
Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease

1
Beijing Institute of Geriatrics, Xuanwu Hospital of Capital University of Medical Sciences, No.45 changchun St., Xicheng District, Beijing 100053, China
2
Parkinson's disease Center of Beijing Institute for Brain Disorders, Beijing 100053, China
*
Author to whom correspondence should be addressed.
Biomolecules 2015, 5(2), 1122-1142; https://doi.org/10.3390/biom5021122
Submission received: 15 March 2015
/
Accepted: 29 April 2015
/
Published: 5 June 2015
(This article belongs to the Special Issue Exploring the Mechanisms by which α-Synuclein Kills Cells in Parkinson Disease)
{kind=link}
{kind=link}
Abstract
:Parkinson’s disease (PD) is a very common neurodegenerative disorder characterized by the accumulation of α-synuclein (α-syn) into Lewy body (LB) inclusions and the loss of neuronmelanin (NM) containing dopamine (DA) neurons in the substantia nigra (SN). Pathological α-syn and NM are two prominent hallmarks in this selective and progressive neurodegenerative disease. Pathological α-syn can induce dopaminergic neuron death by various mechanisms, such as inducing oxidative stress and inhibiting protein degradation systems. Therefore, to explore the factors that trigger α-syn to convert from a non-toxic protein to toxic one is a pivotal question to clarify the mechanisms of PD pathogenesis. Many triggers for pathological α-syn aggregation have been identified, including missense mutations in the α-syn gene, higher concentration, and posttranslational modifications of α-Syn. Recently, the role of NM in inducing α-syn expression and aggregation has been suggested as a mechanism for this pigment to modulate neuronal vulnerability in PD. NM may be responsible for PD and age-associated increase and aggregation in α-syn. Here, we reviewed our previous study and other recent findings in the area of interaction between NM and α-syn.
1. Pathological Characteristics and Aetiology of Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder. The clinical symptoms of PD include motor and non-motor symptoms. The main motor symptoms of PD are bradykinesia (resting tremor), rigidity, and postural instability [1,2,3]. It is believed that the progressive loss of neuromelanin (NM)-containing dopaminergic neurons in the substantia nigra pars compacta (SNpc) is the cause of motor symptoms in PD. Signaling between the SN and the striatum is involved in controlling muscle movements; therefore the resultant loss of nigro-striatal pathway signaling can explain the classical motor symptoms of PD [2,3]. “Dopamine (DA) replacement” pharmacotherapy is relatively effective at reducing the motor disturbances [4]. The non-motor symptoms include autonomic dysfunction (orthostatic hypotension, sphincter disturbances, and/or constipation), cognitive changes, psychiatric effects (depression, psychosis, and/or impulse control disorder), sensory symptoms (pain and/or aching), restlessness, and sleep disturbances [5,6]. These non-motor symptoms may result from disturbances of other neurotransmitter pathways, such as cholinergic, serotonergic, or GABA-ergic and they respond relatively poorly to dopaminergic therapies [7].
The aetiology of PD is often explained by the interaction between environmental and genetic factors. Environmental studies have identified significant risk factors for PD, such as the exposure to pesticides, herbicide, and metal irons [8]. Many of the recent studies reported that some rare genetic causes of PD relate to the mutations in genes. These genes include SNCA [9,10], the leucine-rich repeat kinase 2 (LRRK2) [11], glucocerebrosidase (GBA) [12], Parkin [13], DJ-1 [14], PINK1 [15] HLA [16] and MAPT [17] genes. In these genetics risk factors for PD, the most important is SNCA; the gene responsible for the expression of α-syn. To date, six PD-linked point mutations in SNCA have been identified, comprising the A30P [18], A53T [19], E46K [20], H50Q [21], G51D [22], and A53E [23].
On the other hand, aging is the biggest known risk factor for the development of idiopathic PD and it is crucial to understand its role. With advancing age, a number of processes essential for the function of SN neurons including DA metabolism, wild type mitochondrial DNA copy number and protein degradation decline [24]. A decline in wild type mtDNA copy number will lead to a decrease in ATP production and a reduction in efficient protein degradation will affect the function of neurons [25]. DA metabolism generates a significant amount of reactive oxygen species that will affect a number of different processes within the neurons [26]. In young and healthy SN neurons, toxic DA metabolic products could be formed in the dark pigment of NM. NM accumulates in double membrane autophagic vacuoles, preventing neurotoxic effects of free neuromelanin in cells exposed to this pigment [27]. However, in aging SN neurons, there is an age-related high content or overall accumulation of NM in the SN. The accumulation NM, which overload of toxic metals or compounds, can potentially result in increased oxidative stress and decreased transformation of toxic DA metabolic products into NM [28]. Moreover, the accumulation of NM may induce the expression and aggregation of α-syn, increasing toxic insults to NM containing SN neurons [29]. NM that leaks from degenerating neurons may contribute to the degeneration of DA neurons in PD by activating microglia [30]. Those toxic mechanisms cause the loss of vulnerable neurons, once this cell loss reaches a certain level, the symptoms of PD develop.
2. Parkinson’s Disease and α-Synuclein
2.1. α-Synuclein is Linked to the Pathogenesis of Parkinson’s Disease
The involvement of α-syn in PD was initially identified through genetic linkage studies in a small number of families [9], including mutations as well as gene duplications [31] and triplications [32]. Up to now, six missense mutations in α-syn gene have been linked to the pathogenesis of PD, comprising A30P, A53T, E46K, H50Q, G51D, and A53E. The A53T and A30P mutations affect the response to oxidative stress, with expression of these mutant isoforms significantly increasing cytotoxicity induced by hydrogen peroxide and 1-methyl-4-phenylpyridinium (MPP+) in comparison to cells expressing wild-type α-syn and control cells [33]. Moreover, the A30P, A53T and H50Q mutations result in increased oligomerization and fibril formation compared to wild-type [34,35], conversely, G51D and A53E mutations reduce the aggregation of α-syn [36,37]. E46K mutation disrupts macroautophagy via inactivation of JNK1-Bcl-2 pathway [38]. Recently, strong association was shown between α-syn and sporadic PD in GWAS [39,40]. α-Syn is also a major component of LBs [41]. These arguments illustrate that α-syn is a central player in the pathogenesis of PD.
Although the mechanisms by which the genetic variants affect protein pathology remain to be resolved, the processes by which α-syn protein can become pathological are better understood. Under certain conditions, α-syn monomers interact to form prefibrillar or protofibrils, which in turn can form insoluble fibrils [42,43,44]. A widely accepted hypothesis for α-syn toxicity proposes that protofibrils of α-syn are cytotoxic, whereas the fibrillar aggregates of the protein could represent a cytoprotective mechanism in PD [45]. Supporting this hypothesis, α-syn protofibrils are increased in the brains of patients with PD and dementia with Lewy bodies (DLB) [46], and have been associated with neurotoxicity in α-syn overexpressing cells and mouse models [47,48].
2.2. α-Synuclein and Cytotoxicity
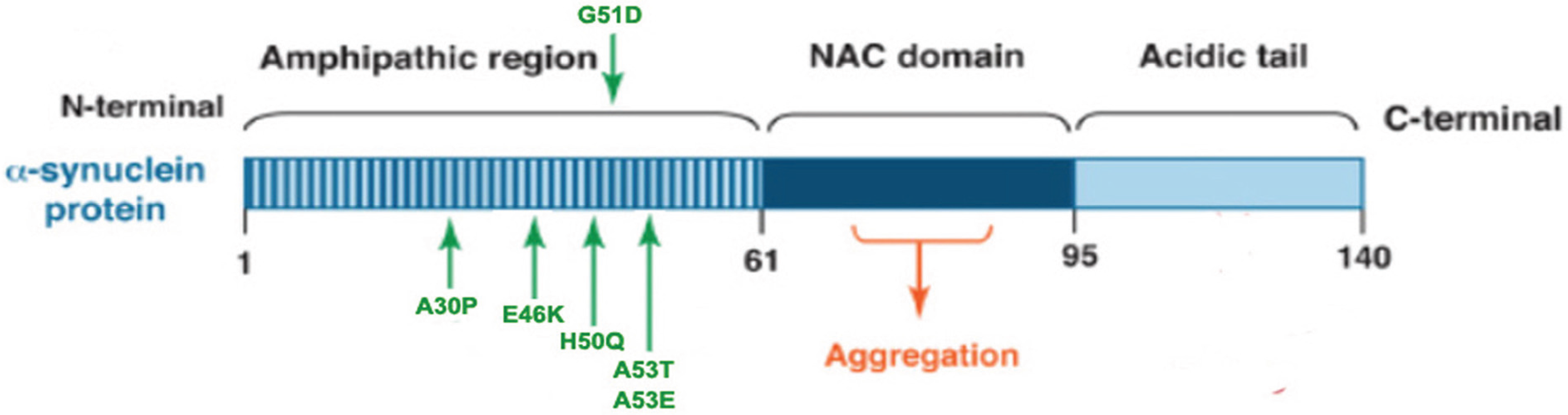
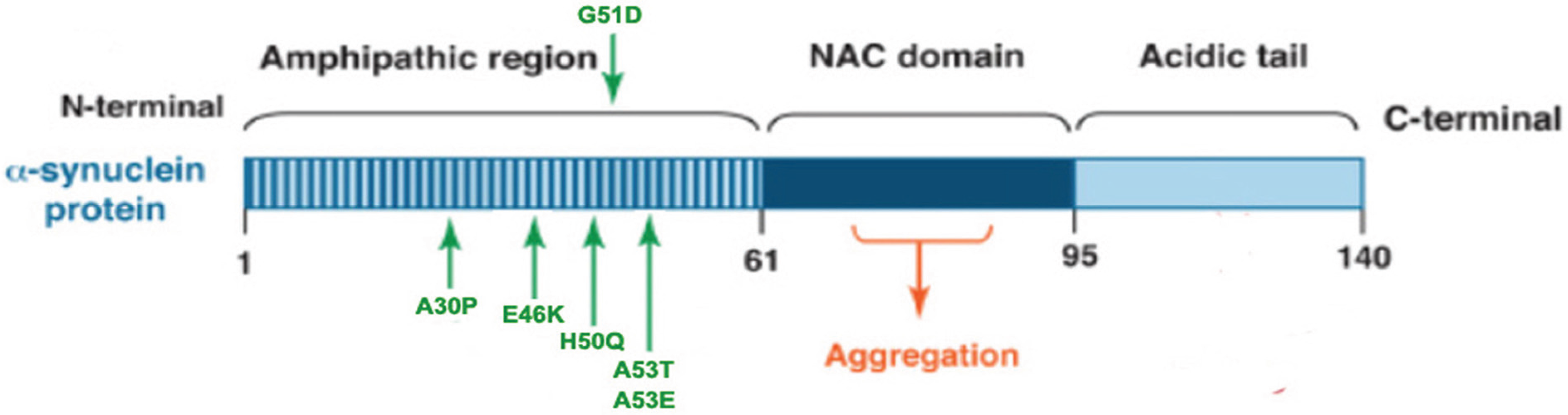
α-Syn is a 14 kDa natively unfolded protein encoded by the SNCA gene that is highly conserved in vertebrate species, and the small protein is abundantly expressed in the brain as well as in multiple other central and peripheral tissues [49]. Maroteaux et al. [50] first described the localization of α-syn to the nucleus and the presynaptic terminal. Although the exact function of α-syn remains unknown, substantial evidence suggest that α-syn associates with vesicular and membranous structures and plays a role in synaptic vesicle recycling, and storage and compartmentalization of neurotransmitters [51]. The α-syn peptide has 140 amino acids, with three distinct regions: a N-terminal (1–60 residues) that contain four imperfect repeats of KTKEGV motifs; a NAC region (61–95 residues), with three additional KTKEGV repeats; and the hydrophobic and amyloidogenic NAC region, a C-terminus (96–140 residues) that is enriched in acidic and proline residues, and that facilitates interactions with different proteins [52] (see Figure 1). All three regions are necessary for the misfolding of the protein.
The conformation of α-syn is highly dependent on environmental conditions. Indeed, despite the overwhelming evidence that α-syn is a disordered monomer in solution, two recent reports suggest that the native protein exists as a helical tetramer with a molecular weight of about 58 kDa under physiological conditions, with reduced aggregation tendencies, and that the dissociation of the tetramer into monomeric subunits promotes toxic aggregation [53,54]. What is more certain is the α-syn protein adopts oligomeric and/or fibrillar conformations in certain pathological conditions [55,56,57] (such as mutations in the SNCA gene, structural modification produced by environmental changes, mistakes on the post-translational modifications and induction of protein misfolding by seeding or cross-seeding mechanisms).
Figure 1.
α-Synuclein (α-syn) protein domain structure. α-Syn is a 140 amino acid protein and its sequence can be divided into three regions with distinct structural characteristics. The highly conserved N-terminal domain encodes for a series of imperfect 11 amino acid repeats with a consensus motif of KTKEGV reminiscent of the lipid-binding domain of apolipoproteins, which in certain conditions forms amphipathic helices. The six missense mutations known to cause familial PD (A30P, E46K, H50Q, G51D, A53E and A53T) lie in the amphipathic region, suggesting an important function for this region of the protein. The central hydrophobic region (non-amyloid-β component or NAC domain) of α-synuclein is associated with an increased propensity of the protein to form fibrils [58]. The acidic C-terminal tail contains mostly negatively charged residues and is largely unfolded.
Figure 1.
α-Synuclein (α-syn) protein domain structure. α-Syn is a 140 amino acid protein and its sequence can be divided into three regions with distinct structural characteristics. The highly conserved N-terminal domain encodes for a series of imperfect 11 amino acid repeats with a consensus motif of KTKEGV reminiscent of the lipid-binding domain of apolipoproteins, which in certain conditions forms amphipathic helices. The six missense mutations known to cause familial PD (A30P, E46K, H50Q, G51D, A53E and A53T) lie in the amphipathic region, suggesting an important function for this region of the protein. The central hydrophobic region (non-amyloid-β component or NAC domain) of α-synuclein is associated with an increased propensity of the protein to form fibrils [58]. The acidic C-terminal tail contains mostly negatively charged residues and is largely unfolded.
Up to now, although the precise mechanisms of cytotoxicity of α-syn are still not fully understood, abundant evidence suggests that the toxic effect of α-syn involves many different mechanisms: (i) loss of the normal function of α-syn in neurotransmission release, especially in the regulating DA release [59,60,61,62,63], leads to the selective dysfunction or degeneration of DA neurons in PD; (ii) α-Syn impairs mitochondrial structure and inhibits complex I activity, promoting the production of reactive oxygen species (ROS) [64,65,66,67,68]; (iii) α-Syn disrupts ER-Golgi vesicular transport and results in toxic ER stress [69,70,71]; (iv) α-Syn inhibits protein degradation systems, including ubiquitin-proteasomal system (UPS) and Autophagy-lysosome system [72], interferes with the normal physiology of the cell, and eventually leads to cell injury and death; (v) α-Syn promotes the permeabilization of membranes and disrupts cellular homeostasis [73]; (vi) α-Syn induces the increase of ROS by direct or indirect interaction with oxidative stress [74]; and (vii) “prion-like” propagation, by which pathological α-syn aggregates are propagated between connected brain regions via a cell-to-cell transmission mechanism, which is an important mechanism to induce progressive DA neurons loss in PD [75].
2.3. α-Synuclein Post-Translational Modifications
Several post-translational modifications of α-syn may occur, such as phosphorylation, truncation, ubiquitination, nitration. Studies investigating the phosphorylation of α-syn in diseased and aged brains have shown that α-syn can be phosphorylated at serines (S87, S129) as well as at several tyrosines, including Y125, Y133, and Y136. The pS129 modification is most often correlated with PD pathology. This notion is primarily supported by the finding that the majority of α-syn in LBs in postmortem PD brains is phosphorylated at S129 (pS129) [76,77]. The S129 phosphorylation of α-syn in aggregates has also been observed in animal models of PD [78,79]. Although a few studies reported that overexpression of the phosphorylated serine 129 isoform in animal models does not produce toxicity [80,81], most evidence suggests that phosphorylation of α-syn at serine 129 to promote its aggregation and neurotoxicity [82]. Mechanistic studies have shown that aggregated forms of α-syn are more prone to phosphorylation and that pS129 phosphorylated aggregates accumulate as the disease progresses [83,84,85], suggesting that the degree of α-syn pS129 phosphorylation is an indicator of disease progression. The link between S129 phosphorylation and PD pathology has fueled an interest in modulating α-syn phosphorylation at S129 as a potential therapy for PD. Multiple kinases have been identified which phosphorylate α-syn at S129, with most evidence pointing to polo-like kinase 2 (PLK2) as the primary phosphorylated of α-syn S129 [86]. A straightforward therapeutic approach based on reducing α-syn phospho-S129 would be to inhibit PLK 2 kinase activity; however some contradictory findings should be taken into account. For instance, overexpression of PLK 2 in rat brain using adeno-associated viral vectors can suppress α-syn toxicity by promoting autophagy-mediated degradation of phospho-S129 α-syn [87]. Therefore, therapies based on modulating α-syn phospho-S129 appear to require an optimal phosphorylation level rather than a complete dephosphorylation.
The majority of ubiquinated α-syn is mono- to tri-ubiquinated, and while poly-ubiquitination serves as a signal for α-syn degradation by the proteasome, it does not seem to be required for α-syn fibrillation and LB formation [88]. Nevertheless, an interplay between phosphorylation and ubiquitination may render the protein more susceptible to aggregation [89]. Small amounts of various C-terminal truncated forms of α-syn have been detected in LBs, which exhibit greater fibrillation capacity [90]. Oxidation leads to other common post-translational modifications, including nitrosylation, and it has been shown that oxidative stress can stabilize oligomeric α-syn species via the formation of di-tyrosine cross-links [91].
2.4. α-Synuclein and Oxidative Stress
Abundant evidence suggests that there is a potential interaction between α-syn and oxidative stress. Some studies have found that oxidative stress induces up-regulation of the expression of α-syn, and promotes its fibrillization and aggregation [92]. Conversely, a high degree of fibrillization and aggregation of α-syn results in an increase of reactive oxygen species (ROS) and neurotoxicity [93]. It is believed that a vicious cycle between α-syn and oxidative stress may play a pivotal role in the progressive loss of SN dopaminergic neurons in PD. For example, incubation of recombinant α-syn with cytochrome c in the presence of H2O2 [94], or exposure of cells in culture to H2O2 and ferrous iron, MPP+, NO and superoxide promotes α-syn aggregation [58,95,96]. Additionally, oxidative stress can cause nuclear membrane modifications and α-syn translocation to the nucleus where it can form complexes with histones leading to its oligomerization into insoluble fibrils [97,98,99]. On the other hand, aggregation and high levels of α-syn have been shown to induce oxidant production or increase the level of oxidative stress. As a result, experiments with cultured cells or animal models have demonstrated that over-expression of wild-type or mutant α-syn increases sensitivity to DA, MPTP and 6-OHDA toxicity [100,101,102,103]. Conversely, mice lacking α-syn demonstrate marked resistance to MPTP and other mitochondrial toxins such as malonate and 3-nitropropionic acid [104,105]. Addition of exogenous α-syn fibrils in culture medium or injecting it into the brain can led to selective decreases in synaptic proteins, progressive impairments in neuronal excitability and connectivity, and eventually, neuron death. These results suggest that oxidative stress promotes α-syn aggregation, which in turn increases oxidative stress level, creating a vicious cycle leading to neurodegeneration.
3. Parkinson’s Disease and Neuromelanin
3.1. Neuromelanin Structure and Biosynthesis
NM is the dark insoluble macromolecule that confers the black (SN) or grey (locus coeruleus) color to monoaminergic basal ganglia. It is a polymer pigment synthesized that contains catecholamine-based compounds such as oxidized DA, DA metabolites as well as proteins and lipids. Histological studies showed that NM granules were located in the neuronal perikaryon and were surrounded by a double membrane [106]. In the SN, NM accumulates during aging and is found after the first two to three years of life [107]. X-ray diffraction studies have shown that NM has a multilayer (graphite-like) three-dimensional structure similar to synthetic and naturally occurring melanin [108]. The three-dimensional structure is derived from planar overlapped sheets consisting of cyclic molecules of indole benzothiazine rings. However, these sheets are stacked much higher in NM than in any other synthetic and naturally occurring melanin [108].
Biosynthesis of NM may due to tyrosine hydroxylase-mediated oxidation of DA [109] or auto-oxidation of DA [110]. Although the process of NM formation is obscure, a recent in vitro study has clearly established some steps of this complex process [111]. The study demonstrated that NM synthesis was induced in rat SN neurons and PC12 cell cultures by exposure to L-dopa. The pigment produced in this model contains a stable free radical; in addition, both light and electron microscopy have shown that the pigment synthesized in these cells appears to be identical to human NM, and the granules are surrounded by a double membrane, similar to the naturally occurring NM of the SN. In this model, NM synthesis was shown to be driven by an excess of cytosolic catecholamines.
3.2. Neuromelanin Involved in the Pathogenesis of Parkinson’s Disease
Although the role of NM remains unclear, especially in the SN, both under physiological conditions and in the pathogenesis of PD, it is a fact that NM containing neurons in SN are more vulnerable than the non-pigmented ones in PD. Normally, NM is considered to play a protective role intracellular by binding toxic metabolite produced in SN cells such as oxidized DA, DA metabolites and metals [112,113,114] and serve as an antioxidant [115,116]. However, it has been also suggested that NM can be potentially toxic to DA neurons, by directly inhibiting proteasomes function [117], and catalyzing the production of free radicals [118]. NM might also become a source of free radicals by reaction with hydrogen peroxide [119,120]. Changed structure and density of NM have been found in early PD, suggesting that NM may play role in the pathogenesis in PD. Analysis of NM in the SN of PD patients has shown an early accumulation and overload of iron, which can potentially result in increased oxidative stress [121]. The interaction of NM with α-syn has also been suggested as a mechanism for this pigment to modulate neuronal vulnerability [74]. α-Syn is over-expressed in individual melanized neurons [29], and its aggregates redistribute to NM in the SN early in PD but not in healthy controls [121]. Moreover, NM that leaks from degenerating neurons may contribute to the neurodegenerative process by activating microglia [122]. The activated microglia produces proinflammatory cytokines such as TNF-α, IL-6 and NO and may involve in PD pathogenesis [123].
3.3. Interaction of Neuromelanin with Organic or Inorganic Molecules
NM interacts with numerous organic and inorganic molecules including lipids, pesticides, toxic compounds and metal ions [124]. NM might reduce the toxicity of MPTP by accumulating its toxic metabolite MPP+ in vivo [125]. The herbicide paraquat has a molecular structure similar to that of MPTP, and has been proposed as a Parkinson’s disease inducing agent. The pesticide is accumulated in NM containing nerve cells, where it appears that the NM adsorbed intraneuronal paraquat, protecting the neurons from consequent damage [126]. NM can also accumulate chlorpromazine, haloperidol, and imipramine, thereby contributing to the control of the intraneuronal concentration of these molecules. Because higher intraneuronal concentrations of dopaminergic drugs might be toxic to substantia nigra neurons, NM can influence this toxicity [127]. The association of NM with lipids has been described in several studies [128,129,130]. Although previous studies proposed that lipids were part of the NM molecule, recent work has shown that NM contains about 20% adsorbed lipids. Cholesterol is a minor component in this lipid mixture, with the major component being a new class of polyunsaturated lipid with a high molecular mass, low volatility, and low oxygen content [129]. It may be that NM itself catalyzes the synthesis of this type of lipid. Alternatively, NM could originate from lipofuscin by an enzymatic reaction occurring in lysosomes [131], although this hypothesis is not supported by previous observations [123]. In this case, high molecular mass lipids could be derived from a lysosomal metabolic pathway and might interact with NM within these organelles. The ability of NM to bind different types of organic compounds may influence the intracellular role of lipids and proteins.
High concentrations of iron and other non-alkaline metals are present in several brain nuclei. NM from the SN can interact with many heavy metal ions such as zinc, copper, manganese, chromium, cobalt, mercury, lead, and cadmium; in addition, it binds iron particularly strongly [132,133]. In the course of PD, the concentration of iron in the SN increases by 30%–35%. This accumulation of SN iron seems to occur within the NM granules: the concentration of iron in these granules is higher in patients with PD than in normal subjects [134]. It seems that the amount of iron bound to NM determines whether this molecule acts as a protective agent blocking redox active metal ions or whether in the presence of excess iron it promotes the formation of cytotoxic radicals [135,136]. The ability of NM to chelate other redox active metals such as copper, manganese, chromium, and toxic metals including cadmium, mercury, and lead [133] strengthens the hypothesis that NM may be a high capacity storage trapping system for metal ions and, as such, may prevent neuronal damage. The capacity of NM to form stable complexes with toxic exogenous metals and redox active metals seems to play a protective role even if such a capability can in principle be saturated by high cytosolic concentrations of metals.
4. Interaction between α-Synuclein and Neuromelanin
4.1. Accumulation of Nenromelanin Increases the Level of α-Synuclein in SN Neurons
NM is considered to play a protective role as a scavenger to clear toxic metabolite produced in SN neurons. However, it has been also suggested that NM can be potentially toxic to DA neurons, by directly inhibiting proteasomal function and catalyzing the production of free radicals [127]. Recently, several studies suggested NM induces the expression or aggregation of α-syn in DA neurons of SN in aging or PD brain [29,115,137,138]. Fasano et al. reported that NM directly binds with α-syn in residual SN neurons of PD [139]. The fact that α-syn is expressed in melanoma and nevus, but not in non-melanocytic cutaneous carcinoma and normal skin, suggests that melanin may induce the expression of α-syn [138]. Moreover, α-syn mRNA levels were detected significantly elevated in individual NM-containing DA neurons from PD midbrain when compared to those from matched controls, further supporting the notion of NM induces the expression of α-syn [137]. NM also increases the accumulation of α-syn in DA neurons. Halliday GM et al. reported that more vulnerable SN A9 neurons with normal morphological appearance exhibited significantly increased NM density associated with a concentration of α-syn to the lipid component of NM in early PD patients [115]. The increased concentration of neuronal NM and α-syn in normal A9 neurons may already predispose these neurons to precipitate α-syn around NM-associated lipid under oxidative conditions. These changes may trigger a cascade of events leading to larger intracellular aggregates of α-syn and eventually lead to the death of those NM-containing DA neurons in PD [115]. In addition to PD brain, our previous study found that NM accumulation also induces expression of α-syn during aging in NM-containing DA neurons and leads to age related loss of DA neurons [29]. In our previous study, the marked increase in NM and α-syn in aged individuals suggests that NM content may be essential for α-syn over-expression [29]. We found that accumulation of NM in SN starts very early in life [140], whereas the detectable pathogenic accumulation of α-syn begins around middle age [141] in the same area. Although both regions contain DA neurons and are closely localized to one another, it has been reported that α-syn accumulates with age in the SN but not in the ventral tegmental area (VTA) [141]. An important difference between the VTA and the SN is that the dopaminergic neurons in the SN contain significantly more NM than those in the VTA. This further supports the notion that NM may be responsible for age-associated increases in α-syn.
Although growing evidence suggests that excessive accumulation of NM may initiate gathering of α-syn, the precise mechanism is still unclear. Findings to date suggested that iron saturated NM produces many free radical species and increases oxidative insults [142], which may initiate gathering of α-syn. Alternatively, accumulated NM could induce proteasome inhibition [111,143] and result in a reduction of α-syn clearance. Further, NM may be synergistic with other factors that promote age-related “autophagic stress” [144] and thereby influence the degradation of α-syn [145].
4.2. α-Synuclein Induces the Biosynthesis of Neuromelanin
Pan et al. [146] reported that over-expressed α-syn increased the content of NM in SH-SY5Y and PC12 dopaminergic cells, suggesting α-syn may promote the biosynthesis of NM in DA neurons. However, the precise mechanisms of α-syn induce biosynthesis of NM is still unclear. At present, it has been accepted that the formation of NM is due to either enzymatically mediated or auto-oxidation of DA pathway [147]. So far, tyrosinase, tyrosine hydroxylase (TH), peroxidase, prostaglandin H synthase, and macrophage migration inhibitory factor were suggested to be involved in NM synthesis [148]. However, there is currently no general agreement on the role eventually played by any of these enzymes in the synthesis of NM in SN neurons [147]. Although the role of these enzymes in the synthesis of NM is not known for sure, current studies indicate that α-syn modulates the activities of some of them, such as tyrosinase [149], tyrosine hydroxylase (TH) [150], and peroxidase [151]. α-Syn may affect the biosynthesis of NM by regulating these enzyme activities.
On the other hand, NM could derive from non-enzymatic oxidation. The auto-oxidation of catechols to quinones with the subsequent addition of thiol groups has been demonstrated in the brain [152]. An in vitro study clarified some steps of the complex biosynthesis of NM in cultured dopaminergic neuronal cells [123]. NM synthesis could be induced in rat SN and in PC12 cells in culture by exposure to L-DOPA, which is rapidly converted in DA in the cytosol. The pigment produced in this model was chemically identical to human NM as demonstrated by EPR. Moreover, it was localized in characteristic organelles surrounded by a double membrane, as is similar to naturally occurring NM [123]. Moreover, NM synthesis could be inhibited by the adenoviral-mediated over-expression of the synaptic vesicular monoamine transporter 2 (VMAT2), which sequesters monoamines from the cytosol into synaptic vesicles for subsequent neurotransmission [153], suggesting that an excess of cytosolic free catecholamines is an essential factor for the synthesis of NM. The high concentration of NM in SN neurons seems to be linked to the presence of considerable amounts of cytosolic dopamine that have not been sequestered into synaptic vesicles [123].
Abundant evidence has proved that α-syn is involved in regulating DA homeostasis and affecting the levels of cytosolic DA. α-Syn may modulate DA homeostasis through multiple mechanisms, such as synthesis, storage, release and reuptake [101]. α-Syn regulates the biosynthesis of DA by inhibiting the activity of tyrosine hydroxylase (TH) [154], the rate-limiting enzyme responsible for converting tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) in the DA synthesis pathway. Overexpression of α-syn has also been reported to decrease the rate of DA release, both in mouse and cell culture models [155,156]. α-Syn can direct bind to DAT via its C-terminal domain and enhance extracellular DA uptake by increasing the number of functional transporters at the cell surface [157,158]. Enhanced extracellular DA uptake increases the levels of cytosolic DA. More importantly, α-syn may affect the storage of DA by regulating vesicle docking and recycling at the nerve terminal, increasing permeability of secretory vesicles and decreasing the levels of VMAT2. This could prevent incorporation of newly synthesized and newly taken up DA into vesicles and lead to increase the level of cytosolic DA [159]. The notion is supported by some studies that found α-syn mutation or overexpression lead to elevated cytosolic DA [160,161]. The cytosolic DA can be metabolized in mitochondria by monoamine oxidase (MAO). If these processes are insufficient to control cytosolic DA homeostasis, excess cytoslic DA can be oxidized to quinones and semiquinones via iron catalysis in the cytosol. These quinones react with Cys residues in proteins, such as α-syn [162], to form DA-Cys-protein adducts, which are phagocytized in autophagic vacuoles (AG). Because of a low level of lysosomal fusion and/or to a buildup of undegradable DA-adducts, even in the presence of lysosomal hydrolases, over time the vacuoles become NM granules [148]. So, α-syn may induce the biosynthesis of NM by increasing the levels of cytosolic DA.
5. Conclusions
The pathological characteristics of PD are progressive loss of neuromelanin (NM)-containing dopaminergic neurons in SNpc and α-syn positive LBs in survival neurons. Abundant evidence has proved that α-syn play a pivotal role in the pathogenesis of PD. Missense mutations in α-syn gene could result in early onset familial PD. The toxicity of abnormal α-syn forms to DA neurons and loss of normal functions of this protein are important causes in the pathogenesis in sporadic PD. NM is a dark, complex, and insoluble pigment granule in several types of neurons of the central nervous system that is particularly concentrated in the DA neurons of SN and in the noradrenergic neurons of locus coeruleus [127]. Although the mechanism is not clear, SN and the locus coeruleus are the two brain areas mostly affected by PD. This fact draws a close attention to NM and its possible involvement in neurodegenerative processes. Recently, several studies suggested that the interaction of NM with α-syn may be a mechanism for this pigment to modulate neuronal vulnerability. α-Syn is over-expressed in individual melanized neurons [29,137,138] and its aggregates redistribute to NM in the SN early in PD but not in healthy controls [115]. On the other hand, α-syn also induces the biosynthesis of NM by increasing the levels of cytosolic DA [146].
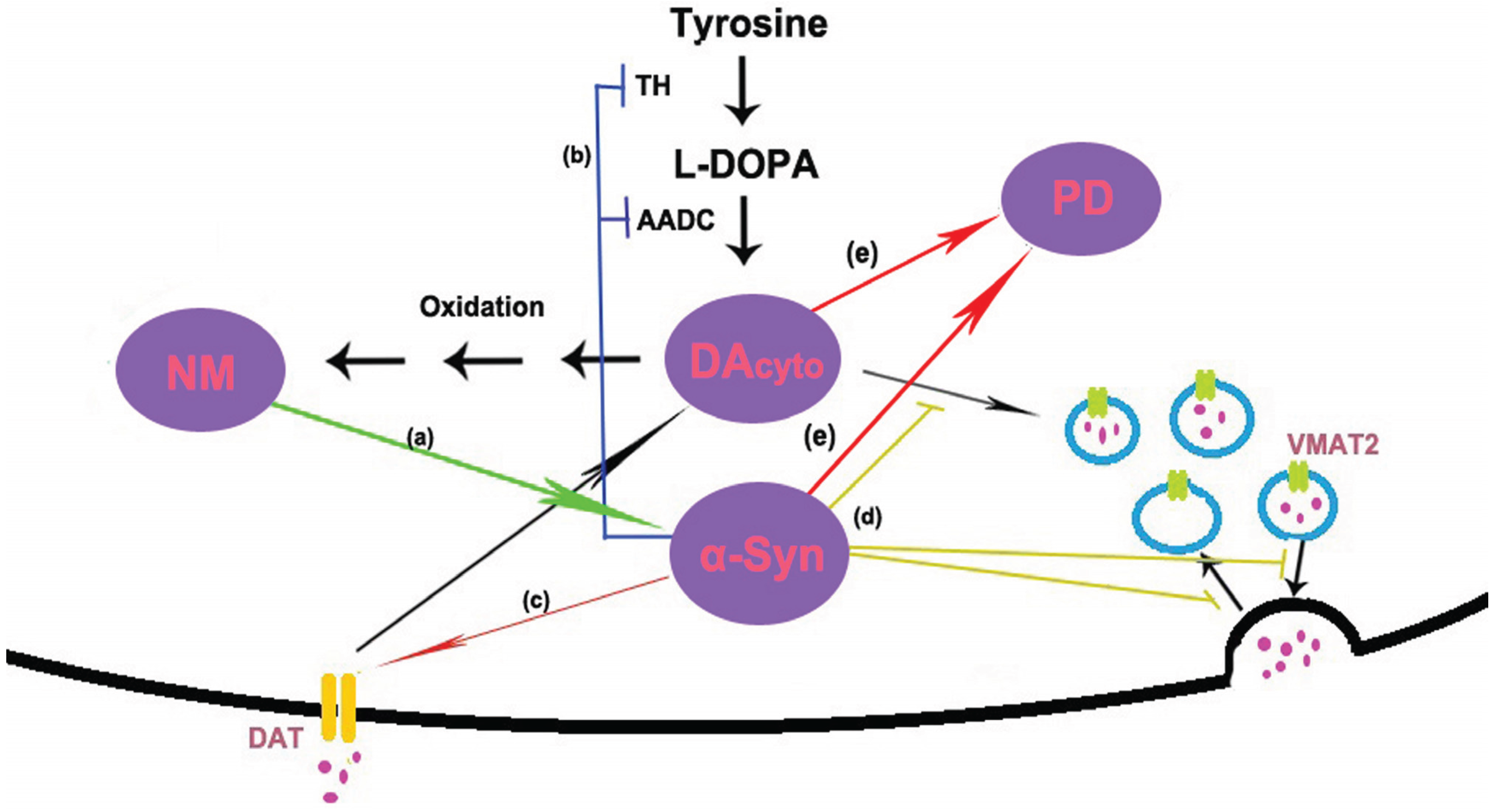
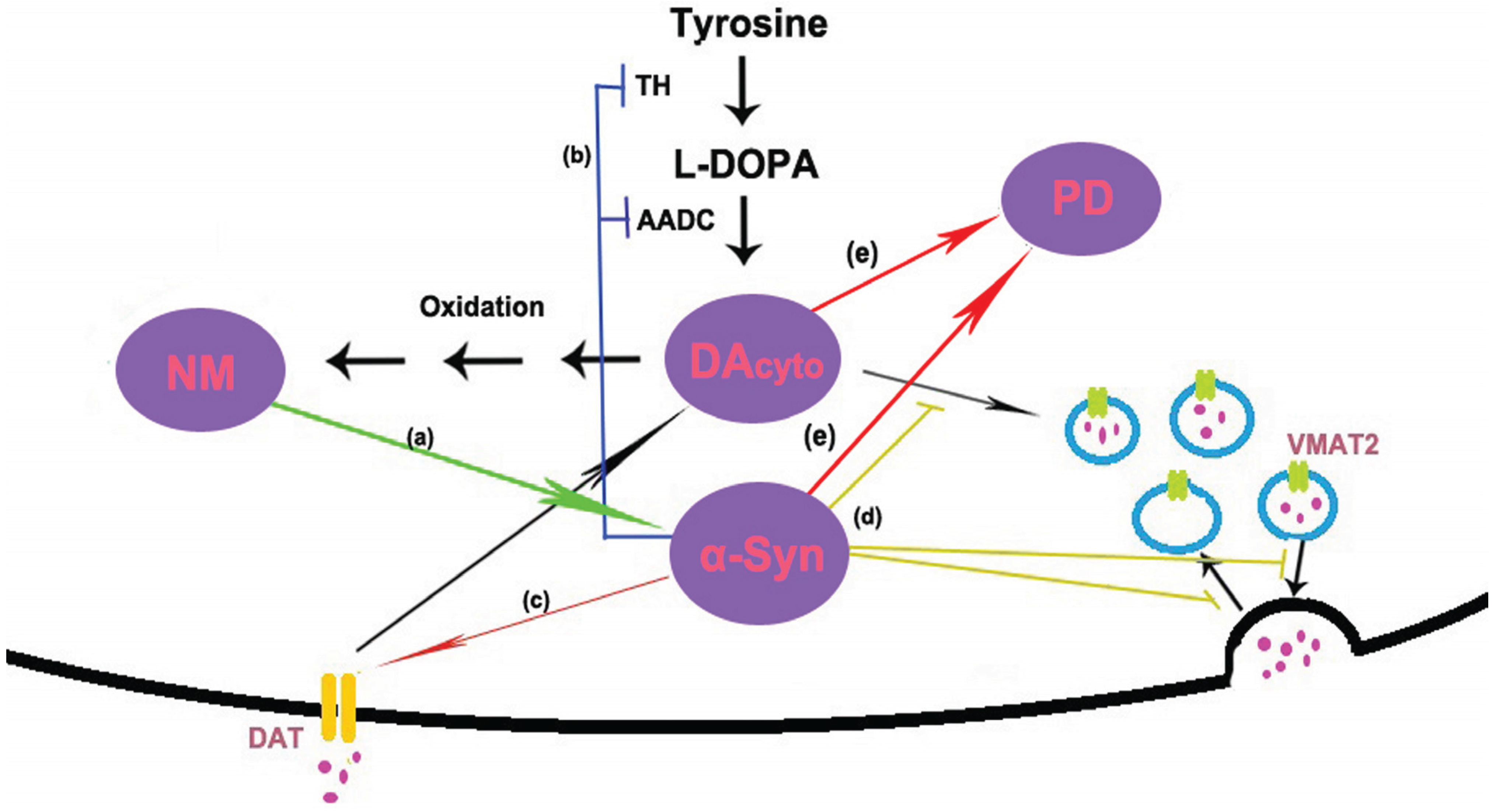
In conclusion, no matter what is the trigger, NM and α-syn are likely to form a vicious cycle of mutual promotion, eventually the untoward cycle results in the death of DA neurons in PD (see Figure 2). In sporadic PD, age-related accumulation of NM induces α-syn expression and aggregation. On the other hand, accumulated α-syn may promote the biosynthesis of NM by increasing the levels of cytosolic DA. In familial PD, the mutants of α-syn also can trigger the accumulation of NM and start this vicious cycle.
Figure 2.
Model of possible interactions between α-synuclein (α-syn) and neuromelanin (NM). (a) NM induces the expression and aggregation of α-syn; (b) dopamine (DA) is synthesized in the cytoplasm by the action of tyrosine hydroxylase (TH) and amino acid decarboxylase (AADC). α-Syn has been shown to regulate the activity of TH and AADC; (c) DA is reuptake via the dopamine transporter (DAT). Studies in cell culture systems have shown that α-syn is necessary for the trafficking of DAT to the cell surface. (d) Once synthesized, DA is immediately sequestered into vesicles by the vesicular monoamine transporter 2 (VMAT2). Several lines of evidence suggest that α-syn is involved in regulating storage, release and presynaptic vesicle cycling. (e) High levels of α-syn and cytosolic DA cause selective DA neuron death and eventually lead to PD.
Figure 2.
Model of possible interactions between α-synuclein (α-syn) and neuromelanin (NM). (a) NM induces the expression and aggregation of α-syn; (b) dopamine (DA) is synthesized in the cytoplasm by the action of tyrosine hydroxylase (TH) and amino acid decarboxylase (AADC). α-Syn has been shown to regulate the activity of TH and AADC; (c) DA is reuptake via the dopamine transporter (DAT). Studies in cell culture systems have shown that α-syn is necessary for the trafficking of DAT to the cell surface. (d) Once synthesized, DA is immediately sequestered into vesicles by the vesicular monoamine transporter 2 (VMAT2). Several lines of evidence suggest that α-syn is involved in regulating storage, release and presynaptic vesicle cycling. (e) High levels of α-syn and cytosolic DA cause selective DA neuron death and eventually lead to PD.
Acknowledgments
We are grateful to the National Natural Science Foundation for financial support (No. 81171298).
Author Contributions
Both Shengli Xu and Piu Chan contributed to reviewing the literature, and to the writing of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Sulzer, D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx 2004, 1, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 7, 229–238. [Google Scholar]
- Gonera, E.G.; Van’t Hof, M.; Berger, H.J.; van Weel, C.; Horstink, M.W. Symptoms and duration of the prodromal phase in Parkinson’s disease. Mov. Disord. 1997, 12, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, T.; Reichmann, H. Non-motor dysfunction in Parkinson’s disease. Park. Relat. Disord. 2007, 13, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Maass, A.; Reichmann, H. Sleep and non-motor symptoms in Parkinson’s disease. J. Neural Transm. 2013, 120, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Morris, H.R.; Robbins, T.W.; Goedert, M.; Hardy, J.; Ben-Shlomo, Y.; Bolam, P.; Burn, D.; Hindle, J.V.; Brooks, D. Parkinson’s disease—The debate on the clinical phenomenology, aetiology, pathology and pathogenesis. J. Parkinsons Dis. 2013, 3, 1–11. [Google Scholar] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Westbroek, W.; Gustafson, A.M.; Sidransky, E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 2011, 17, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Gouider-Khouja, N.; Larnaout, A.; Amouri, R.; Sfar, S.; Belal, S.; Ben Hamida, C.; Ben Hamida, M.; Hattori, N.; Mizuno, Y.; Hentati, F. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Park. Relat. Disord. 2003, 9, 247–251. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Skipper, L.; Wilkes, K.; Toft, M.; Baker, M.; Lincoln, S.; Hulihan, M.; Ross, O.A.; Hutton, M.; Aasly, J.; Farrer, M. Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am. J. Hum. Genet. 2004, 75, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Wavrant-De Vrieze, F.; Crook, R.; Boles, L.; Perez-Tur, J.; Hardy, J.; Johnson, W.G.; Steele, J.; Maraganore, D.; Gwinn, K.; et al. Low frequency of alpha-synuclein mutations in familial Parkinson’s disease. Ann. Neurol. 1998, 43, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein cause selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Park. Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Mosharov, E.; Talloczy, Z.; Zucca, F.A.; Simon, J.D.; Zecca, L. Neuronal pigmented autophagic vacuoles: Lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J. Neurochem. 2008, 106, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Q.; Xu, S.L.; Lu, D.H.; Yu, S.; Zhou, M.; Uéda, K.; Cui, Y.Q.; Zhang, B.Y.; Chan, P. Increased expression of α-synuclein in aged human brain associated with neuromelanin accumulation. J. Neural Transm. 2011, 118, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef] [PubMed]
- Kanda, S.; Bishop, J.F.; Eglitis, M.A.; Yang, Y.; Mouradian, M.M. Enhanced viability to oxidative stress by alpha-synuclein mutations and C-terminal truncation. Neuroscience 2000, 97, 279–284. [Google Scholar] [CrossRef]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances α-synuclein aggregation, secretion and toxicity. J. Biol. Chem. 2014, 289, 21856–21876. [Google Scholar]
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α-synuclein. J. Neurochem. 2014, 131, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant α-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Saykin, A.J. Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2013, 18, 145–175. [Google Scholar]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.Y.; Tang, Z.; Liu, C.W. α-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: Understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J. Biol. Chem. 2008, 283, 20288–20298. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Grillo-Bosch, D. In vitro preparation of prefibrillar intermediates of amyloid-beta and alpha-synuclein. Methods Mol. Biol. 2005, 299, 19–33. [Google Scholar] [PubMed]
- Cole, N.B.; Murphy, D.D.; Lebowitz, J.; Di Noto, L.; Levine, R.L.; Nussbaum, R.L. Metal-catalyzed oxidation of alpha-synuclein: Helping to define the relationship between oligomers, protofibrils, and filaments. J. Biol. Chem. 2005, 280, 9678–9690. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Fagerqvist, T.; Lindström, V.; Nordström, E.; Lord, A.; Tucker, S.M.; Su, X.; Sahlin, C.; Kasrayan, A.; Andersson, J.; Welander, H.; et al. Monoclonal antibodies selective for α-synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and α-synuclein transgenic mice with the disease-causing A30P mutation. J. Neurochem. 2013, 126, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, S.J. Characterization of cytoplasmic alpha-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J. Biol. Chem. 2002, 277, 48976–48983. [Google Scholar] [CrossRef] [PubMed]
- Lindström, V.; Fagerqvist, T.; Nordström, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lavedan, C. The synuclein family. Genome Res. 1998, 8, 871–880. [Google Scholar] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- George, J.M. The synucleins. Genome Biol. 2002. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Harper, J.D.; Williamson, R.E.; Lansbury, P.T. Accelerated oligomerization by Parkinson’s disease linked α-synuclein mutants. Ann. NY Acad. Sci. 2000, 920, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, P.T., Jr. Kinetic stabilization of the α-synuclein protofibril by a dopamine—α-Synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [PubMed]
- Jethva, P.N.; Kardani, J.R.; Roy, I. Modulation of α-synuclein aggregation by dopamine in the presence of MPTP and its metabolite. FEBS J. 2011, 278, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Jenco, J.M.; Rawlingson, A.; Daniels, B.; Morris, A.J. Regulation of phospholipase D2: Selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 1998, 37, 4901–4909. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [PubMed]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [PubMed]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. α-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Uéda, K.; Chan, P.; Yu, S. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell. 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar]
- Oueslati, A.; Ximerakis, M.; Vekrellis, K. Protein transmission, seeding and degradation: key steps for α-synuclein prion-like propagation. Exp. Neurobiol. 2014, 23, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Surgucheva, I.; Newell, K.L.; Burns, J.; Surguchov, A. New α- and γ-synuclein immunopathological lesions in human brain. Acta Neuropathol Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [PubMed]
- Takahashi, T.; Yamashita, H.; Nagano, Y.; Nakamura, T.; Ohmori, H.; Avraham, H.; Avraham, S.; Yasuda, M.; Matsumoto, M. Identification and characterization of a novel Pyk2/related adhesion focal tyrosine kinase-associated protein that inhibits alpha-synuclein phosphorylation. J. Biol. Chem. 2003, 278, 42225–42233. [Google Scholar] [CrossRef] [PubMed]
- Azeredo da Silveira, S.; Schneider, B.L.; Cifuentes-Diaz, C.; Sage, D.; Abbas-Terki, T.; Iwatsubo, T.; Unser, M.; Aebischer, P. Phosphorylation does not prompt, nor prevent, the formation of α-synuclein toxic species in a rat model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 872–887. [Google Scholar] [PubMed]
- McFarland, N.R.; Fan, Z.; Xu, K.; Schwarzschild, M.A.; Feany, M.B.; Hyman, B.T.; McLean, P.J. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2009, 68, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kato, T.; Arawaka, S. The role of Ser129 phosphorylation of α-synuclein in neurodegeneration of Parkinson’s disease: Areview of in vivo models. Rev. Neurosci. 2013, 24, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J. Neuropathol. Exp. Neurol. 2008, 67, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Mbefo, M.K.; Paleologou, K.E.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E3945–E3954. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Shani, V.; Bisharat, S.; Engelender, S. α-Synuclein ubiquitination and novel therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yahya, M.; Fauvet, B.; Herman-Bachinsky, Y.; Hejjaoui, M.; Bavikar, S.N.; Karthikeyan, S.V.; Ciechanover, A.; Lashuel, H.A.; Brik, A. Synthetic polyubiquitinated α-synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. USA 2013, 110, 17726–17731. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Garcia Reitböck, P.; Humby, T.; Lambourne, S.L.; O’Connell, M.; Ghetti, B.; Gossage, H.; Emson, P.C.; Wilkinson, L.S.; Goedert, M.; et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human α-synuclein (1–120): Implications for Lewy body disorders. J. Neurosci. 2006, 26, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. α-Synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Hashimoto, M.; Takeda, A.; Hsu, L.J.; Takenouchi, T.; Masliah, E. Role of cytochrome C as a stimulator of alpha-synuclein aggregation in Lewy body disease. J. Biol. Chem. 1999, 274, 28849–28852. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Högen, T.; Hillmer, A.S.; Bader, B.; Schmidt, F.; Kamp, F.; Kretzschmar, H.A.; Bötzel, K.; Giese, A. Generation of ferric iron links oxidative stress to α-synuclein oligomer formation. J. Park. Dis. 2011, 1, 205–216. [Google Scholar] [CrossRef]
- Kim, K.S.; Choi, S.Y.; Kwon, H.Y.; Won, M.H.; Kang, T.C.; Kang, J.H. Aggregation of α-synuclein induced by the Cu, Zn-superoxide dismutase and hydrogen peroxide system. Free Radic. Biol. Med. 2002, 32, 544–550. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhou, M.; Yu, S.; Cai, Y.; Zhang, A.; Uéda, K.; Chan, P. Oxidative stress induces nuclear translocation of C-terminus of alpha-synuclein in dopaminergic cells. Biochem. Biophys. Res. Commun. 2006, 342, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, S.; Mi, J.; Uéda, K.; Chan, P. Nuclear translocation of alpha-synuclein increases susceptibility of MES23.5 cells to oxidative stress. Brain Res. 2013, 1500, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Matsuzaki-Kobayashi, M.; Takeda, A.; Sugeno, N.; Kikuchi, A.; Furukawa, K.; Perry, G.; Smith, M.A.; Itoyama, Y. Alpha-synuclein facilitates the toxicity of oxidized catechol metabolites: Implications for selective neurodegeneration in Parkinson’s disease. FEBS Lett. 2006, 580, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Uéda, K.; Chan, P. α-Synuclein and dopamine metabolism. Mol. Neurobiol. 2005, 31, 243–254. [Google Scholar] [CrossRef]
- Schlüter, O.M.; Fornai, F.; Alessandrí, M.G.; Takamori, S.; Geppert, M.; Jahn, R.; Südhof, T.C. Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuroscience 2003, 118, 985–1002. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, S.L.; Chen, B. Dual effects of different concentrations of alpha-synuclein on the neurotoxicity of 6-hydroxydopamine in SH-SY5Y cells. Sheng Li Xue Bao 2009, 61, 324–330. [Google Scholar] [PubMed]
- Thomas, B.; Mandir, A.S.; West, N.; Liu, Y.; Andrabi, S.A.; Stirling, W.; Dawson, V.L.; Dawson, T.M.; Lee, M.K. Resistance to MPTP-neurotoxicity in α-synuclein knockout mice is complemented by human α-synuclein and associated with increased β-synuclein and Akt activation. PLoS ONE 2011, 6, e16706. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Duffy, P.E.; Tennyson, V.M. Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and locus coeruleus in Parkinson’s disease. J. Neuropthol. Exp. Neurol. 1965, 24, 398–414. [Google Scholar] [CrossRef]
- Fenichel, G.M.; Bazelon, M. Studies on neuromelanin. II. Melanin in the brainstems of infants and children. Neurology 1968, 18, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Moss, S.C.; Eisner, M. X-ray characterization of melanins–II. Pigment Cell Res. 1994, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Haavik, J. L-DOPA is a substrate for tyrosine hydroxylase. J. Neurochem. 1997, 69, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.R. The role of peroxidase in neuromelanin synthesis: A review. Physiol. Chem. Phys. Med. NMR 1997, 29, 15–22. [Google Scholar] [PubMed]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, R.J.; Lipman, Z.P.; Snyder, S.H. Selectivity of the parkinsonian neurotoxin MPTP: Toxic metabolite MPP+ binds to neuromelanin. Science 1986, 231, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, N.G.; Larsson, B.S.; Lydén-Sokolowski, A. Neuromelanin and its possible protective and destructive properties. Pigment Cell Res. 1987, 1, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Pietra, R.; Goj, C.; Mecacci, C.; Radice, D.; Sabbioni, E. Iron and other metals in neuromelanin, substantia nigra, and putamen of human brain. J. Neurochem. 1994, 62, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Fornstedt, B.; Brun, A.; Rosengren, E.; Carlsson, A. The apparent autoxidation rate of catechols in dopamine-rich regions of human brains increases with the degree of depigmentation of substantia nigra. J. Neural Transm. Park. Dis. Dement Sect. 1989, 1, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Wilczok, T.; Stepien, K.; Dzierzega-Lecznar, A.; Zajdel, A.; Wilczok, A. Model neuromelanins as antioxidative agents during lipid peroxidation. Neurotox. Res. 1999, 1, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Shamoto-Nagai, M.; Maruyama, W.; Akao, Y.; Osawa, T.; Tribl, F.; Gerlach, M.; Zucca, F.A.; Zecca, L.; Riederer, P.; Naoi, M. Neuromelanin inhibits enzymatic activity of 26S proteasome in human dopaminergic SH-SY5Y cells. J. Neural Transm. 2004, 111, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Zareba, M.; Bober, A.; Korytowski, W.; Zecca, L.; Sarna, T. The effect of a synthetic neruomelanin on yield of free hydroxyl radicals generated in model systems. Biochim. Biophys. Acta 1995, 1271, 343–348. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Fujikawa, K.; Zucca, F.A.; Zecca, L.; Ito, S. The structure of neuromelanin as studied by chemical degradative methods. J. Neurochem. 2003, 86, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Chemical degradation of melanins: Application to identification of dopamine-melanin. Pigment Cell Res. 1998, 11, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Ophof, A.; Broe, M.; Jensen, P.H.; Kettle, E.; Fedorow, H.; Cartwright, M.I.; Griffiths, F.M.; Shepherd, C.E.; Double, K.L. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain 2005, 128, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yan, Z.F.; Gao, J.H.; Sun, L.; Huang, X.Y.; Liu, Z.; Yu, S.Y.; Cao, C.J.; Zuo, L.J.; Chen, Z.J.; et al. Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 2014, 49, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Wilms, H.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Zecca, L.; Lucius, R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: Implications for Parkinson’s disease. FASEB J. 2003, 17, 500–502. [Google Scholar] [PubMed]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ostergren, A.; Annas, A.; Skog, K.; Lindquist, N.G.; Brittebo, E.B. Longterm retention of neurotoxic beta-carbolines in brain neuromelanin. J. Neural Transm. 2004, 111, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, N.G.; Larsson, B.S.; Lyden-Sokolowski, A. Autoradiography of [14C]paraquat or [14C]diquat in frogs and mice: Accumulation in neuromelanin. Neurosci. Lett. 1988, 93, 1–6. [Google Scholar] [CrossRef]
- Zucca, F.A.; Giaveri, G.; Gallorini, M.; Albertini, A.; Toscani, M.; Pezzoli, G.; Lucius, R.; Wilms, H.; Sulzer, D.; Ito, S.; et al. The neuromelanin of human substantia nigra: Physiological and pathogenic aspects. Pigment Cell Res. 2004, 17, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Mecacci, C.; Seraglia, R.; Parati, E. The chemical characterization of melanin contained in substantia nigra of human brain. Biochim. Biophys. Acta 1992, 1138, 6–10. [Google Scholar] [CrossRef]
- Zecca, L.; Costi, P.; Mecacci, C.; Ito, S.; Terreni, M.; Sonnino, S. Interaction of human substantia nigra neuromelanin with lipids and peptides. J. Neurochem. 2000, 74, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Fasano, M.; Bergamasco, B.; Lopiano, L.; Valente, G. Evidence for a glycidic-lipidic matrix in human neuromelanin, potentially responsible for the enhanced iron sequestering ability of substantia nigra. J. Neurochem. 1994, 62, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Barden, H. The histochemical relationship of neuromelanin and lipofuscin. J. Neuropathol. Exp. Neurol. 1969, 28, 419–441. [Google Scholar] [CrossRef] [PubMed]
- Swartz, H.M.; Sarna, T.; Zecca, L. Modulation by neuromelanin of the availability and reactivity of metal ions. Ann. Neurol. 1992, 32, S69–S75. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Bellei, C.; Giannelli, S.; Terreni, M.R.; Gallorini, M.; Rizzio, E.; Pezzoli, G.; Albertini, A.; Zecca, L. Neuromelanin and iron in human locus coeruleus and substantia nigra during aging: Consequences for neuronal vulnerability. J. Neural. Transm. 2006, 113, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Martin, M.E.; Beaumont, C.; Hauw, J.J.; Agid, Y.; Hirsch, E.C. Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2003, 86, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Gerlach, M.; Schunemann, V.; Trautwein, A.X.; Zecca, L.; Gallorini, M.; Youdim, M.B.; Riederer, P.; Ben-Shachar, D. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 2003, 66, 489–494. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Iron-melanin interaction and lipid peroxidation: Implications for Parkinson’s disease. J. Neurochem. 1991, 57, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Gründemann, J.; Schlaudraff, F.; Liss, B. UV-laser microdissection and mRNA expression analysis of individual neurons from postmortem Parkinson’s disease brains. Methods Mol. Biol. 2011, 755, 363–374. [Google Scholar] [PubMed]
- Matsuo, Y.; Kamitani, T. Parkinson’s disease-related protein, α-synuclein, in malignant melanoma. PLoS ONE 2010, 5, e10481. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Giraudo, S.; Coha, S.; Bergamasco, B.; Lopiano, L. Residual substantia nigra neuromelanin in Parkinson’s disease is cross-linked to alpha-synuclein. Neurochem. Int. 2003, 42, 603–606. [Google Scholar] [CrossRef]
- Fedorow, H.; Halliday, G.M.; Rickert, C.H.; Gerlach, M.; Riederer, P.; Double, K.L. Evidence for specific phases in the development of human neuromelanin. Neurobiol. Aging 2006, 27, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kordower, J.H. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Ben-Shachar, D.; Youdim, M.B.; Zecca, L.; Riederer, P.; Gerlach, M. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotoxicol. Teratol. 2002, 24, 621–628. [Google Scholar] [CrossRef]
- Shamoto-Nagai, M.; Maruyama, W.; Yi, H.; Akao, Y.; Tribl, F.; Gerlach, M.; Osawa, T.; Riederer, P.; Naoi, M. Neuromelanin induces oxidative stress in mitochondria through release of iron: Mechanism behind the inhibition of 26S proteasome. J. Neural Transm. 2006, 113, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Zhu, J.; Hwu, W.J.; Jankovic, J. The role of alpha-synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells. PLoS ONE 2012, 7, e45183. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Zucca, F.A.; Costi, P.; Tampellini, D.; Gatti, A.; Gerlach, M.; Riederer, P.; Fariello, R.G.; Ito, S.; Gallorini, M.; et al. The neuromelanin of human substantia nigra: Structure, synthesis and molecular behaviour. J. Neural Transm. Suppl. 2003, 65, 145–155. [Google Scholar] [PubMed]
- Wakamatsu, K.; Murase, T.; Zucca, F.A.; Zecca, L.; Ito, S. Biosynthetic pathway to neuromelanin and its aging process. Pigment Cell Melanoma Res. 2012, 25, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Tessari, I.; Bisaglia, M.; Valle, F.; Samorì, B.; Bergantino, E.; Mammi, S.; Bubacco, L. The reaction of alpha-synuclein with tyrosinase: Possible implications for Parkinson disease. J. Biol. Chem. 2008, 283, 16808–16817. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Gao, N.; Ye, Y.W.; Li, X.; Yu, S.; Yang, H.; Uéda, K.; Chan, P. α-Synuclein functions as a negative regulator for expression of tyrosine hudroxylase. Acta Neurol. Belg. 2011, 111, 130–135. [Google Scholar] [PubMed]
- Koo, H.J.; Yang, J.E.; Park, J.H.; Lee, D.; Paik, S.R. α-Synuclein-mediated defense against oxidative stress via modulation of glutathione peroxidase. Biochim. Biophys. Acta 2013, 1834, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Fornstedt, B.; Rosengren, E.; Carlsson, A. Occurrence and distribution of 5-S-cysteinyl derivatives of dopamine, dopa and dopac in the brains of eight mammalian species. Neuropharmacology 1986, 25, 451–454. [Google Scholar] [CrossRef]
- Liang, C.L.; Nelson, O.; Yazdani, U.; Pasbakhsh, P.; German, D.C. Inverse relationship between the contents of neuromelanin pigment and the vesicular monoamine transporter-2: Human midbrain dopamine neurons. J. Comp. Neurol. 2004, 473, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zuo, X.; Li, Y.; Zhang, C.; Zhou, M.; Zhang, Y.A.; Uéda, K.; Chan, P. Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci. Lett. 2004, 367, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Navarria, L.; Falarti, E.; Zaltieri, M.; Bono, F.; Collo, G.; Spillantini, M.G.; Missale, C.; Spano, P. Redistribution of DAT/α-synuclein complexes visualized by “in situ” proximity ligation assay in transgenic mice modelling early Parkinson’s disease. PLoS ONE 2011, 6, e27959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oaks, A.W.; Marsh-Armstrong, N.; Jones, J.M.; Credle, J.J.; Sidhu, A. Synucleins antagonize endoplasmic reticulum function to modulate dopamine transporter trafficking. PLoS ONE 2013, 8, e70872. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Barg, S.; Wiekop, P.; Lundberg, C.; Raymon, H.K.; Brundin, P. Effect of mutant α-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem. 2002, 277, 38884–38894. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Staal, R.G.; Bove, J.; Prou, D.; Hananiya, A.; Markov, D.; Poulsen, N.; Larsen, K.E.; Moore, C.M.; Troyer, M.D.; et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J. Neurosci. 2006, 26, 9304–9311. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.; Chow, A.M.; Cheng, X.R.; Tang, D.W.; Brown, I.R.; Kerman, K. Oxidative stress effect of dopamine on α-synuclein: Electroanalysis of solvent interactions. ACS Chem. Neurosci. 2012, 3, 569–574. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xu, S.; Chan, P. Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease. Biomolecules 2015, 5, 1122-1142. https://doi.org/10.3390/biom5021122
AMA Style
Xu S, Chan P. Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease. Biomolecules. 2015; 5(2):1122-1142. https://doi.org/10.3390/biom5021122
Chicago/Turabian StyleXu, Shengli, and Piu Chan. 2015. "Interaction between Neuromelanin and Alpha-Synuclein in Parkinson’s Disease" Biomolecules 5, no. 2: 1122-1142. https://doi.org/10.3390/biom5021122