
Direct and/or Indirect Roles for SUMO in Modulating Alpha-Synuclein Toxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Parkinson’s Disease
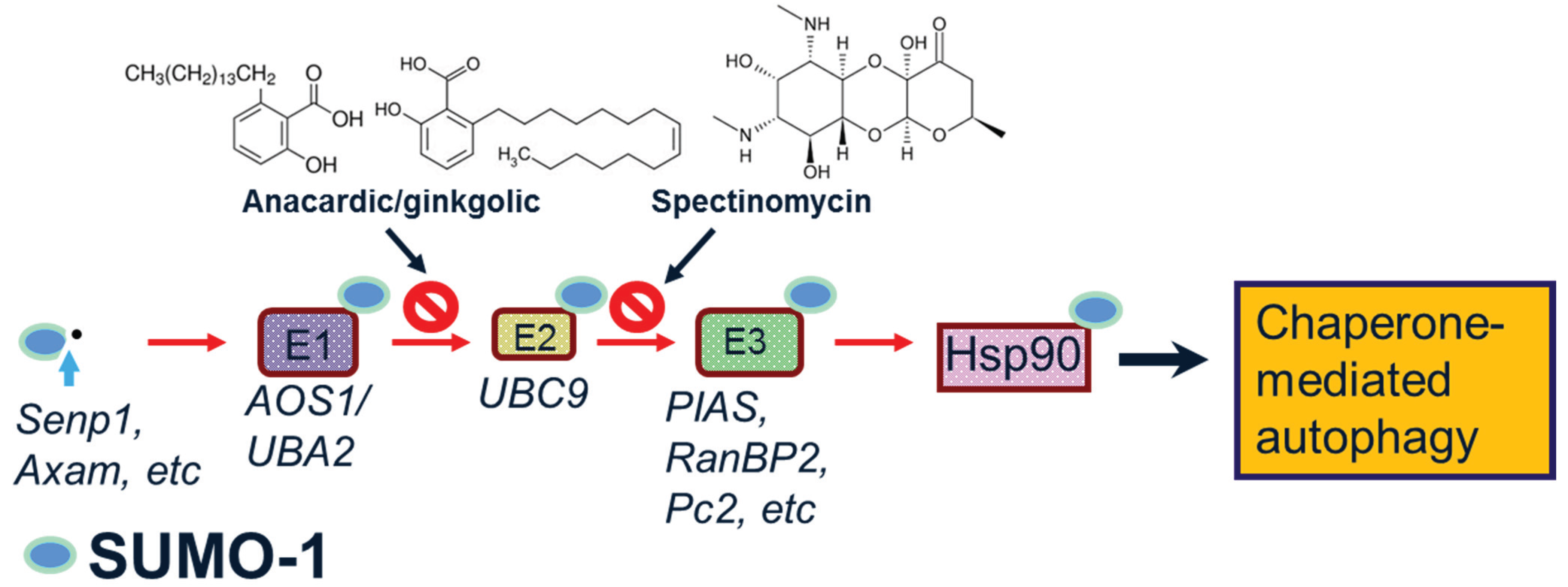
1.2. SUMO-1
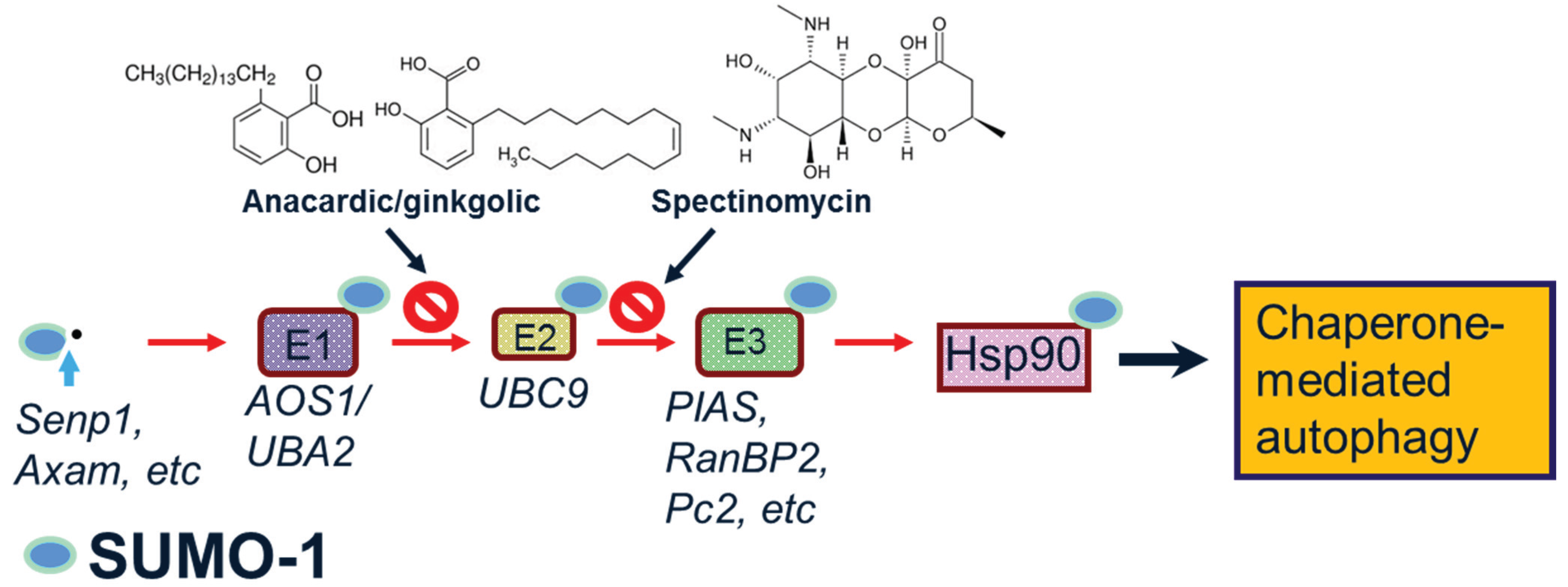
2. Potential Roles of SUMO-1 in α-Synuclein Aggregation, Degradation and Neuroprotection
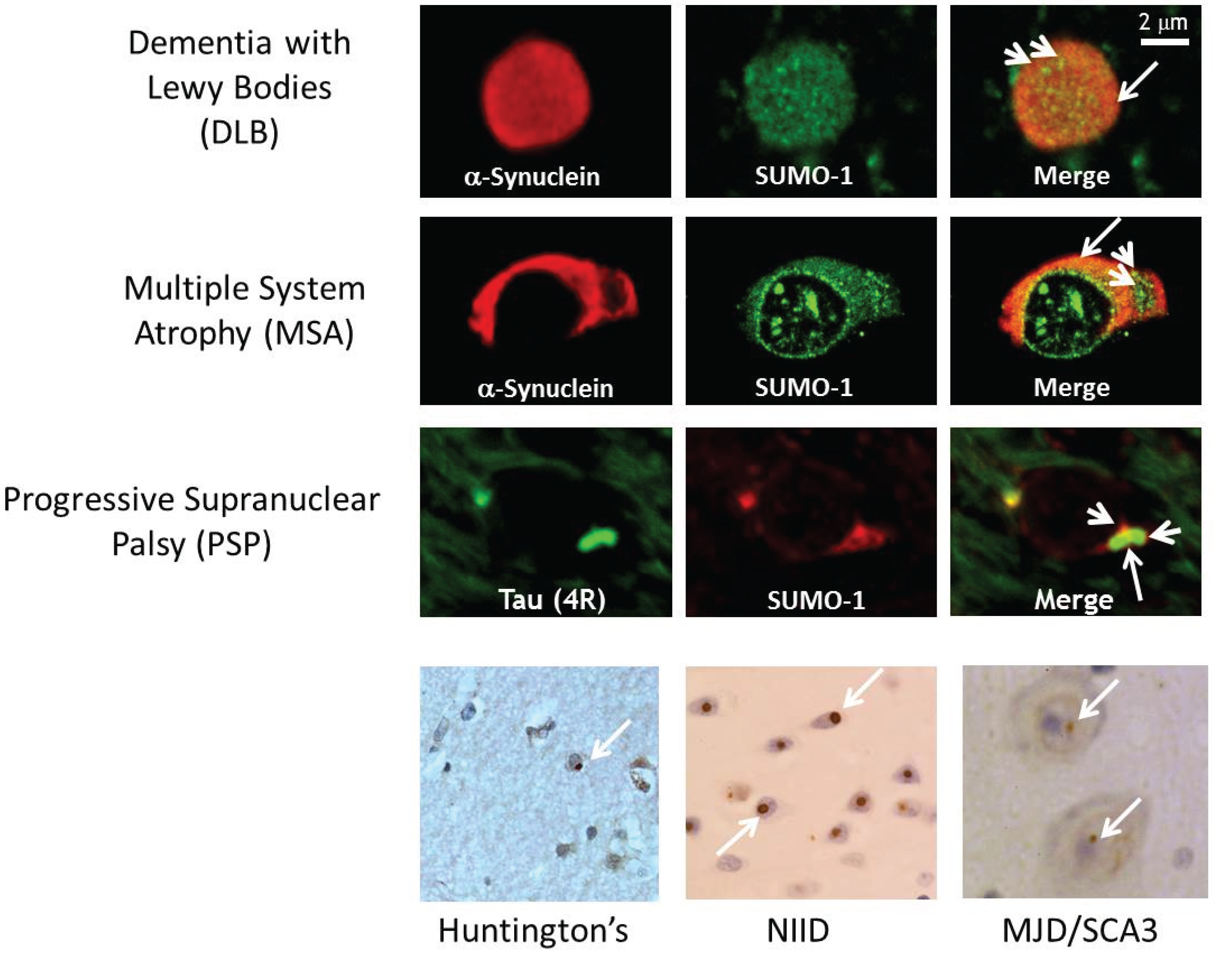
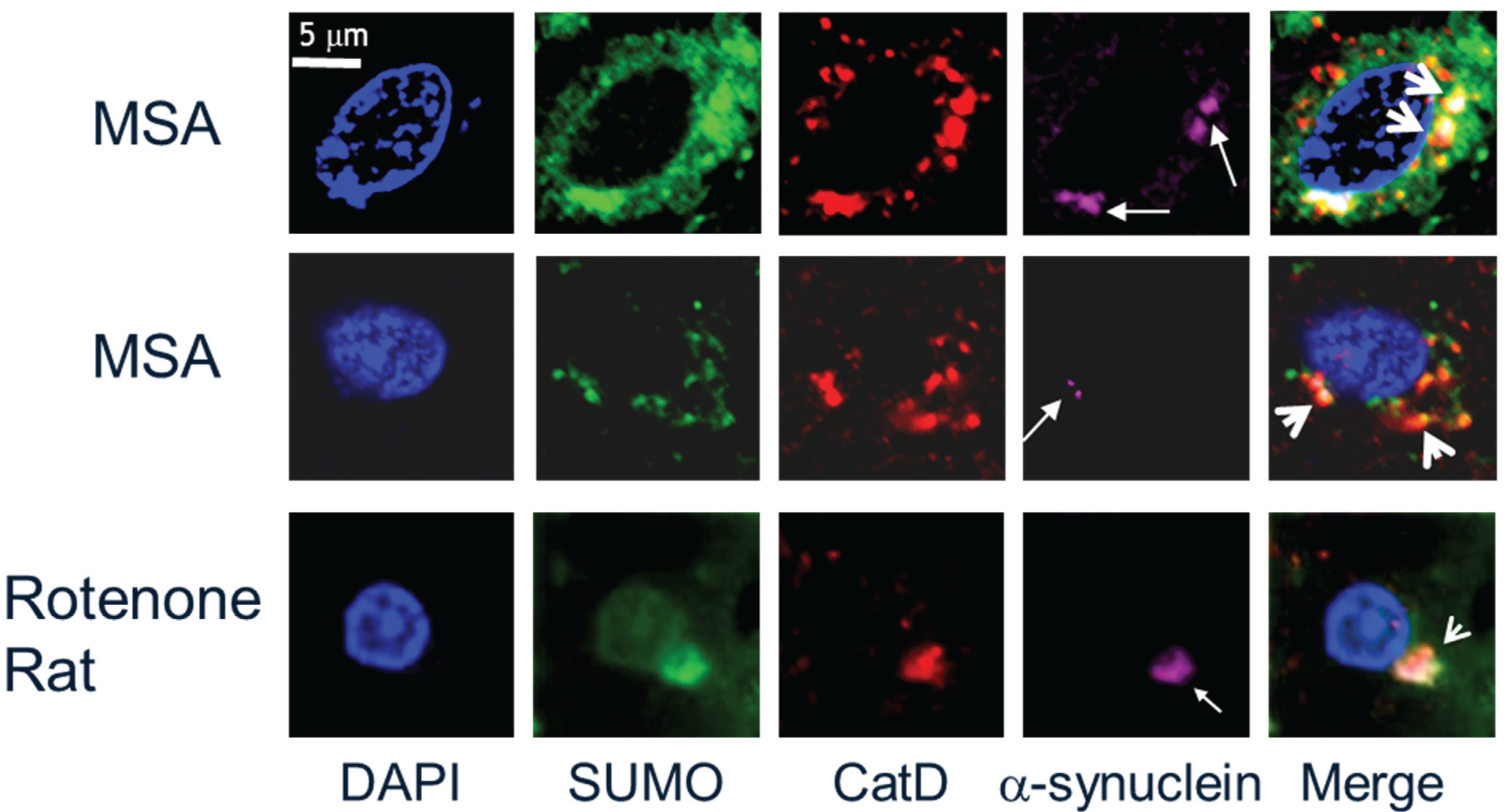
2.1. SUMO-1 in α-Synuclein Disease
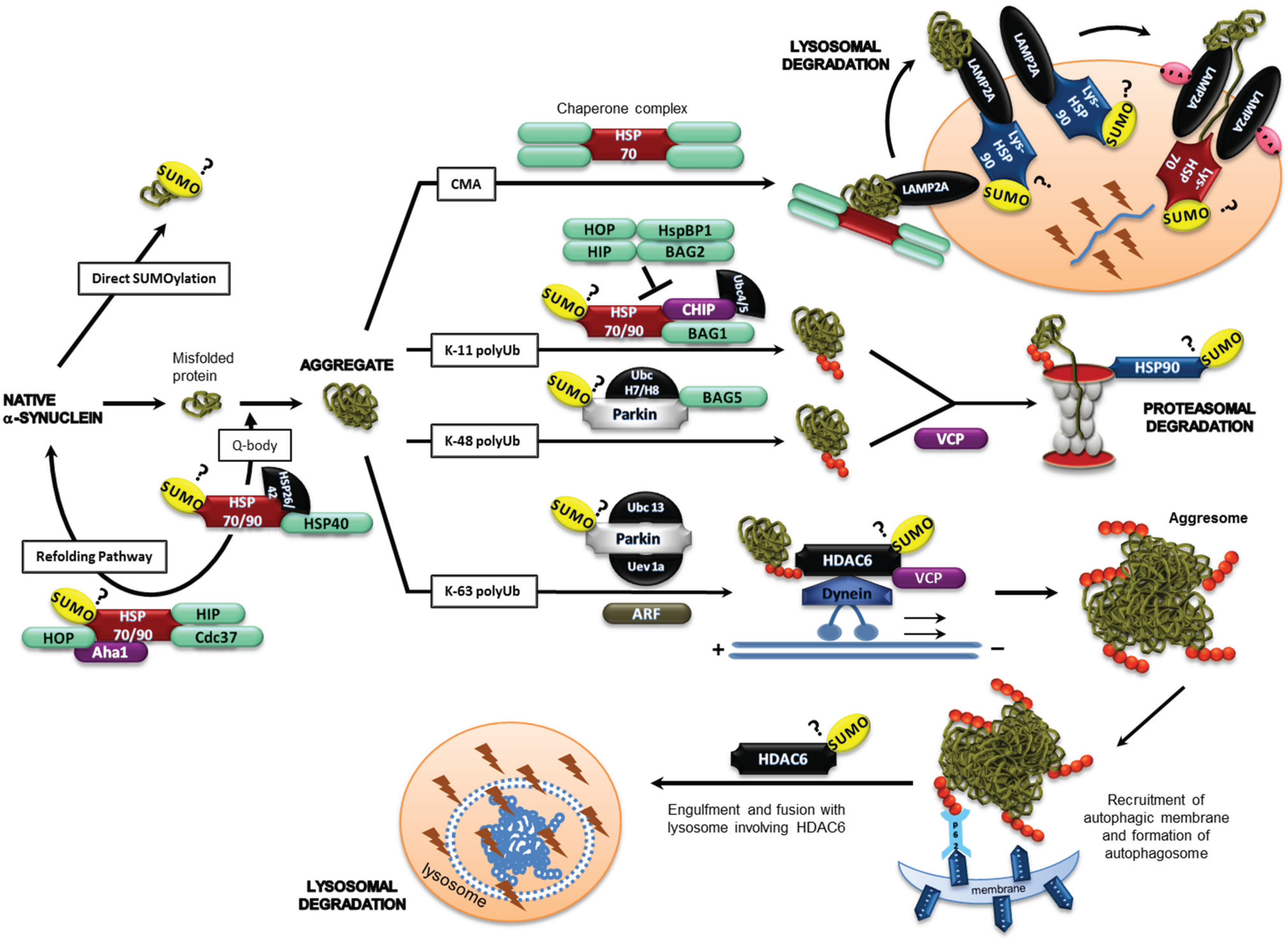
2.2. SUMO-1 in α-Synuclein Aggregate Clearance
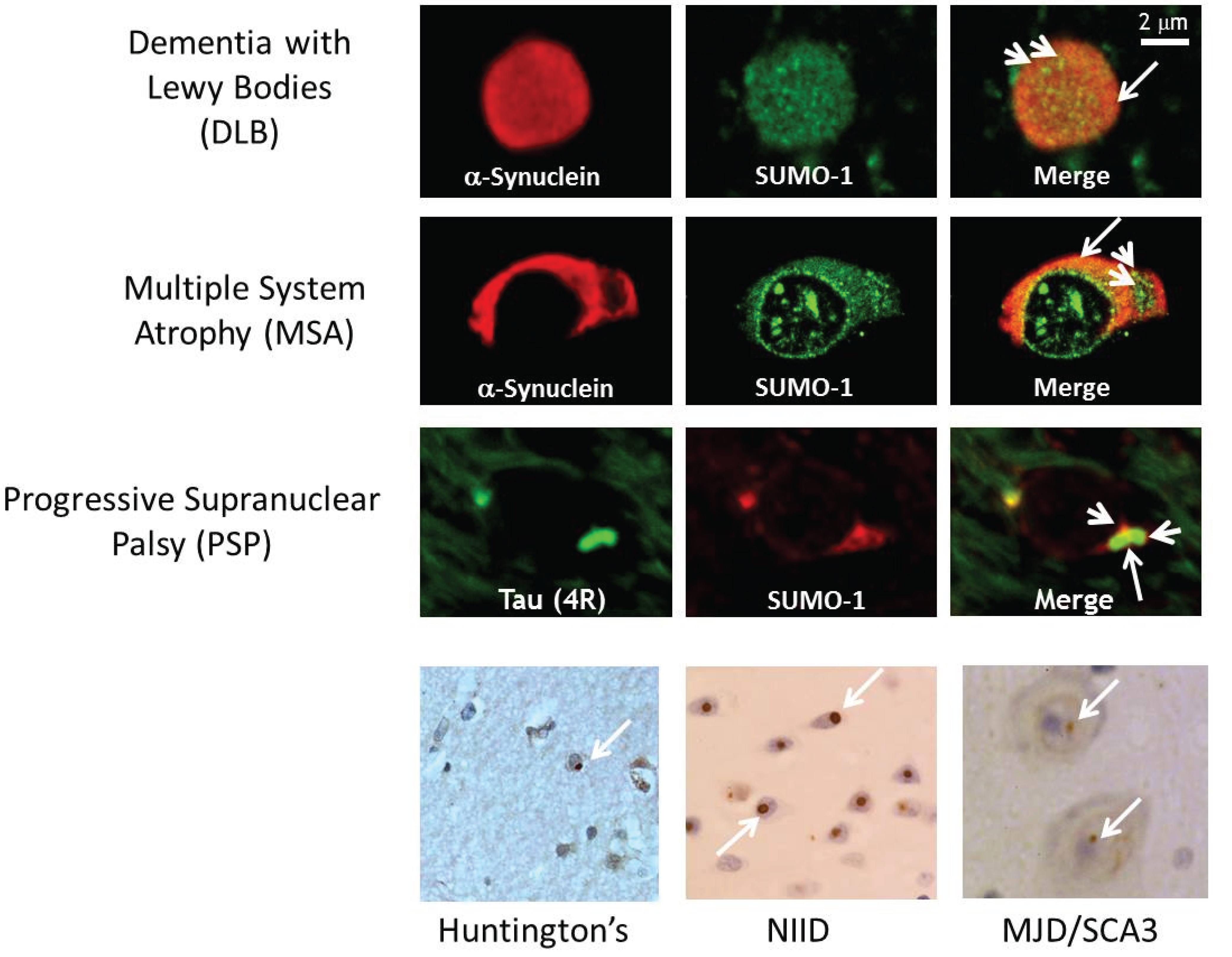
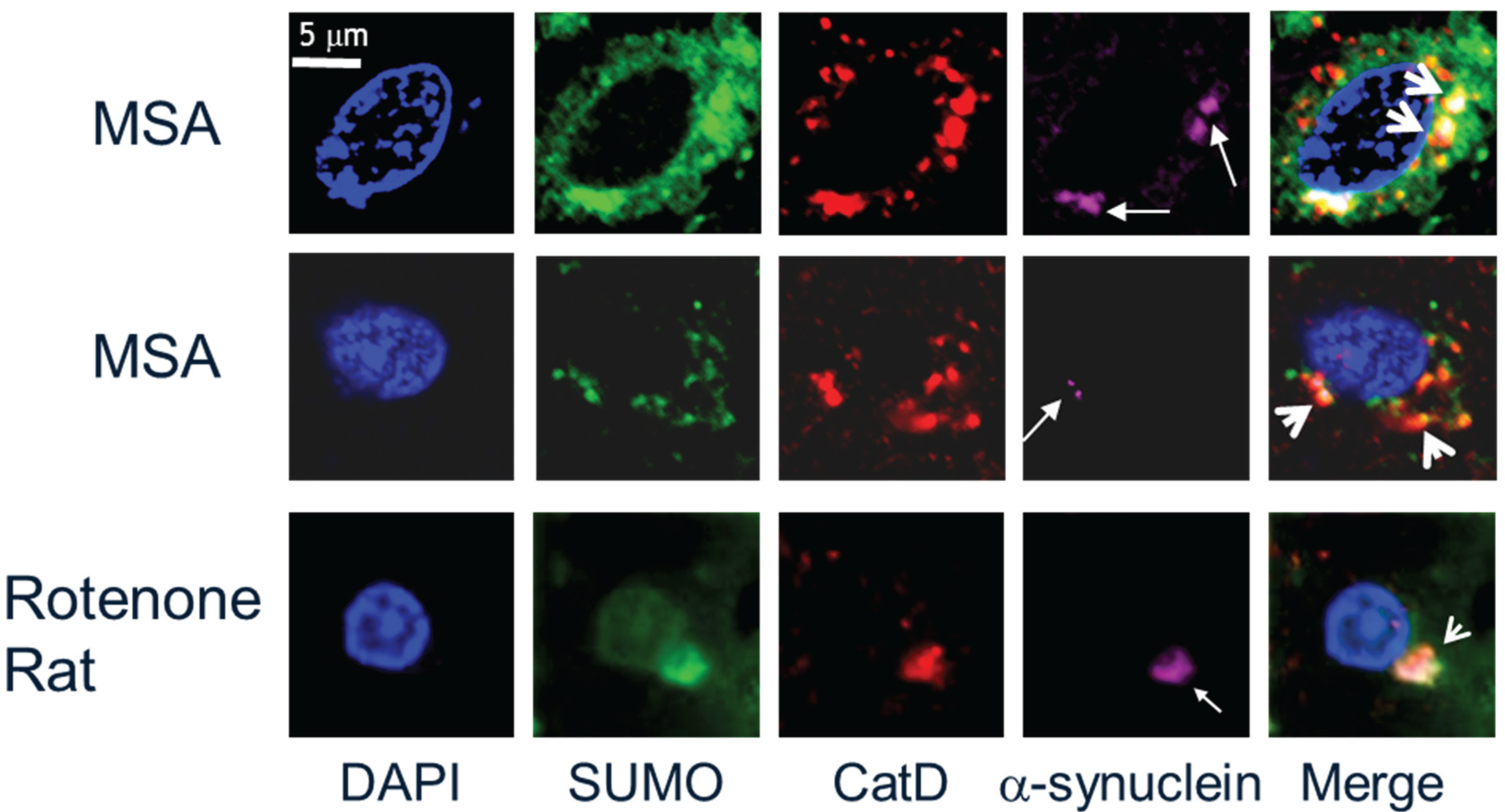
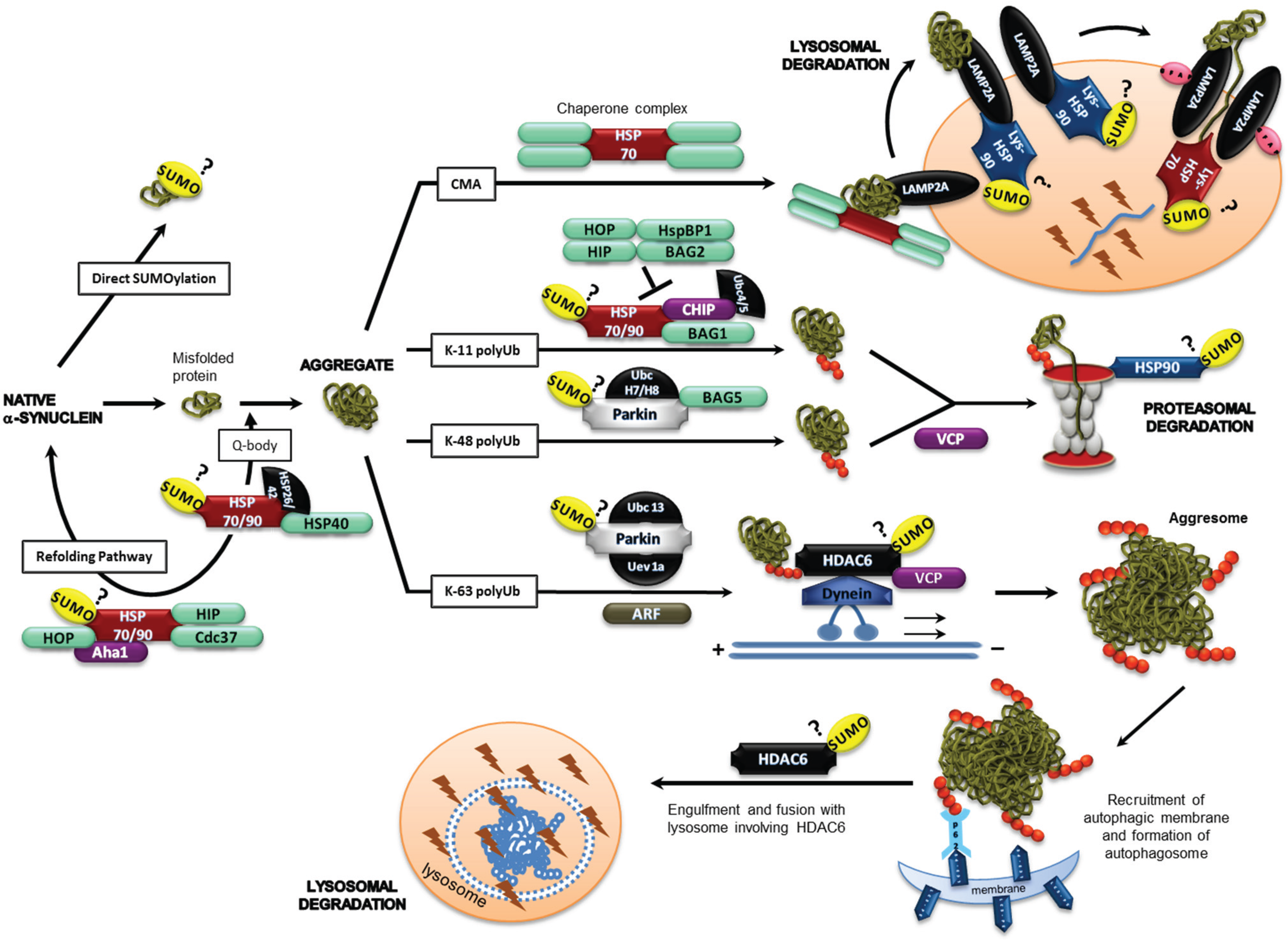
2.3. α-Synuclein and SUMOylation
2.4. Chaperones, Co-Chaperones and Adaptor Proteins
2.4.1. Hsp90
2.4.2. HDAC6
2.4.3. VCP
2.4.4. Parkin
2.4.5. TRAF6
2.4.6. CHIP
2.4.7. P53 and ARF
3. Therapeutic Implications of SUMO Modulation of Selective Autophagy for Neuroprotection
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Rcom-H’cheo-Gauthier, A.; Goodwin, J.; Pountney, D.L. Interactions between calcium and alpha-synuclein in neurodegeneration. Biomolecules 2014, 4, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Giráldez-Pérez, R.M.; Antolín-Vallespín, M.; Muñoz, M.D.; Sánchez-Capelo, A. Models of α-synuclein aggregation in Parkinson’s. Acta Neuropathol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Kågedal, K.; Halliday, G.M. α-Synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther. 2014. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Formation and development of Lewy pathology: A critical update. J. Neurol. 2009, 256, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Radford, R.; Wong, M.; Pountney, D.L. Neurodegenerative aspects of multiple system atrophy. In Handbook of Neurotoxicity; Kostrezewa, R., Ed.; Springer: Berlin, Germany, 2014; pp. 2157–2180. [Google Scholar]
- Roberts, H.; Brown, D. Seeking a mechanism for the toxicity of oligomeric α-synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Daturpalli, S.; Waudby, C.A.; Meehan, S.; Jackson, S.E. Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 2013, 425, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Marques, O.; Outeiro, T.F. α-Synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.S.; Olzmann, J.A.; Li, L. Aggresome formation and neurodegenerative diseases: Therapeutic implications. Curr. Med. Chem. 2008, 15, 47–60. [Google Scholar] [CrossRef]
- Jackrel, M.E.; Shorter, J. Reversing deleterious protein aggregation with re-engineered protein disaggregases. Cell Cycle 2014, 13, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Sarge, K.D.; Park-Sarge, O.K. SUMO and its role in human diseases. Int. Rev. Cell Mol. Biol. 2011, 288, 167–183. [Google Scholar] [PubMed]
- Eckermann, K. SUMO and Parkinson’s disease. Neuromol. Med. 2013, 15, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.B.; Wilkinson, K.A.; Henley, J.M. Protein SUMOylation in neuropathological conditions. Drug News Perspect. 2009, 22, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.A.; Nakamura, Y.; Henley, J.M. Targets and consequences of protein SUMOylation in neurons. Brain Res. Rev. 2010, 64, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Dorval, V.; Fraser, P.E. SUMO on the road to neurodegeneration. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Henley, J.M.; Craig, T.J.; Wilkinson, K.A. Neuronal SUMOylation: Mechanisms, physiology, and roles in neuronal dysfunction. Physiol. Rev. 2014, 94, 1249–1285. [Google Scholar] [CrossRef] [PubMed]
- Wasik, U.; Filipek, A. Non-nuclear function of sumoylated proteins. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2878–2885. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Henley, J.M. Wrestling with stress: Roles of protein SUMOylation and deSUMOylation in cell stress response. IUBMB Life 2014, 66, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Droescher, M.; Chaugule, V.K.; Pichler, A. SUMO rules: Regulatory concepts and their implication in neurologic functions. Neuromol. Med. 2013, 15, 639–660. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Bourboulia, D.; Beebe, K.; Woodford, M.R.; Polier, S.; Hoang, A.; Chelluri, R.; Li, Y.; Guo, A.; Lee, M.J.; et al. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol. Cell 2014, 53, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Goodwin, J.; Norazit, A.; Meedeniya, A.B.; Richter-Landsberg, C.; Gai, W.P.; Pountney, D.L. SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox. Res. 2013, 23, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Pountney, D.L.; Huang, Y.; Burns, R.J.; Haan, E.; Thompson, P.D.; Blumbergs, P.C.; Gai, W.P. SUMO-1 marks the nuclear inclusions in familial neuronal intranuclear inclusion disease. Exp. Neurol. 2003, 184, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Pountney, D.L.; Chegini, F.; Shen, X.; Blumbergs, P.C.; Gai, W.P. SUMO-1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neurosci. Lett. 2005, 381, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jang, W.H.; Quezado, M.M.; Oh, Y.; Chung, K.C.; Junn, E.; Mouradian, M.M. Proteasome inhibition induces α-synuclein SUMOylation and aggregate formation. J. Neurol. Sci. 2011, 307, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Pountney, D.; Raftery, M.; Chegini, F.; Blumbergs, P.; Gai, W. NSF, Unc-18-1, dynamin-1 and Hsp90 are inclusion body components in neuronal intranuclear inclusion disease identified by anti-SUMO-1-immunocapture. Acta Neuropathol. 2008, 116, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Weetman, J.; Wong, M.B.; Sharry, S.; Rcom-H’cheo-Gauthier, A.; Gai, W.P.; Meedeniya, A.; Pountney, D.L. Increased SUMO-1 expression in the unilateral rotenone-lesioned mouse model of Parkinson’s disease. Neurosci. Lett. 2013, 544, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Abeywardana, T.; Pratt, M.R. Extent of inhibition of α-synuclein aggregation in vitro by SUMOylation is conjugation site-and SUMO isoform-selective. Biochemistry 2015, 54, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits α-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Weishaupt, J.H. Sumoylation in neurodegenerative diseases. Cell. Mol. Life Sci. 2013, 70, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Kim, Y.M.; Mouradian, M.M.; Chung, K.C. Human Polycomb protein 2 promotes α-synuclein aggregate formation through covalent SUMOylation. Brain Res. 2011, 1381, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: Therapeutic implications. Apoptosis 2010, 15, 1292–1311. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagy basics. Microbiol. Immunol. 2011, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martinez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-mediated autophagy at a glance. J. Cell Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Cuervo, A.M. Entering the lysosome through a transient gate by chaperone-mediated autophagy. Autophagy 2008, 4, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Reggiori, F.; Peter, M. Selective types of autophagy in yeast. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.P. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer 2010, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R.; et al. Global changes to the ubiquitin system in Huntington’s disease. Nature 2007, 448, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Dawson, V.L.; Dawson, T.M. Parkin-mediated lysine 63-linked polyubiquitination: A link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol. Aging 2006, 27, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.; Wong, E.S.; Dawson, V.L.; Dawson, T.; Lim, K.L. Lysine 63-linked polyubiquitin potentially partners with p62 to promote the clearance of protein inclusions by autophagy. Autophagy 2008, 4, 251–253. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Batlevi, Y.; Baehrecke, E.H.; Taylor, J.P. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system, and neurodegeneration. Autophagy 2007, 3, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- McLean, P.J.; Kawamata, H.; Ribich, S.; Hyman, B.T. Membrane association and protein conformation of α-synuclein in intact neurons effect of Parkinson’s disease-linked mutations. J. Biol. Chem. 2000, 275, 8812–8816. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. α-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449. [Google Scholar] [CrossRef] [PubMed]
- Kunadt, M.; Eckermann, K.; Stuendl, A.; Gong, J.; Russo, B.; Strauss, K.; Rai, S.; Kügler, S.; Falomir Lockhart, L.; et al. Extracellular vesicle sorting of α-synuclein is regulated by sumoylation. Acta Neuropathol. 2015, 129, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.M.; Balch, W.E. Q-bodies monitor the quinary state of the protein fold. Nat. Cell Biol. 2013, 10, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Riedel, M.; Goldbaum, O.; Schwarz, L.; Schmitt, S.; Richter-Landsberg, C. 17-AAG induces cytoplasmic alpha-synuclein aggregate clearance by induction of autophagy. PLoS ONE 2010, 18, e8753. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, A.; Himstedt, F.; Funk, M.; Korn, B.; Gaestel, M.; Niedenthal, R. Ubc9 fusion-directed SUMOylation identifies constitutive and inducible SUMOylation. Nucleic Acids Res. 2007. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; di Prospero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Przedborski, S. Mitophagy: The latest problem for Parkinson’s disease. Trends Mol. Med. 2011, 17, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T. Diversity in the regulation of autophagy and mitophagy: Lessons from Parkinson’s disease. Parkinson’s Dis. 2011. [Google Scholar] [CrossRef]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Neptune, M.A.; DePinho, R.A. SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J. Biol. Chem. 2002, 277, 23658–23663. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Sharrocks, A.D. SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 2004, 13, 611–617. [Google Scholar] [CrossRef]
- Kirsh, O.; Seeler, J.S.; Pichler, A.; Gast, A.; Müller, S.; Miska, E.; Mathieu, M.; Harel-Bellan, A.; Kouzarides, T.; Melchior, F.; et al. The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J. 2002, 21, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Girdwood, D.; Bumpass, D.; Vaughan, O.A.; Thain, A.; Anderson, L.A.; Snowden, A.W.; Garcia-Wilson, E.; Perkins, N.D.; Hay, R.T. P300 transcriptional repression is mediated by SUMO modification. Mol. Cell 2003, 11, 1043–1054. [Google Scholar] [CrossRef]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.; Prodromou, C.; Workman, P. The Hsp90 molecular chaperone: An open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Regulation of signaling protein function and trafficking by the Hsp90/Hsp70-based chaperone machinery. Exp. Biol. Med. 2003, 228, 111–133. [Google Scholar]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology 2011, 31, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Sasagawa, Y.; Ogura, T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Boyault, C.; Gilquin, B.; Zhang, Y.; Rybin, V.; Garman, E.; Meyer-Klaucke, W.; Matthias, P.; Müller, C.W.; Khochbin, S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006, 25, 3357–3366. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.S.; Miller, S.E.; Hanson, P.I.; Weihl, C.C. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J. Biol. Chem. 2008, 283, 30289–30299. [Google Scholar] [CrossRef] [PubMed]
- Dargemont, C.; Ossareh-Nazari, B. Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Bergink, S.; Ammon, T.; Kern, M.; Schermelleh, L.; Leonhardt, H.; Jentsch, S. Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51–Rad52 interaction. Nat. Cell Biol. 2013, 15, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Chung, K.C. Functional modulation of Parkin through physical interaction with SUMO-1. J. Neurosci. Res. 2006, 84, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.G.; Chua, D.S.; Basil, A.H.; Chan, H.Y.; Chai, C.; Arumugam, T.; Lim, K.L. Cytosolic pten-induced putative kinase 1 is stabilized by NF-κB pathway and promotes non-selective mitophagy. J. Biol. Chem. 2015, 290, 16882–16893. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.L.; Hao, R.; Chen, L.F.; Lai, C.H.; Kapur, M.; Shaughnessy, P.J.; Chou, D.; Yan, J.; Taylor, J.P.; Engelender, S.; et al. Convergence of Parkin, PINK1, and α-synuclein on stress-induced mitochondrial morphological remodeling. J. Biol. Chem. 2015, 290, 13862–13874. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. The three “P”s of mitophagy: Parkin, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.K.; Kumar, A. TRAF6 coordinates the activation of autophagy and ubiquitin-proteasome systems in atrophying skeletal muscle. Autophagy 2011, 7, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.V.; Zhou, H.J.; Lin-Lee, Y.C.; Tamayo, A.T.; Yoshimura, L.C.; Fu, L.; Darnay, B.G.; Ford, R.J. Nuclear tumor necrosis factor receptor-associated factor 6 in lymphoid cells negatively regulates c-Myb-mediated transactivation through small ubiquitin-related modifier-1 modification. J. Biol. Chem. 2008, 283, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Li, H.; Mei, W.; Cheng, J. SUMOylation attenuates human β-Arrestin 2 inhibition of IL-1R/TRAF6 signaling. J. Biol. Chem. 2015, 290, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- McDonough, H.; Patterson, C. CHIP: A link between the chaperone and proteasome systems. Cell Stress Chaperones 2003, 8, 303–308. [Google Scholar] [CrossRef]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.; Hrstka, R.; Lane, D.; Vojtesek, B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 2013, 32, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ambasta, R.K.; Veereshwarayya, V.; Rosen, K.M.; Kosik, K.S.; Band, H.; Mestril, R.; Patterson, C.; Querfurth, H.W. CHIP and Hsps interact with β-APP in a proteasome-dependent manner and influence Aβ metabolism. Hum. Mol. Genet. 2007, 16, 848–864. [Google Scholar] [CrossRef] [PubMed]
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005, 94, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Sun, X.; Xiang, B.; Cang, H.; Kang, X.; Chen, Y.; Li, H.; Shi, G.; Yeh, E.T.; Wang, B.; et al. Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO J. 2010, 29, 3773–3786. [Google Scholar] [CrossRef] [PubMed]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Tago, K.; Chiocca, S.; Sherr, C.J. Sumoylation induced by the Arf tumor suppressor: A p53-independent function. Proc. Natl. Acad. Sci. USA 2005, 102, 7689–7694. [Google Scholar] [CrossRef] [PubMed]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, M.; Li, H.Y. Tumor suppressor ARF promotes non-classic proteasome-independent polyubiquitination of COMMD1. J. Biol. Chem. 2008, 283, 11453–11460. [Google Scholar] [CrossRef] [PubMed]
- Ivanschitz, L. PML, SUMOylation, and senescence. Front. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Murphy, M.E. Interaction of the ARF tumor suppressor with cytosolic Hsp70 contributes to its autophagy function. Cancer Biol. Ther. 2011, 12, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.R.; Lakhter, A.J.; Androphy, E.J. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle 2012, 11, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vijayakumaran, S.; Wong, M.B.; Antony, H.; Pountney, D.L. Direct and/or Indirect Roles for SUMO in Modulating Alpha-Synuclein Toxicity. Biomolecules 2015, 5, 1697-1716. https://doi.org/10.3390/biom5031697
Vijayakumaran S, Wong MB, Antony H, Pountney DL. Direct and/or Indirect Roles for SUMO in Modulating Alpha-Synuclein Toxicity. Biomolecules. 2015; 5(3):1697-1716. https://doi.org/10.3390/biom5031697
Chicago/Turabian StyleVijayakumaran, Shamini, Mathew B. Wong, Helma Antony, and Dean L. Pountney. 2015. "Direct and/or Indirect Roles for SUMO in Modulating Alpha-Synuclein Toxicity" Biomolecules 5, no. 3: 1697-1716. https://doi.org/10.3390/biom5031697