
A Mechanistic Review of Mitophagy and Its Role in Protection against Alcoholic Liver Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Alcohol Metabolism
3. ALD Pathogenesis
4. Mitophagy Protects against Alcohol-Induced Liver Injury and Steatosis by Selectively Removing Damaged Mitochondria
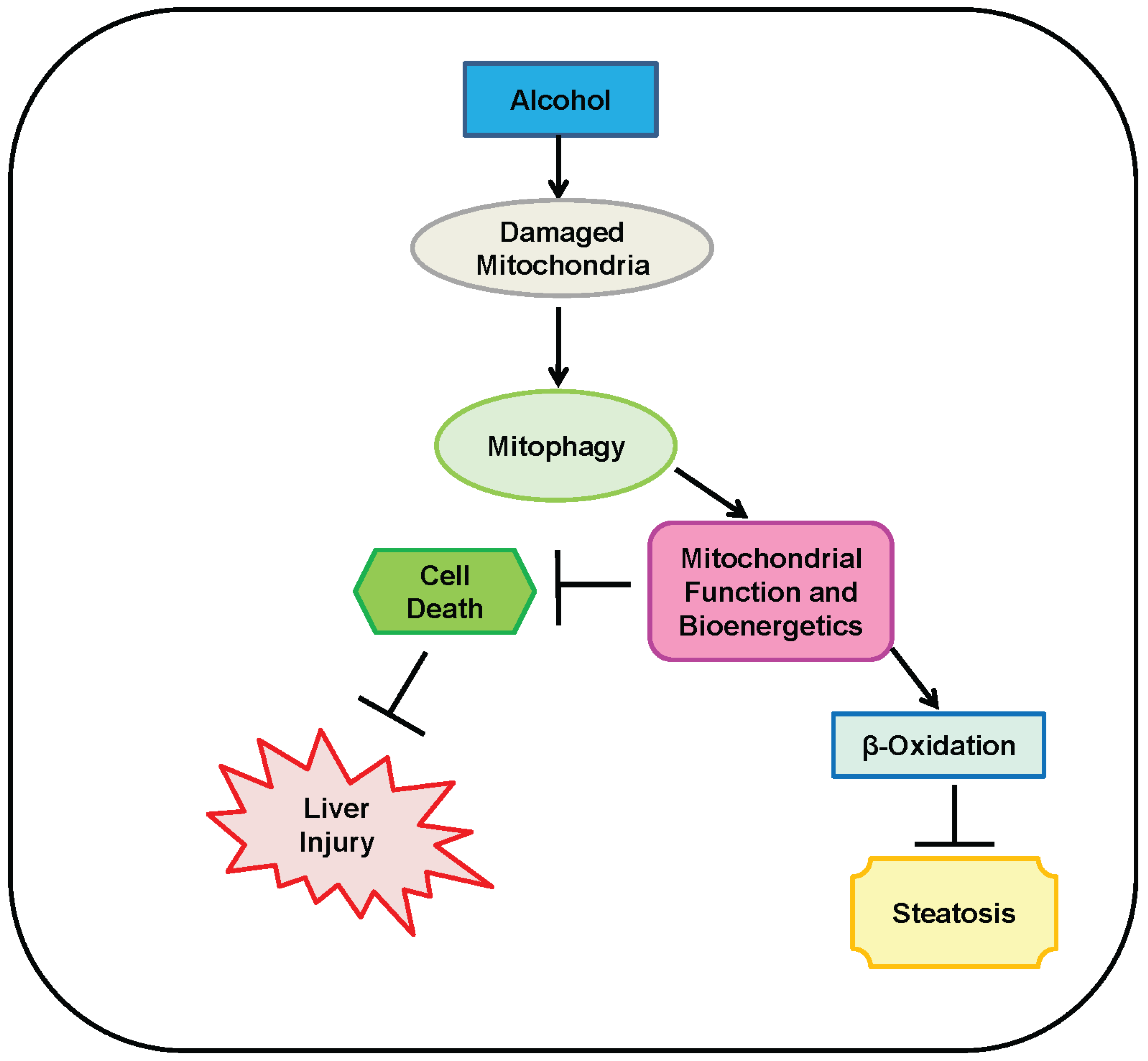
5. Parkin-Dependent Mitophagy
6. Role of Parkin and Parkin-Dependent Mitophagy in Protection against Alcohol-Induced Liver Injury and Steatosis in Mice
7. Parkin-Independent Mitophagy
8. Mitochondrial Spheroids May Be a Novel Mechanism of Protection against Alcohol-Induced Liver Injury
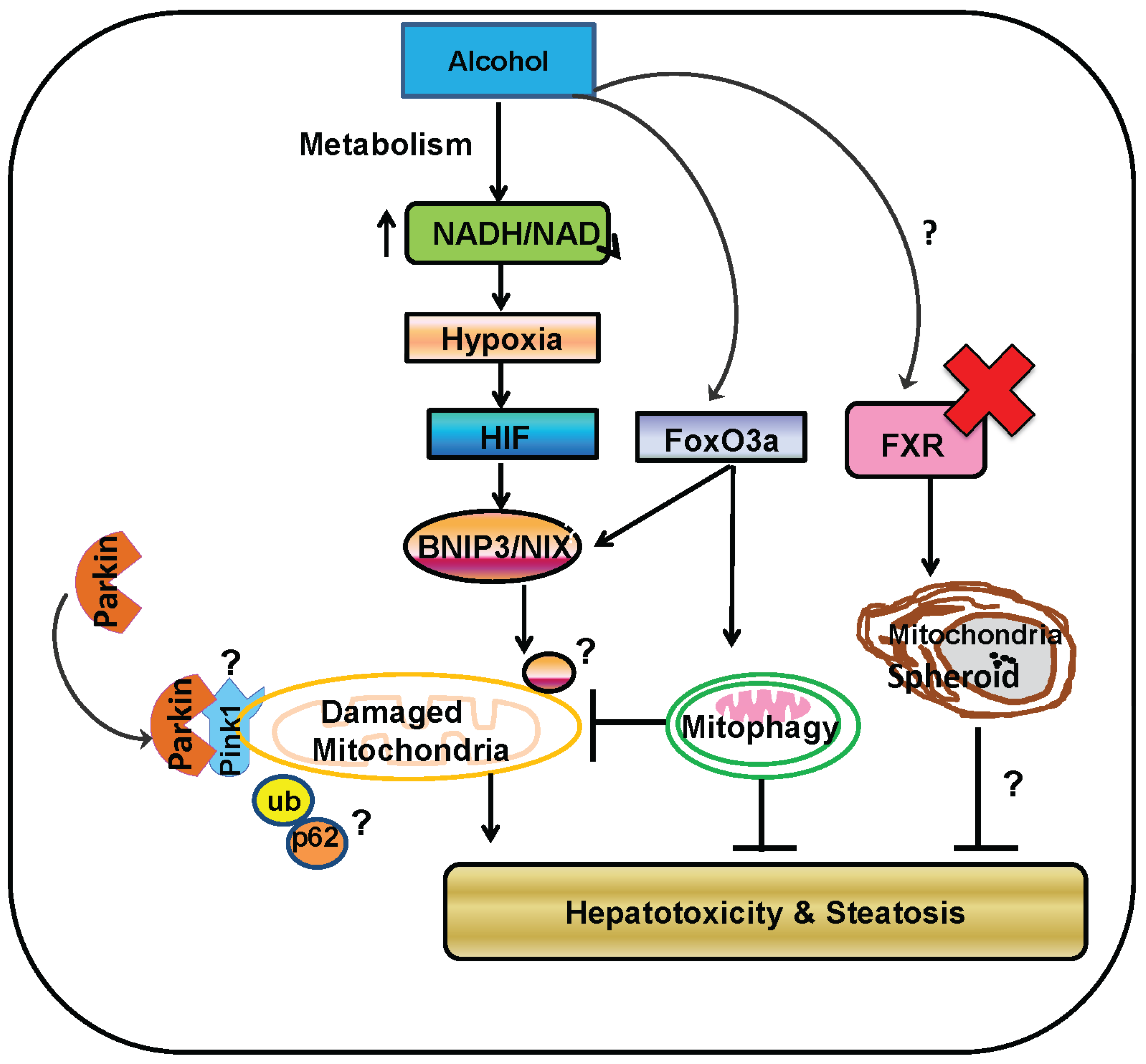
9. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALD | Alcoholic Liver Disease |
| HCC | hepatocellular carcinoma |
| TNFα | tumor necrosis factor alpha |
| ADH | alcohol dehydrogenase |
| CYP2E1 | Cytochrome p450 2E1 |
| ALDH | aldehyde dehydrogenase |
| ROS | reactive oxygen species |
| FAEE | fatty acid ethyl ester |
| PINK1 | phosphatase and tensin homolog-induced putative kinase 1 |
| CCCP | carbonyl cyanide m-chlorophenyl hydrazine |
| PARL | presenilin-associated rhomboid-like |
| UBL | ubiquitin-like |
| MFN1/2 | mitofusin 1 and 2 |
| TOM20 | translocase of outer mitochondrial membrane 20 |
| VDAC | voltage-dependent anion channel |
| UBA | ubiquitin-associated domain |
| LC3 | microtubule-associated protein light chain 3 |
| LIR | LC3 interacting region |
| BNIP3L | BCL2/adenovirus E1B 19 kDa interaction protein 3-like |
| NBR1 | neighbor of BRCA gene 1 |
| USP | ubiquitin-specific peptidase |
| KO | knock out |
| WT | wild-type |
| BNIP3 | BCL2/adenovirus E1B 19 kDa interaction protein 3 |
| FUNDC1 | Fun14 domain containing-1 |
| Mul1 | mitochondrial ubiquitin ligase 1 |
| BH3 | Bcl-2 homology 3 |
| Hif-1α | hypoxia inducing factor alpha |
| HRE | Hif-1 response element |
| PGAM5 | phosphoglycerate mutase family member 5 |
| FXR | Farnesoid X receptor |
References
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Williams, J.A.; Manley, S.; Ding, W.X. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World J. Gastroenterol. 2014, 20, 12908–12933. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Am. J. Gastroenterol. 2010, 105, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, T.F.; McCullough, A.J. Do corticosteroids reduce mortality from alcoholic hepatitis? A meta-analysis of the randomized trials. Ann. Intern. Med. 1990, 113, 299–307. [Google Scholar] [CrossRef] [PubMed]
- De, B.K.; Gangopadhyay, S.; Dutta, D.; Baksi, S.D.; Pani, A.; Ghosh, P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: A randomized controlled trial. World J. Gastroenterol. 2009, 15, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Louvet, A.; Duhamel, A.; Nahon, P.; Carbonell, N.; Boursier, J.; Anty, R.; Diaz, E.; Thabut, D.; Moirand, R.; et al. Prednisolone with versus without pentoxifylline and survival of patients with severe alcoholic hepatitis: A randomized clinical trial. JAMA 2013, 310, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, A.M. Liver disease in alcohol abusers: Clinical perspective. Alcohol 2002, 27, 7–11. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Cjaza, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Komatsu, M.; Finley, K.; Simonsen, A. Selective types of autophagy. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Li, M.; Yin, X.M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy 2011, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Manley, S.; Ni, H.M. The emerging role of autophagy in alcoholic liver disease. Exp. Biol. Med. 2011, 236, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Thomes, P.G.; Ding, W.X.; Lemasters, J.J.; Donohue, T.M., Jr. Autophagy in alcohol-induced liver diseases. Alcohol. Clin. Exp. Res. 2012, 36, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M., Jr.; Thomes, P.G. Ethanol-induced oxidant stress modulates hepatic autophagy and proteasome activity. Redox Biol. 2014, 3, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, S.; Ni, H.M.; Huang, H.; Ding, W.X. Autophagy in alcohol-induced multiorgan injury: Mechanisms and potential therapeutic targets. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shvets, E.; Elazar, Z. Lipophagy: Selective catabolism designed for lipids. Dev. Cell 2009, 16, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Russmann, S.; Diogo, C.; Bonfrate, L.; Oliveira, P.J.; Wang, D.Q.; Portincasa, P. Mitochondria in chronic liver disease. Curr. Drug Targets 2011, 12, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Ishii, H. Role of mitochondria in alcoholic liver injury. Free Radic. Biol. Med. 2002, 32, 487–491. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Haynes, C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011, 36, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Kaguni, L.S. Matrix proteases in mitochondrial DNA function. Biochim. Biophys. Acta 2012, 1819, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 2011, 23, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.M.; Ding, Y.; Ding, W.X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Guo, F.; Ni, H.M.; Bockus, A.; Manley, S.; Stolz, D.B.; Eskelinen, E.L.; Jaeschke, H.; Yin, X.M. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J. Biol. Chem. 2012, 287, 42379–42388. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Li, M.; Biazik, J.M.; Morgan, D.G.; Guo, F.; Ni, H.M.; Goheen, M.; Eskelinen, E.L.; Yin, X.M. Electron microscopic analysis of a spherical mitochondrial structure. J. Biol. Chem. 2012, 287, 42373–42378. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M.; Ding, W.X. The reciprocal roles of PARK2 and mitofusins in mitophagy and mitochondrial spheroid formation. Autophagy 2013, 9, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Zelner, I.; Matlow, J.N.; Natekar, A.; Koren, G. Synthesis of fatty acid ethyl esters in mammalian tissues after ethanol exposure: A systematic review of the literature. Drug Metab. Rev. 2013, 45, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.Y.; Ingelman-Sundberg, M.; Neve, E.; Matsumoto, H.; Nishitani, Y.; Minowa, Y.; Fukui, Y.; Bailey, S.M.; Patel, V.B.; Cunningham, C.C.; et al. Ethanol and oxidative stress. Alcohol. Clin. Exp. Res. 2001, 25, S237–S243. [Google Scholar] [CrossRef]
- Wu, H.; Cai, P.; Clemens, D.L.; Jerrells, T.R.; Ansari, G.A.; Kaphalia, B.S. Metabolic basis of ethanol-induced cytotoxicity in recombinant HepG2 cells: Role of nonoxidative metabolism. Toxicol. Appl. Pharmacol. 2006, 216, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, J.; Bataller, R. Alcoholic liver disease: Pathogenesis and new targets for therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Teli, M.R.; Day, C.P.; Burt, A.D.; Bennett, M.K.; James, O.F. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995, 346, 987–990. [Google Scholar] [CrossRef]
- Singal, A.K.; Kamath, P.S.; Francisco Ziller, N.; DiCecco, S.; Shoreibah, M.; Kremers, W.; Charlton, M.R.; Heimbach, J.K.; Watt, K.D.; Shah, V.D. Nutritional status of patients with alcoholic cirrhosis undergoing liver transplantation: Time trends and impact on survival. Transpl. Int. 2013, 26, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Basra, S.; Anand, B.S. Definition, epidemiology and magnitude of alcoholic hepatitis. World J. Hepatol. 2011, 3, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Streba, L.A.; Vere, C.C.; Streba, C.T.; Ciurea, M.E. Focus on alcoholic liver disease: From nosography to treatment. World J. Gastroenterol. 2014, 20, 8040–8047. [Google Scholar] [CrossRef] [PubMed]
- Stinson, F.S.; Grant, B.F.; Dufour, M.C. The critical dimension of ethnicity in liver cirrhosis mortality statistics. Alcohol. Clin. Exp. Res. 2001, 25, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.H. Racial and ethnic differences in alcohol-associated aspartate aminotransferase and gamma-glutamyltransferase elevation. Arch. Intern. Med. 2002, 162, 2236–2239. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Lindros, K.O.; Baraona, E.; Ikejima, K.; Mezey, E.; Järveläinen, H.A.; Ramchandani, V.A. Sex difference in alcohol-related organ injury. Alcohol. Clin. Exp. Res. 2001, 25, S40–S45. [Google Scholar] [CrossRef]
- Moshage, H. Alcoholic liver disease: A matter of hormones? J. Hepatol. 2001, 35, 130–133. [Google Scholar] [CrossRef]
- Baraona, E.; Abittan, C.S.; Dohmen, K.; Moretti, M.; Pozzato, G.; Chayes, Z.W.; Schaefer, C.; Lieber, C.S. Gender differences in pharmacokinetics of alcohol. Alcohol. Clin. Exp. Res. 2001, 25, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.J.; McCullough, A.J. The impact of obesity and metabolic syndrome on alcoholic liver disease. Clin. Liver Dis. 2014, 18, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J.C. Excess weight risk factor for alcoholic liver disease. Hepatology 1997, 25, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, M.; Rafiq, N.; Younossi, Z.M. Components of metabolic syndrome are independent predictors of mortality in patients with chronic liver disease: A population-based study. Gut 2010, 59, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lai, K.K.; Verlinsky, A.; Lugea, A.; French, S.W.; Cooper, M.P.; Ji, C.; Tsukamato, H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 2011, 55, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Degos, F. Hepatitis C and alcohol. J. Hepatol. 1999, 31, S113–S118. [Google Scholar] [CrossRef]
- Monto, A.; Patel, K.; Bostrom, A.; Pianko, S.; Pockros, P.; McHutchison, J.G.; Wright, T.L. Risks of a range of alcohol intake on hepatitis C-related fibrosis. Hepatology 2004, 39, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.R.; Gonin, R.; Alter, H.J.; Wright, E.C.; Buskell, Z.J.; Hollinger, F.B.; Seeff, L.B.; National Heart, Lung, and Blood Institute Study Group. The relationship of acute transfusion-associated hepatitis to the development of cirrhosis in the presence of alcohol abuse. Ann. Intern. Med. 2001, 134, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Monzoni, A.; Masutti, F.; Saccoccio, G.; Bellentani, S.; Tiribelli, C.; Giacca, M. Genetic determinants of ethanol-induced liver damage. Mol. Med. 2001, 7, 255–262. [Google Scholar] [PubMed]
- Zintzaras, E.; Stefanidis, I.; Santos, M.; Vidal, F. Do alcohol-metabolizing enzyme gene polymorphisms increase the risk of alcoholism and alcoholic liver disease? Hepatology 2006, 43, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Lim, R.W. Epigenetic effects of ethanol on the liver and gastrointestinal system. Alcohol Res. 2013, 35, 47–55. [Google Scholar] [PubMed]
- Altamirano, J.; Bataller, R. Cigarette smoking and chronic liver diseases. Gut 2010, 59, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Corrao, G.; Lepore, A.R.; Torchio, P.; Valenti, M.; Galatola, G.; D’Amicis, A.; Aricò, S.; di Orio, F.; The Provincial Group for the Study of Chronic Liver Disease. The effect of drinking coffee and smoking cigarettes on the risk of cirrhosis associated with alcohol consumption. Eur. J. Epidemiol. 1994, 10, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Pessione, F.; Ramond, M.J.; Peters, L.; Pham, B.N.; Batel, P.; Rueff, B.; Valla, D.C. Five-year survival predictive factors in patients with excessive alcohol intake and cirrhosis. Effect of alcoholic hepatitis, smoking and abstinence. Liver Int. 2003, 23, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Stroffolini, T.; Cotticelli, G.; Medda, E.; Niosi, M.; del Vecchio-Blanco, C.; Addolorato, G.; Petrelli, E.; Salemo, M.T.; Picardi, A.; Bernardi, M.; et al. Interaction of alcohol intake and cofactors on the risk of cirrhosis. Liver Int. 2010, 30, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, J.; Kangastupa, P.; Laatikainen, T.; Aalto, M.; Niemela, O. Dose- and gender-dependent interactions between coffee consumption and serum GGT activity in alcohol consumers. Alcohol Alcohol. 2013, 48, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Gores, G.J. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front. Biosci. 2005, 10, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Ziol, M.; Tepper, M.; Lohez, M.; Arcangeli, G.; Ganne, N.; Christidis, C.; Trinchet, J.C.; Beaugrand, M.; Guillet, J.G.; Guettier, C. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J. Hepatol. 2001, 34, 254–260. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R., Jr.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kompare, M.; Rizzo, W.B. Mitochondrial fatty-acid oxidation disorders. Semin. Pediatr. Neurol. 2008, 15, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; le Bloc’h, J.; Silliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Coleman, W.B.; Cunningham, C.C. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim. Biophys. Acta 1991, 1058, 178–186. [Google Scholar] [CrossRef]
- Zhong, Z.; Ramshesh, V.K.; Rehman, H.; Liu, Q.; Theruvath, T.P.; Krishnasamy, Y.; Lemasters, J.J. Acute ethanol causes hepatic mitochondrial depolarization in mice: Role of ethanol metabolism. PLoS ONE 2014, 9, e91308. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Narendra, D.P.; Tanaka, A.; Manfredi, G.; Youle, R.J. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11835–11840. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zheng, K.; Clark, J.; Swerdlow, R.H.; Pulst, S.M.; Sutton, J.P.; Shinobu, L.A.; Simon, D.K. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum. Mol. Genet. 2014, 23, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Andringa, K.K.; Udoh, U.S.; Landar, A.; Bailey, S.M. Proteomic analysis of 4-hydroxynonenal (4-HNE) modified proteins in liver mitochondria from chronic ethanol-fed rats. Redox Biol. 2014, 2, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Kaplowitz, N.; Fernandez-Checa, J.C. Role of mitochondria in alcoholic liver disease. Curr. Pathobiol. Rep. 2013, 1, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zelickson, B.R.; Benavides, G.A.; Johnson, M.S.; Chacko, B.K.; Venkatraman, A.; Landar, A.; Betancourt, A.M.; Bailey, S.M.; Darley-Usmar, V.M. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim. Biophys. Acta 2011, 1807, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- King, A.L.; Swain, T.M.; Mao, Z.; Udoh, U.S.; Oliva, C.R.; Betancourt, A.M.; Griquer, C.E.; Crowe, D.R.; Lesort, M.; Bailey, S.M. Involvement of the mitochondrial permeability transition pore in chronic ethanol-mediated liver injury in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G265–G277. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, Parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with Parkin and associates with LC3 in mitophagy. FEBS Lett. 2010, 584, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Matsumine, H.; Saito, M.; Shimoda-Matsubayashi, S.; Tanaka, H.; Ishikawa, A.; Nakagawa-Hattori, Y.; Yokochi, M.; Kobayashi, T.; Igarashi, S.; Takano, H.; et al. Localization of a gene for an autosomal recessive form of juvenile parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 1997, 60, 588–596. [Google Scholar] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, M.; Kujuro, Y.; Okatsu, K.; Koyano, F.; Kosako, H.; Kimura, M.; Suzuki, N.; Uchiyama, S.; Tanaka, K.; Matsuda, N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 2013, 288, 22019–22032. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Vollmer, S.; Golombek, S.; Kahle, P.J. The ubiquitin-conjugating enzymes UBE2N, UBE2L3 and UBE2D2/3 are essential for Parkin-dependent mitophagy. J. Cell Sci. 2014, 127, 3280–3293. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/Parkin pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Lemasters, J.J. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am. J. Physiol. Cell Physiol. 2011, 300, C308–C317. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Williams, J.A.; Ding, W.X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp. Biol. Med. 2013, 238, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [PubMed]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes Parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, T.; Haddad, D.; Wauters, F.; van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; de Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in Parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [PubMed]
- Periquet, M.; Corti, O.; Jacquier, S.; Brice, A. Proteomic analysis of Parkin knockout mice: Alterations in energy metabolism, protein handling and synaptic function. J. Neurochem. 2005, 95, 1259–1276. [Google Scholar] [CrossRef] [PubMed]
- Stichel, C.C.; Zhu, X.R.; Bader, V.; Linnartz, B.; Schmidt, S.; Lübbert, H. Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum. Mol. Genet. 2007, 16, 2377–2393. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Maemura, K.; Otsuki, Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: An immunohistochemical and electron microscopic study. J. Mol. Histol. 2013, 44, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Otsuki, Y. The autophagic response to alcohol toxicity: The missing layer. J. Hepatol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.; Xu, M.; Wang, H.; Bertola, A.; Gao, B. Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: Pathophysiology, translational relevance, and challenges. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G819–G823. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Pruett, S.B.; Szabo, G.; Arteel, G.E. Binge ethanol and liver: New molecular developments. Alcohol. Clin. Exp. Res. 2013, 37, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Park, O.; Gao, B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: A critical role for E-selectin. Hepatology 2013, 58, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Ybanez, M.D.; Johnson, H.S.; McDonald, J.N.; Mesropyan, L.; Sancheti, H.; Martin, G.; Martin, A.; Lim, A.M.; Dara, L.; et al. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: Biogenesis, remodeling, and functional alterations. J. Biol. Chem. 2012, 287, 42165–42179. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Mitsui, T.; Kunishige, M.; Shono, M.; Akaike, M.; Azuma, H.; Matsumoto, T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum. Mol. Genet. 2006, 15, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Rothfuss, O.; Fischer, H.; Hasegawa, T.; Maisel, M.; Leitner, P.; Miesel, F.; Sharma, M.; Bornemann, A.; Berg, D.; Gasser, T.; et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 2009, 18, 3832–3850. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noquichi, A.; Springer, D.; Bochargov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Invest. 2011, 121, 3701–3712. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.M.; Haynes, A.; Manley, S.; Li, Y.; Jaeschke, H.; Ding, W.X. Chronic deletion and acute knockdown of Parkin have differential responses to acetaminophen-induced mitophagy and liver injury in mice. J. Biol. Chem. 2015, 290, 10934–10946. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Theodorakis, P.; Subramanian, T.; Chinnadurai, G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem. 1998, 273, 12415–12421. [Google Scholar] [CrossRef] [PubMed]
- Galvez, A.S.; Brunskill, E.W.; Marreez, Y.; Benner, B.J.; Regula, K.M.; Kirschenbaum, L.A.; Dorn, G.W., 2nd. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J. Biol. Chem. 2006, 281, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Aerts, L.; de Strooper, B.; Morais, V.A. PINK1 activation-turning on a promiscuous kinase. Biochem. Soc. Trans. 2015, 43, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Aerts, L.; Craessaerts, K.; de Strooper, B.; Morais, V.A. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Cizeau, J.; Vande Velde, C.; Park, J.H.; Bozek, G.; Bolton, J.; Shi, L.; Dubik, D.; Greenberg, A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J. Biol. Chem. 1999, 274, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opfermam, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Chatelain, E.H.; Lavie, J.; Mahfouf, W.; Jose, C.; Obre, E.; Goorden, S.; Priault, M.; Elgersma, Y.; Rezvani, H.R.; et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013, 17, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Schlattner, U.; Tokarska-Schlattner, M.; Rousseau, D.; Boissan, M.; Mannella, C.; Epand, R.; Lacombe, M.L. Mitochondrial cardiolipin/phospholipid trafficking: The role of membrane contact site complexes and lipid transfer proteins. Chem. Phys. Lipids 2014, 179, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Puri, R.; Yang, H.; Lizzio, M.A.; Wu, C.; Sheng, Z.H.; Guo, M. MUL1 acts in parallel to the PINK1/Parkin pathway in regulating mitofusin and compensates for loss of PINK1/Parkin. Elife 2014. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Xi, X.P. Incomplete compensation of enhanced hepatic oxygen consumption in rats with alcoholic centrilobular liver necrosis. Hepatology 1989, 9, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Lemasters, J.J.; Christenson, V.; Thurman, R.G. Periportal and pericentral pyridine nucleotide fluorescence from the surface of the perfused liver: Evaluation of the hypothesis that chronic treatment with ethanol produces pericentral hypoxia. Proc. Natl. Acad. Sci. USA 1982, 79, 5415–5419. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Raleigh, J.A.; Bradford, B.U.; Thurman, R.G. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: Role of Kupffer cells. Am. J. Physiol. 1996, 271, G494–G500. [Google Scholar] [PubMed]
- Manley, S.; Ni, H.M.; Williams, J.A.; Kong, B.; DiTacchio, L.; Guo, G.; Ding, W.X. Farnesoid X receptor regulates forkhead Box O3a activation in ethanol-induced autophagy and hepatotoxicity. Redox Biol. 2014, 2, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/Parkin-mediated mitophagy. Autophagy 2015, 11, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Manley, S.; Ni, H.M.; Kong, B.; Apte, U.; Guo, G.; Ding, W.X. Suppression of autophagic flux by bile acids in hepatocytes. Toxicol. Sci. 2014, 137, 478–490. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, J.A.; Ding, W.-X. A Mechanistic Review of Mitophagy and Its Role in Protection against Alcoholic Liver Disease. Biomolecules 2015, 5, 2619-2642. https://doi.org/10.3390/biom5042619
Williams JA, Ding W-X. A Mechanistic Review of Mitophagy and Its Role in Protection against Alcoholic Liver Disease. Biomolecules. 2015; 5(4):2619-2642. https://doi.org/10.3390/biom5042619
Chicago/Turabian StyleWilliams, Jessica A., and Wen-Xing Ding. 2015. "A Mechanistic Review of Mitophagy and Its Role in Protection against Alcoholic Liver Disease" Biomolecules 5, no. 4: 2619-2642. https://doi.org/10.3390/biom5042619