
Hepatitis C, Innate Immunity and Alcohol: Friends or Foes?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ethanol and HCV Replication
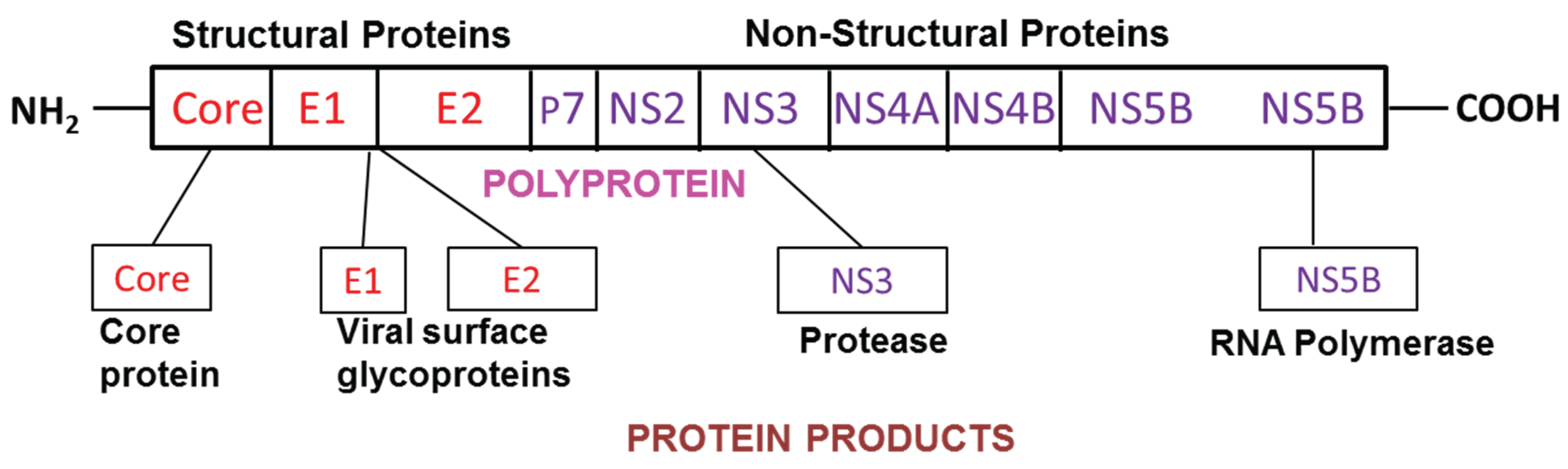
3. HCV-Impaired Innate Immunity
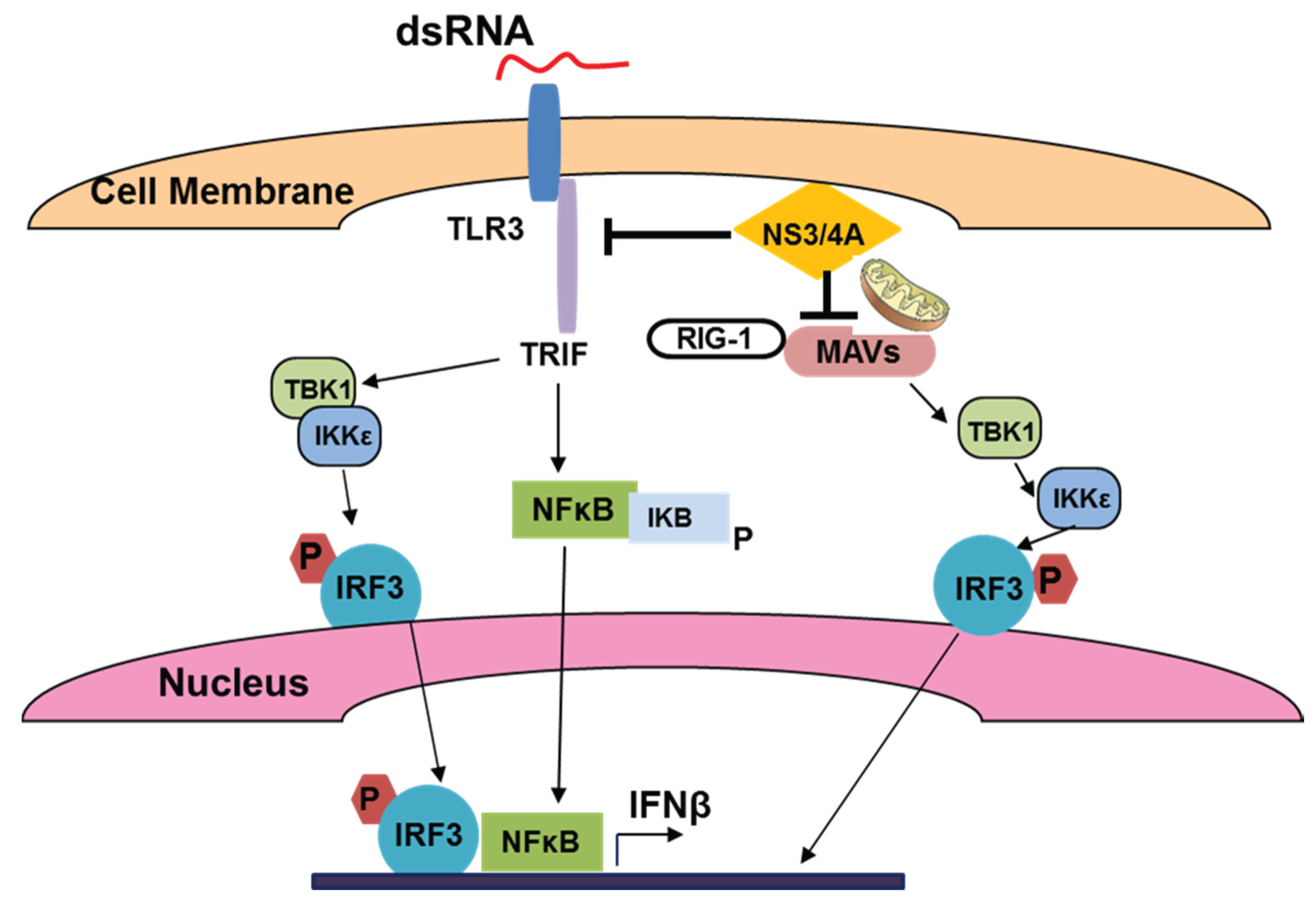
3.1. HCV and Viral dsRNA Recognition
3.2. HCV and ssRNA Recognition
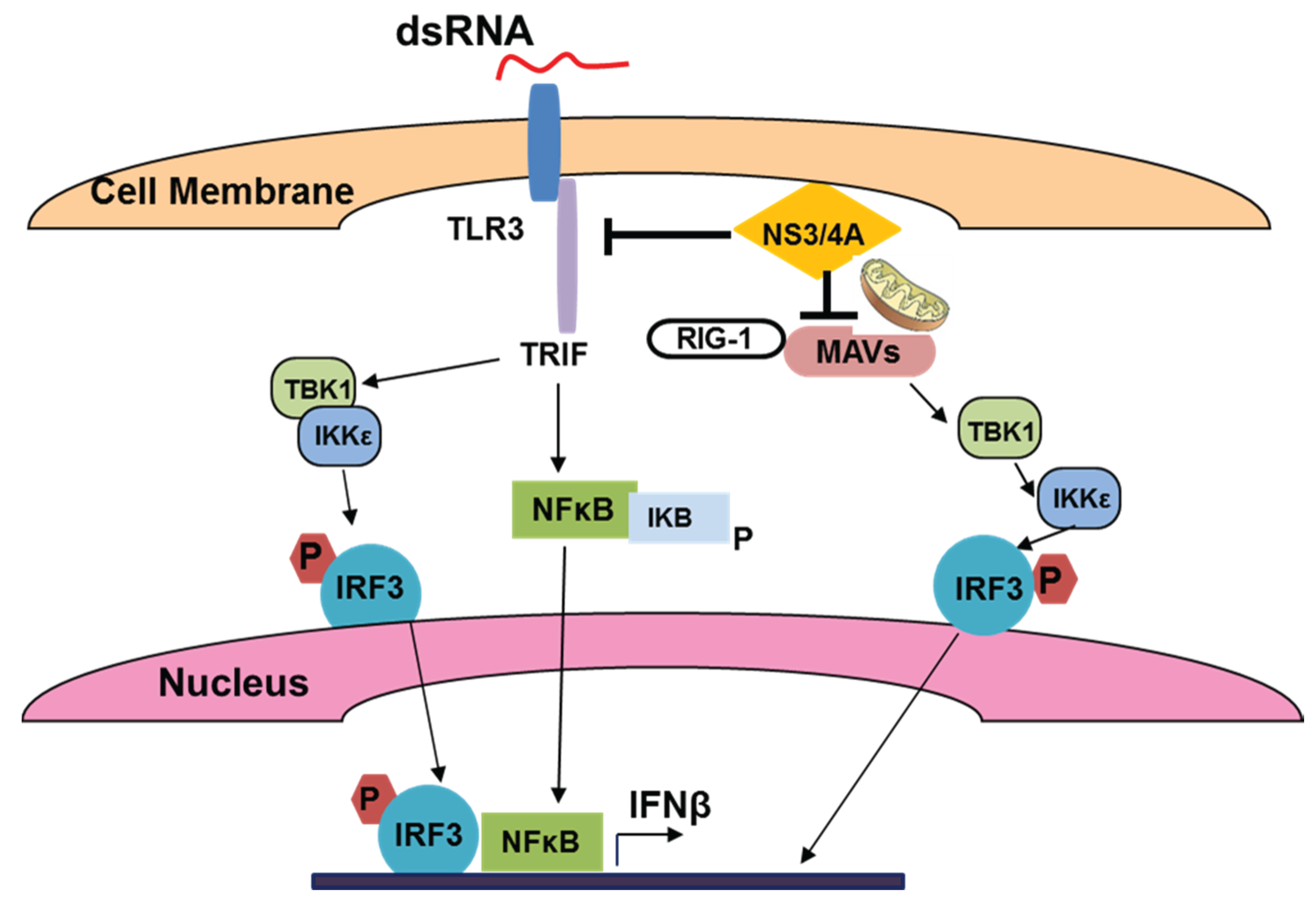
3.3. HCV, TLR Expression and Innate Immunity in Hepatocytes
3.4. HCV, TLR Expression and Innate Immunity in Immune Cells
3.5. Interferons and Their Role in Innate Immunity
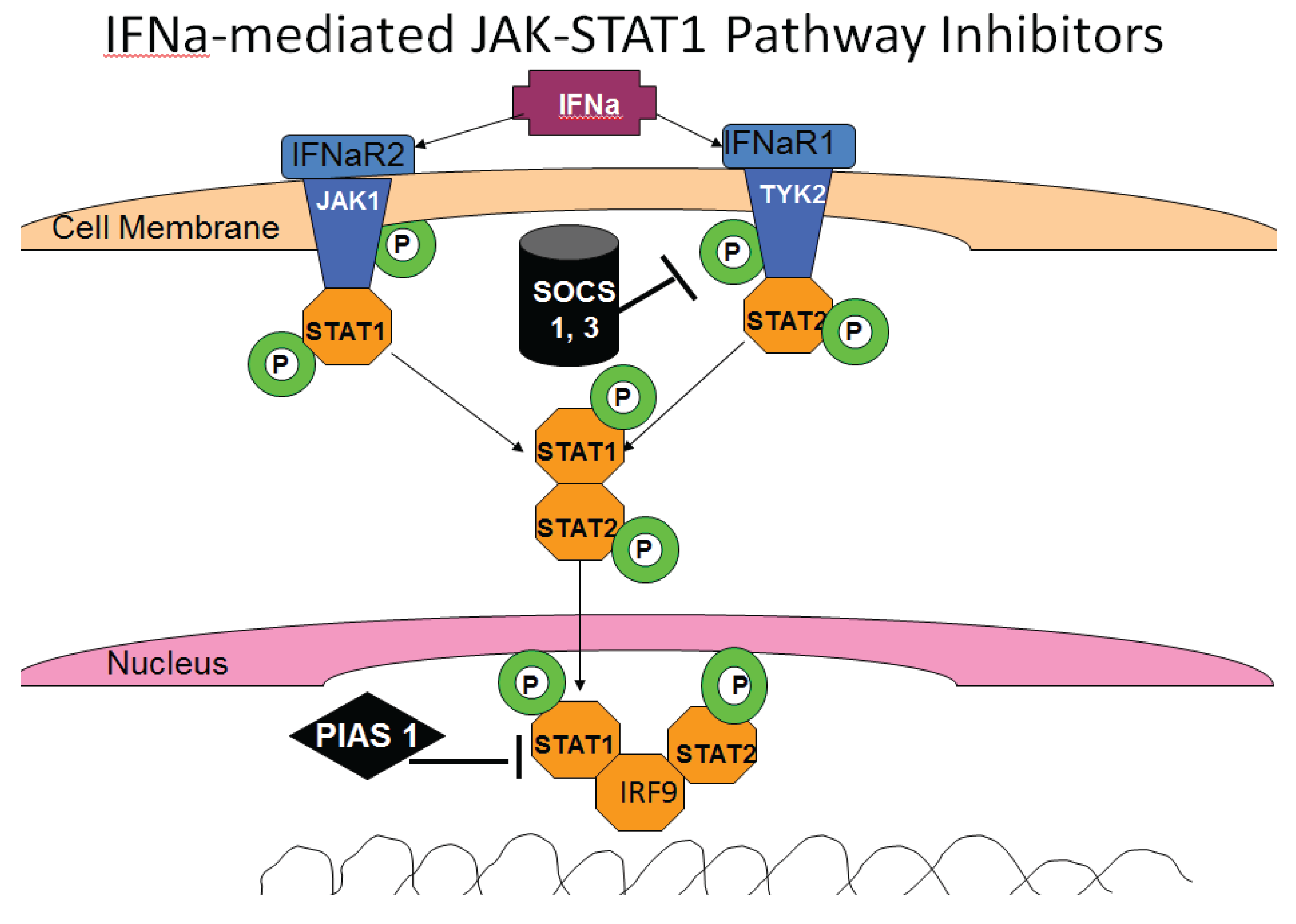
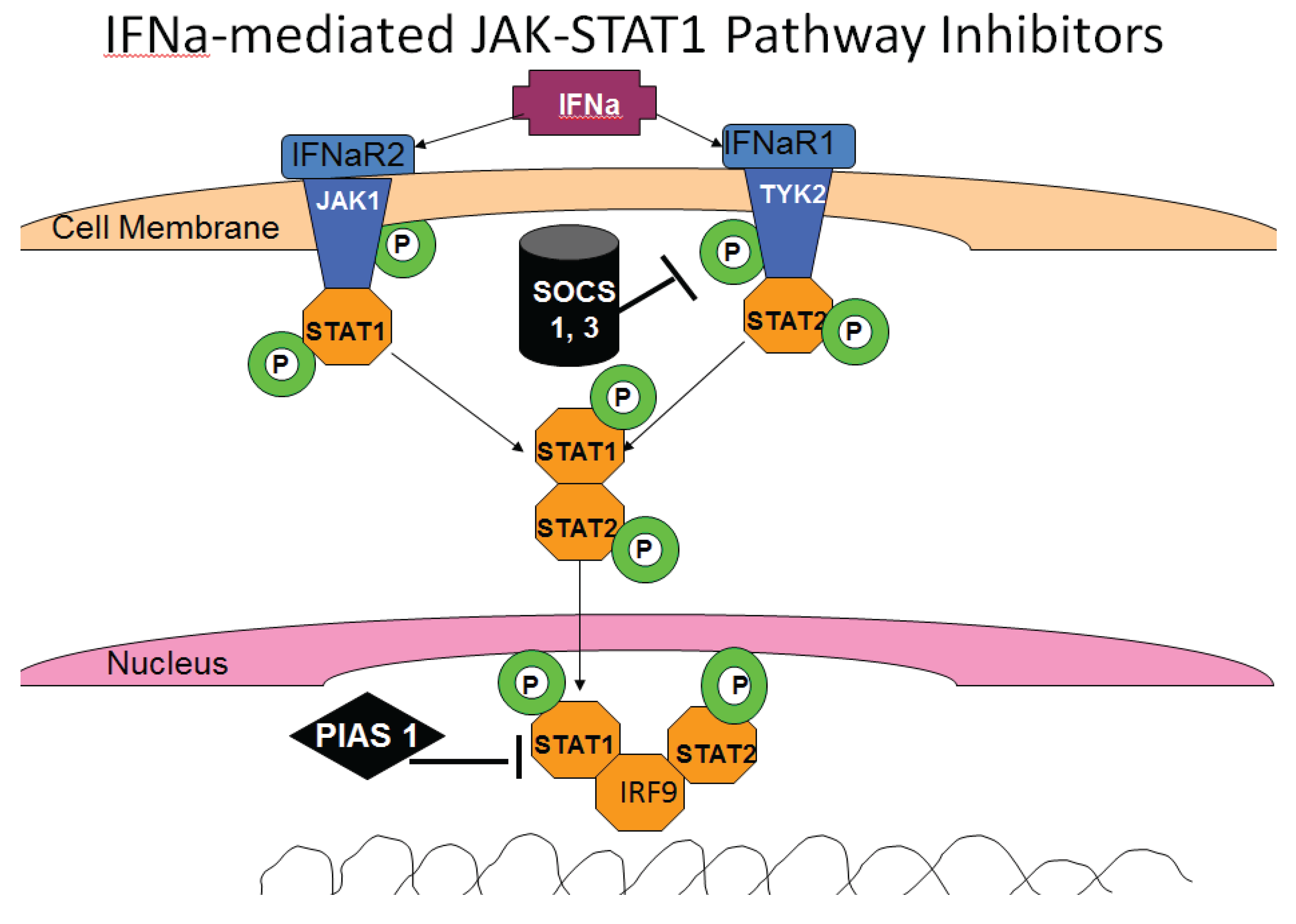
3.6. HCV and IFN Signaling
3.7. HCV and ISG Activation
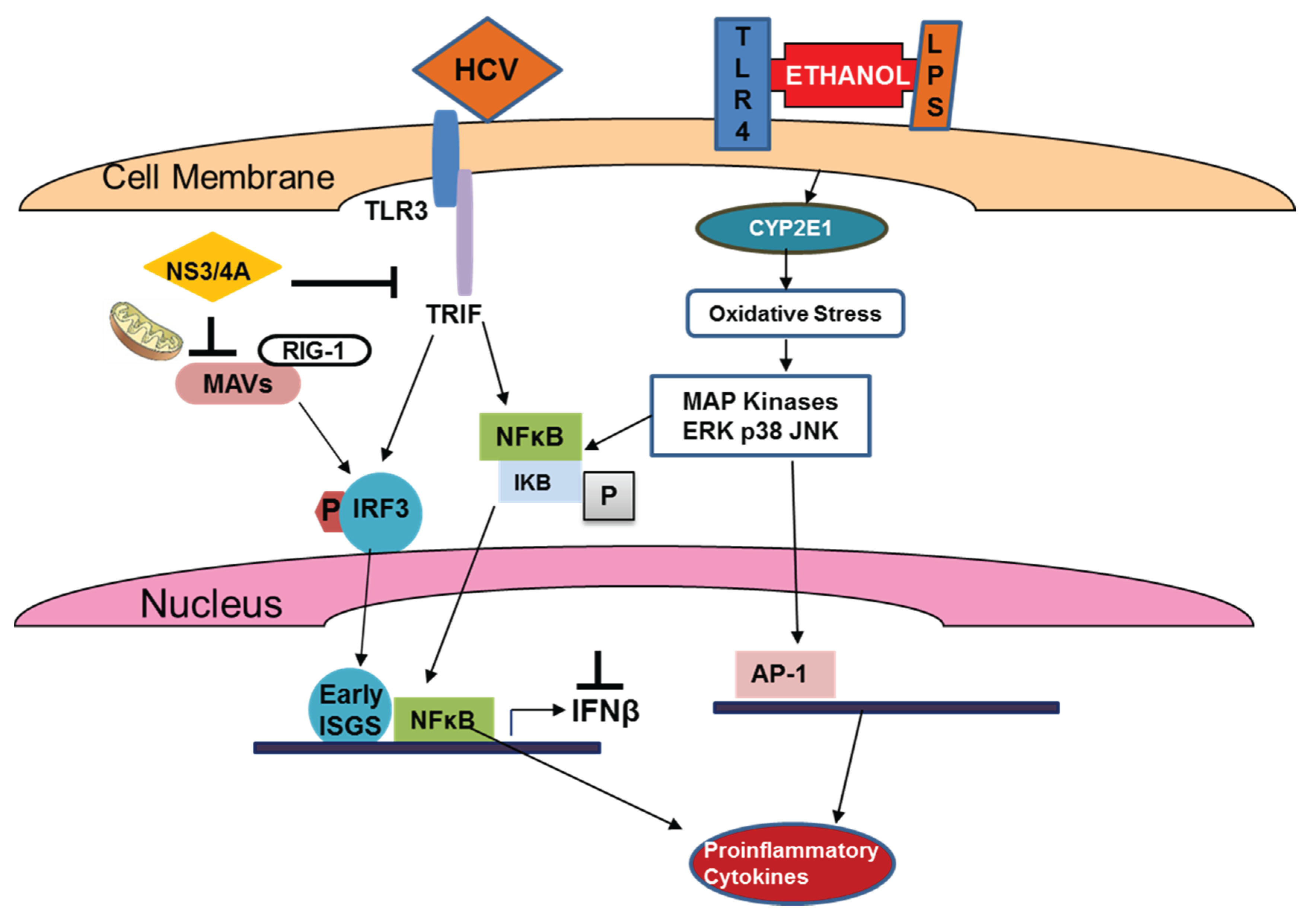
4. Ethanol-Regulated HCV Innate Immunity
4.1. Ethanol, IRFs and TLRs
4.2. Ethanol and TLR-Related Outcomes of HCV-Infection

4.3. Ethanol, HCV and IFN Signaling
4.4. Ethanol and Adaptive Immune Response
4.5. Treatment of HCV-Infection in Alcohol-Abused Patients
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ye, L.; Wang, S.; Wang, X.; Zhou, Y.; Li, J.; Persidsky, Y.; Ho, W. Alcohol impairs interferon signaling and enhances full cycle hepatitis C virus JFH-1 infection of human hepatocytes. Drug Alcohol Depend. 2010, 112, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.M.; Morgan, T.R. Liver disease in alcohol and hepatitis C. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Aloman, C.; Polyak, S.J.; Weinman, S.A.; Wands, J.; Zakhari, S. Hepatitis C infection and alcohol use: A dangerous mix for the liver and antiviral immunity. Alcohol. Clin. Exp. Res. 2006, 30, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; Yatsuhashi, H. Effect of alcohol consumption on the progression of hepatitis C virus infection and risk of hepatocellular carcinoma in Japanese patients. Alcohol Alcohol. 2000, 35, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; Bedossa, P.; Opolon, P.; The Obsvirc, Metavir, Clinivir, and Dosvirc Groups. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997, 349, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Yoshihara, H.; Suzuki, K.; Yamada, Y.; Tsujimura, T.; Kawano, K.; Abe, H. Efficacy of interferon therapy in patients with chronic hepatitis C: Comparison between non-drinkers and drinkers. Scand. J. Gastroenterol. 1994, 29, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Hepatitis C and alcohol. J. Clin. Gastroenterol. 2003, 36, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Otani, K.; Korenaga, M.; Beard, M.R.; Li, K.; Qian, T.; Showalter, L.A.; Singh, A.K.; Wang, T.; Weinman, S.A. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology 2005, 128, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Shoji, I.; Ogawa, W.; Kaneda, S.; Soga, T.; Jiang, D.P.; Ide, Y.H.; Hotta, H. Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J. Virol. 2011, 85, 8556–8568. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; White, R.L.; Krutik, V.M.; Wang, T.; Weinman, S.A.; Donohue, T.M., Jr. Proteasome activation by hepatitis C core protein is reversed by ethanol-induced oxidative stress. Gastroenterology 2008, 134, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.; Foont, J.; Wands, J.R. Hepatitis C virus and alcohol. Semin. Liver Dis. 2009, 29, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Anand, B.S. Mechanisms of synergy between alcohol and hepatitis C virus. J. Clin. Gastroenterol. 2007, 41, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Sherman, K.E.; Rouster, S.D.; Mendenhall, C.; Thee, D. Hepatitis crna quasispecies complexity in patients with alcoholic liver disease. Hepatology 1999, 30, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Takahashi, T.; Takahashi, S.; Watanabe, K.; Boku, S.; Matsui, S.; Arai, F.; Asakura, H. Difference in quasispecies of the hypervariable region 1 of hepatitis C virus between alcoholic and non-alcoholic patients. J. Gastroenterol. Hepatol. 2001, 16, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Seronello, S.; Montanez, J.; Presleigh, K.; Barlow, M.; Park, S.B.; Choi, J. Ethanol and reactive species increase basal sequence heterogeneity of hepatitis C virus and produce variants with reduced susceptibility to antivirals. PLOS ONE 2011, 6, e27436. [Google Scholar] [CrossRef] [PubMed]
- McCartney, E.M.; Semendric, L.; Helbig, K.J.; Hinze, S.; Jones, B.; Weinman, S.A.; Beard, M.R. Alcohol metabolism increases the replication of hepatitis C virus and attenuates the antiviral action of interferon. J. Infect. Dis. 2008, 198, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Seronello, S.; Ito, C.; Wakita, T.; Choi, J. Ethanol enhances hepatitis C virus replication through lipid metabolism and elevated NADH/NAD+. J. Biol. Chem. 2010, 285, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Seronello, S.; Sheikh, M.Y.; Choi, J. Redox regulation of hepatitis C in nonalcoholic and alcoholic liver. Free Radic. Biol. Med. 2007, 43, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Bukong, T.N.; Kodys, K.; Szabo, G. Alcohol facilitates HCV RNA replication via up-regulation of miR-122 expression and inhibition of cyclin G1 in human hepatoma cells. Alcohol. Clin. Exp. Res. 2013, 37, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Hou, W.; Kodys, K.; Szabo, G. Ethanol facilitates hepatitis C virus replication via up-regulation of GW182 and heat shock protein 90 in human hepatoma cells. Hepatology 2013, 57, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Randall, G.; Panis, M.; Cooper, J.D.; Tellinghuisen, T.L.; Sukhodolets, K.E.; Pfeffer, S.; Landthaler, M.; Landgraf, P.; Kan, S.; Lindenbach, B.D.; et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. USA 2007, 104, 12884–12889. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; French, S.W.; French, B.A.; Seitz, H.K.; Cohen, L.B.; Mueller, S.; Osna, N.A.; Kharbanda, K.K.; Seth, D.; Bautista, A.; et al. Alcoholic and non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2014, 97, 492–510. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Kharbanda, K.K.; Sun, Y.; Simpson, R.L.; Poluektova, L.E.; Ganesan, M.; Wisecarver, J.L.; Mercer, D.F. Ethanol affects hepatitis C pathogenesis: Humanized SCID Alb-upA mouse model. Biochem. Biophys. Res. Commun. 2014, 450, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Shuhart, M.C. Hepatitis C and alcohol: Interactions, outcomes, and implications. J. Clin. Gastroenterol. 2003, 36, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Pessione, F.; Degos, F.; Marcellin, P.; Duchatelle, V.; Njapoum, C.; Martinot-Peignoux, M.; Degott, C.; Valla, D.; Erlinger, S.; Rueff, B. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology 1998, 27, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.S.; Thornby, J. Alcohol has no effect on hepatitis C virus replication: A meta-analysis. Gut 2005, 54, 1468–1472. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Pietschmann, T. Efficient hepatitis C virus cell culture system: What a difference the host cell makes. Proc. Natl. Acad. Sci. USA 2005, 102, 9739–9740. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Lemon, S.M. Innate immune responses in hepatitis C virus infection. Semin. Immunopathol. 2013, 35, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Funami, K.; Matsumoto, M.; Oshiumi, H.; Akazawa, T.; Yamamoto, A.; Seya, T. The cytoplasmic “linker region” in toll-like receptor 3 controls receptor localization and signaling. Int. Immunol. 2004, 16, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. NY Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. TLR pathways and IFN-regulatory factors: To each its own. Eur. J. Immunol. 2007, 37, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dolganiuc, A.; Mandrekar, P. Pattern recognition receptors: A contemporary view on liver diseases. Hepatology 2006, 44, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dolganiuc, A. Hepatitis C and innate immunity: Recent advances. Clin. Liver Dis. 2008, 12, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. Ipc: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Qashqari, H.; Al-Mars, A.; Chaudhary, A.; Abuzenadah, A.; Damanhouri, G.; Alqahtani, M.; Mahmoud, M.; El Sayed Zaki, M.; Fatima, K.; Qadri, I. Understanding the molecular mechanism(s) of hepatitis C virus (HCV) induced interferon resistance. Infect. Genet. Evol. 2013, 19, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Sarasin-Filipowicz, M.; Oakeley, E.J.; Duong, F.H.; Christen, V.; Terracciano, L.; Filipowicz, W.; Heim, M.H. Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl. Acad. Sci. USA 2008, 105, 7034–7039. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ishikawa, T.; Okumura, A.; Yamauchi, T.; Sato, S.; Ayada, M.; Matsumoto, E.; Hotta, N.; Oohashi, T.; Fukuzawa, Y.; et al. Expression of toll-like receptors in chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2007, 22, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Gale, M., Jr.; Foy, E.M. Evasion of intracellular host defence by hepatitis C virus. Nature 2005, 436, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Waheed, Y.; Manzoor, S.; Bilal, M.; Ashraf, W.; Ali, M.; Ashraf, M. Interaction of hepatitis C virus proteins with pattern recognition receptors. Virol. J. 2012. [Google Scholar] [CrossRef]
- Moriyama, M.; Kato, N.; Otsuka, M.; Shao, R.X.; Taniguchi, H.; Kawabe, T.; Omata, M. Interferon-beta is activated by hepatitis C virus NS5B and inhibited by NS4A, NS4B, AND NS5A. Hepatol. Int. 2007, 1, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Kodys, K.; Szabo, G. Impaired expression and function of toll-like receptor 7 in hepatitis C virus infection in human hepatoma cells. Hepatology 2010, 51, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Yakushijin, T.; Kanto, T.; Inoue, M.; Oze, T.; Miyazaki, M.; Itose, I.; Miyatake, H.; Sakakibara, M.; Kuzushita, N.; Hiramatsu, N.; et al. Reduced expression and functional impairment of toll-like receptor 2 on dendritic cells in chronic hepatitis C virus infection. Hepatol. Res. 2006, 34, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dolganiuc, A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology 2005, 210, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Chang, S.; Kodys, K.; Mandrekar, P.; Bakis, G.; Cormier, M.; Szabo, G. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J. Immunol. 2006, 177, 6758–6768. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Zeisel, M.B.; Jilg, N.; Paranhos-Baccala, G.; Stoll-Keller, F.; Wakita, T.; Hafkemeyer, P.; Blum, H.E.; Barth, H.; Henneke, P.; et al. Toll-like receptor 2 senses hepatitis C virus core protein but not infectious viral particles. J. Innate Immun. 2009, 1, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Shiina, M.; Rehermann, B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology 2008, 47, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.E.; Mitchell, A.; Brownell, J.; Miklin, D.J.; Golden-Mason, L.; Polyak, S.J.; Gale, M.J., Jr.; Rosen, H.R. Hepatitis C virus core protein inhibits interferon production by a human plasmacytoid dendritic cell line and dysregulates interferon regulatory factor-7 and signal transducer and activator of transcription (STAT) 1 protein expression. PLOS ONE 2014, 9, e95627. [Google Scholar] [CrossRef] [PubMed]
- Broering, R.; Wu, J.; Meng, Z.; Hilgard, P.; Lu, M.; Trippler, M.; Szczeponek, A.; Gerken, G.; Schlaak, J.F. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J. Hepatol. 2008, 48, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Hosomura, N.; Kono, H.; Tsuchiya, M.; Ishii, K.; Ogiku, M.; Matsuda, M.; Fujii, H. HCV-related proteins activate kupffer cells isolated from human liver tissues. Dig. Dis. Sci. 2011, 56, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLOS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.F.; Sutaria, R.; Jo, J.; Barnes, A.; Blahova, M.; Meredith, L.W.; Cosset, F.L.; Curbishley, S.M.; Adams, D.H.; Bertoletti, A.; et al. Activated macrophages promote hepatitis C virus entry in a tumor necrosis factor-dependent manner. Hepatology 2014, 59, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Funami, K.; Aly, H.H.; Matsumoto, M.; Seya, T. Multi-step regulation of interferon induction by hepatitis C virus. Arch. Immunol. Ther. Exp. 2013, 61, 127–138. [Google Scholar] [CrossRef]
- Park, S.J.; Hahn, Y.S. Regulation of host innate immunity by hepatitis C virus: Crosstalk between hepatocyte and NK/DC. Rev. Infect. 2010, 1, 151–157. [Google Scholar] [PubMed]
- Rosen, H.R. Emerging concepts in immunity to hepatitis C virus infection. J. Clin. Investig. 2013, 123, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Kanda, T.; Imazeki, F.; Wu, S.; Nakamoto, S.; Tanaka, T.; Arai, M.; Fujiwara, K.; Saito, K.; Roger, T.; et al. Hepatitis C virus nonstructural 5A protein inhibits lipopolysaccharide-mediated apoptosis of hepatocytes by decreasing expression of toll-like receptor 4. J. Infect. Dis. 2011, 204, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.; Qureshi, S.A.; Kerr, I.M.; Darnell, J.E., Jr.; Stark, G.R. Role of STAT2 in the alpha interferon signaling pathway. Mol. Cell. Biol. 1995, 15, 1312–1317. [Google Scholar] [PubMed]
- Gupta, S.; Yan, H.; Wong, L.H.; Ralph, S.; Krolewski, J.; Schindler, C. The SH2 domains of STAT1 and STAT2 mediate multiple interactions in the transduction of IFN-alpha signals. EMBO J. 1996, 15, 1075–1084. [Google Scholar] [PubMed]
- Ramana, C.V.; Chatterjee-Kishore, M.; Nguyen, H.; Stark, G.R. Complex roles of STAT1 in regulating gene expression. Oncogene 2000, 19, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Kramer, O.H.; Knauer, S.K.; Greiner, G.; Jandt, E.; Reichardt, S.; Guhrs, K.H.; Stauber, R.H.; Bohmer, F.D.; Heinzel, T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009, 23, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Balagopal, A.; Thomas, D.L.; Thio, C.L. IL28B and the control of hepatitis C virus infection. Gastroenterology 2010, 139, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Kim, S.S.; Yeung, E.; Kamegaya, Y.; Blackard, J.T.; Kim, K.A.; Holtzman, M.J.; Chung, R.T. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J. Virol. 2006, 80, 9226–9235. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, N.J.; Bourke, N.M.; Ryan, E.J.; Binder, M.; Fanning, L.; Johnston, J.A.; Hegarty, J.E.; Long, A.; O’Farrelly, C. Hepatitis C virus targets the interferon-alpha JAK/STAT pathway by promoting proteasomal degradation in immune cells and hepatocytes. FEBS Lett. 2013, 587, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Kumthip, K.; Chusri, P.; Jilg, N.; Zhao, L.; Fusco, D.N.; Zhao, H.; Goto, K.; Cheng, D.; Schaefer, E.A.; Zhang, L.; et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J. Virol. 2012, 86, 8581–8591. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Ludwig, S.; Ehrhardt, C.; Albrecht, U.; Erhardt, A.; Schaper, F.; Heinrich, P.C.; Haussinger, D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003, 17, 488–490. [Google Scholar] [PubMed]
- Duong, F.H.; Christen, V.; Filipowicz, M.; Heim, M.H. S-adenosylmethionine and betaine correct hepatitis C virus induced inhibition of interferon signaling in vitro. Hepatology 2006, 43, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Duong, F.H.; Christen, V.; Lin, S.; Heim, M.H. Hepatitis C virus-induced up-regulation of protein phosphatase 2A inhibits histone modification and DNA damage repair. Hepatology 2010, 51, 741–751. [Google Scholar] [PubMed]
- Duong, F.H.; Filipowicz, M.; Tripodi, M.; la Monica, N.; Heim, M.H. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology 2004, 126, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.J.; Ni, L.; Zhang, Y.; Zhang, C.L.; Wu, X.Y.; Atia, A.N.; Thayer, P.; Moorman, J.P.; Yao, Z.Q. PD-1 negatively regulates interleukin-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C virus infection. Immunology 2011, 132, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 1999, 285, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.J., Jr.; Korth, M.J.; Tang, N.M.; Tan, S.L.; Hopkins, D.A.; Dever, T.E.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Dabo, S.; Meurs, E.F. DsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 2012, 4, 2598–2635. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidere, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012. [Google Scholar] [CrossRef]
- Jia, Y.; Song, T.; Wei, C.; Ni, C.; Zheng, Z.; Xu, Q.; Ma, H.; Li, L.; Zhang, Y.; He, X.; et al. Negative regulation of MAVS-mediated innate immune response by PSMA7. J. Immunol. 2009, 183, 4241–4248. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, N.; Dabo, S.; Akazawa, D.; Fukasawa, M.; Shinkai-Ouchi, F.; Hugon, J.; Wakita, T.; Meurs, E.F. Hepatitis C virus reveals a novel early control in acute immune response. PLOS Pathog. 2011, 7, e1002289. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H. Innate immunity and HCV. J. Hepatol. 2013, 58, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Heintges, T.; Wands, J.R. Hepatitis C virus: Epidemiology and transmission. Hepatology 1997, 26, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Nath, B.; Hritz, I.; Kodys, K.; Catalano, D.; Kurt-Jones, E.; Mandrekar, P.; Szabo, G. Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology 2011, 53, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Pruett, S.B.; Schwab, C.; Zheng, Q.; Fan, R. Suppression of innate immunity by acute ethanol administration: A global perspective and a new mechanism beginning with inhibition of signaling through TLR3. J. Immunol. 2004, 173, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- Pruett, S.B.; Zheng, Q.; Fan, R.; Matthews, K.; Schwab, C. Acute exposure to ethanol affects toll-like receptor signaling and subsequent responses: An overview of recent studies. Alcohol 2004, 33, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.S.; Suh, Y.G.; Yi, H.S.; Lee, Y.S.; Jeong, W.I. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating kupffer cells and stellate cells to produce interleukin-10 in mice. J. Hepatol. 2013, 58, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Bala, S.; Kodys, K.; Catalano, D.; Szabo, G. Inhibition of TLR8- and TLR4-induced type I IFN induction by alcohol is different from its effects on inflammatory cytokine production in monocytes. BMC Immunol. 2011. [Google Scholar] [CrossRef]
- Testro, A.G.; Visvanathan, K. Toll-like receptors and their role in gastrointestinal disease. J. Gastroenterol. Hepatol. 2009, 24, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Bala, S.; Catalano, D.; Kodys, K.; Szabo, G. The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes. J. Immunol. 2009, 183, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Machida, K. TLRs, alcohol, HCV, and tumorigenesis. Gastroenterol. Res. Pract. 2010. [Google Scholar] [CrossRef]
- Mueller, S.; Millonig, G.; Seitz, H.K. Alcoholic liver disease and hepatitis C: A frequently underestimated combination. World J. Gastroenterol. 2009, 15, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Tsukamoto, H.; Mkrtchyan, H.; Duan, L.; Dynnyk, A.; Liu, H.M.; Asahina, K.; Govindarajan, S.; Ray, R.; Ou, J.H.; et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc. Natl. Acad. Sci. USA 2009, 106, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [PubMed]
- Plumlee, C.R.; Lazaro, C.A.; Fausto, N.; Polyak, S.J. Effect of ethanol on innate antiviral pathways and HCV replication in human liver cells. Virol. J. 2005. [Google Scholar] [CrossRef]
- Nguyen, V.A.; Chen, J.; Hong, F.; Ishac, E.J.; Gao, B. Interferons activate the p42/44 mitogen-activated protein kinase and JAK-STAT (janus kinase-signal transducer and activator transcription factor) signalling pathways in hepatocytes: Differential regulation by acute ethanol via a protein kinase C-dependent mechanism. Biochem. J. 2000, 349, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Clemens, D.L.; Donohue, T.M., Jr. Ethanol metabolism alters interferon gamma signaling in recombinant HEPG2 cells. Hepatology 2005, 42, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr. CYP2E1-catalyzed alcohol metabolism: Role of oxidant generation in interferon signaling, antigen presentation and autophagy. Subcell. Biochem. 2013, 67, 177–197. [Google Scholar] [PubMed]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating micrornas in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat. Immunol. 2013, 14, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Encke, J.; Wands, J.R. Ethanol inhibition: The humoral and cellular immune response to hepatitis C virus NS5 protein after genetic immunization. Alcohol. Clin. Exp. Res. 2000, 24, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Encke, J.; zu Putlitz, J.; Geissler, M.; Wands, J.R. Genetic immunization generates cellular and humoral immune responses against the nonstructural proteins of the hepatitis C virus in a murine model. J. Immunol. 1998, 161, 4917–4923. [Google Scholar] [PubMed]
- Donohue, T.M., Jr.; Osna, N.A.; Trambly, C.S.; Whitaker, N.P.; Thomes, P.G.; Todero, S.L.; Davis, J.S. Early growth response-1 contributes to steatosis development after acute ethanol administration. Alcohol. Clin. Exp. Res. 2012, 36, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; White, R.L.; Thiele, G.M.; Donohue, T.M., Jr. Ethanol metabolism alters major histocompatibility complex class I-restricted antigen presentation in liver cells. Hepatology 2009, 49, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; White, R.L.; Todero, S.; McVicker, B.L.; Thiele, G.M.; Clemens, D.L.; Tuma, D.J.; Donohue, T.M., Jr. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology 2007, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.; McVicker, B.; Poluektova, L.; Ganesan, M.; Kharbanda, K. Mode of oral ethanol feeding affects liver oxidative stress levels and methylation status: Study on NS5A-transgenic mice. Int. J. Biochem. Res. Rev. 2014, 4, 344–357. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Harrington, P.R.; O’Rear, J.J.; Naeger, L.K. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology 2015, 61, 56–65. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osna, N.A.; Ganesan, M.; Kharbanda, K.K. Hepatitis C, Innate Immunity and Alcohol: Friends or Foes? Biomolecules 2015, 5, 76-94. https://doi.org/10.3390/biom5010076
Osna NA, Ganesan M, Kharbanda KK. Hepatitis C, Innate Immunity and Alcohol: Friends or Foes? Biomolecules. 2015; 5(1):76-94. https://doi.org/10.3390/biom5010076
Chicago/Turabian StyleOsna, Natalia A., Murali Ganesan, and Kusum K. Kharbanda. 2015. "Hepatitis C, Innate Immunity and Alcohol: Friends or Foes?" Biomolecules 5, no. 1: 76-94. https://doi.org/10.3390/biom5010076