the liver and not the heart.
5. Conclusions
In this study, by far the largest evaluating the predictive value of hepcidin in patients with manifest CHD, levels of hepcidin were not associated with future MI or cardiovascular death. This implicates a limited, if any, role for hepcidin in secondary cardiovascular risk prediction. Based on findings from other epidemiological studies, we strongly emphasize evaluation of hepcidin as marker of incident CHD in primary-prevention settings.
Author Contributions
Conceptualization, T.Z. and M.K.; Methodology, F.O., M.K. and C.W.; Software, F.O. and S.A.; Validation A.A, R.B.S. and K.J.L.; Formal Analysis, F.O. and S.A.; Investigation, T.Z., A.A. and S.B.; Resources, R.B.S., K.J.L., S.B. and M.K.; Data Curation, T.Z., A.A., F.O., S.A. and J.R.; Writing-Original Draft Preparation, T.Z., A.A. and M.K.; Writing-Review and Editing, T.Z., A.A., S.A., C.W., F.O., R.B.S., J.R., K.J.L., S.B. and M.K.; Visualization, F.O. and S.A.; Supervision, T.Z. and M.K.; Project Administration, T.Z. and M.K.; Funding Acquisition, M.K., S.B. and K.J.L.
Funding
The AtheroGene study was supported by a grant of the ‘Stiftung Rheinland-Pfalz für Innovation’, Ministry for Science and Education (AZ 15202–386261/545), Mainz. This work has been supported by the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement No. HEALTH-F2-2011-278913 (BiomarCaRE) and by the European Research Area Network (ERA-Net) (PREMED-CAD).
Conflicts of Interest
The authors declare no conflict of interest. S.B. reports investigator-initiated grants from SIEMENS, Abbott Diagnostics, and Thermofisher. All other authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Galy, B.; Ferring-Appel, D.; Sauer, S.W.; Kaden, S.; Lyoumi, S.; Puy, H.; Kölker, S.; Gröne, H.J.; Hentze, M.W. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010, 12, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Jankowska, E.A.; van Veldhuisen, D.J.; Ponikswski, P.; Anker, S.D. Iron deficiency and cardiovascular disease. Nat. Rev. Cardiol. 2015, 12, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [PubMed]
- Florian, A.; Ludwig, A.; Rösch, S.; Ylidiz, H.; Klumpp, S.; Sechtem, U.; Yilmaz, A. Positive effect of intravenous iron-oxide administration on left ventricular remodelling in patients with acute ST-elevation myocardial infarction—A cardiovascular magnetic resonance (CMR) study. Int. J. Cardiol. 2014, 73, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. Document Reviewers. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 18, 891–975. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, A.S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Investig. 2013, 123, 2337–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.D.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; Malyszko, J.; Ardehali, H.; Koc-Zorawska, E.; Banasiak, W.; von Haehling, S.; Macdougall, I.C.; Weiss, G.; McMurray, J.J.; Anker, S.D.; et al. Iron status in patients with chronic heart failure. Eur. Heart J. 2013, 34, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Shamayeva, G.; Chow, B.K.; DePalma, R.G. Ferritin and percent transferrin saturation levels predict type 2 diabetes risk and cardiovascular disease outcomes. Curr. Diabetes Rev. 2017, 13, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Depalma, R.G.; Hayes, V.W.; Chow, B.K.; Shamayeva, G.; May, P.E.; Zacharski, L.R. Ferritin levels, inflammatory biomarkers, and mortality in peripheral arterial disease: A substudy of the Iron (Fe) and Atherosclerosis Study (FeAST) Trial. J. Vasc. Surg. 2010, 51, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, U.; Fein, E.; Gehrke, S.G.; Stremmel, W.; Kulaksiz, H. The iron regulatory peptide hepcidin is expressed in the heart and regulated by hypoxia and inflammation. Endocrinology 2007, 148, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Simonis, G.; Mueller, K.; Schwarz, P.; Wiedemann, S.; Adler, G.; Strasser, R.H.; Kulaksiz, H. The iron-regulatory peptide hepcidin is upregulated in the ischemic and in the remote myocardium after myocardial infarction. Peptides 2010, 31, 1786–1790. [Google Scholar] [CrossRef] [PubMed]
- Isoda, M.; Hanawa, H.; Watanabe, R.; Yoshida, T.; Toba, K.; Yoshida, K.; Kojima, M.; Otaki, K.; Hao, K.; Ding, L.; et al. Expression of the peptide hormone hepcidin increases in cardiomyocytes under myocarditis and myocardial infarction. J. Nutr. Biochem. 2010, 21, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Martinelle, N.; Traglia, M.; Campostrini, N.; Biino, G.; Corbella, M.; Sala, C.; Busti, F.; Masciullo, C.; Manna, D.; Previtali, S.; et al. Increased serum hepcidin levels in subjects with the metabolic syndrome: A population study. PLoS ONE 2012, 7, e48250. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; Kasztura, M.; Sokolski, M.; Bronisz, M.; Nawrocka, S.; Oleśkowska-Florek, W.; Zymliński, R.; Biegus, J.; Siwołowski, P.; Banasiak, W.; et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. Eur. Heart J. 2014, 35, 2468–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, C.J.; Howard, L.S.; Busbridge, M.; Ashby, D.; Kondili, E.; Gibbs, J.S.; Wharton, J.; Wilkins, M.R. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: Clinical prevalence, outcomes, and mechanistic insights. J. Am. Coll. Cardiol. 2011, 58, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Galesloot, T.E.; Janss, L.L.; Burgess, S.; Kiemeney, L.A.; den Heijer, M.; de Graaf, J.; Holewijn, S.; Benyamin, B.; Whitfield, J.B.; Swinkels, D.W.; et al. Iron and hepcidin as risk factors in atherosclerosis: What do the genes say? BMC Genet. 2015, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, L.; Maloberti, A.; Signorini, S.; Milano, M.; Cesana, F.; Cappellini, F.; Dongiovanni, P.; Porzio, M.; Soriano, F.; Brambilla, M.; et al. Iron stores, hepcidin, and aortic stiffness in individuals with hypertension. PLoS ONE 2015, 10, e0134635. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Ortegon, M.F.; Arbelaez, A.; Mosquera, M.; Moreno-Navarrete, J.M.; Aguilar-Plata, C.; Fernández-Real, J.M. Circulating hepcidin is independently associated with systolic blood pressure in apparently healthy individuals. Arch. Med. Res. 2015, 46, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Karakas, M.; Schulte, C.; Appelbaum, S.; Ojeda, F.; Lackner, K.J.; Münzel, T.; Schnabel, R.B.; Blankenberg, S.; Zeller, T. Circulating microRNAs strongly predict cardiovascular death in patients with coronary artery disease-results from the large AtheroGene study. Eur. Heart J. 2016, 38, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Calvin, J.E.; Klein, L.W.; Van den Berg, B.J.; Meyer, P.; Condon, J.V.; Snell, R.J.; Ramirez-Morgen, L.M.; Parrillo, J.E. Risk stratification in unstable angina. Prospective validation of the Braunwald classification. JAMA 1995, 273, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Theroux, P. Pathophysiology of coronary artery disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).


{kind=link}