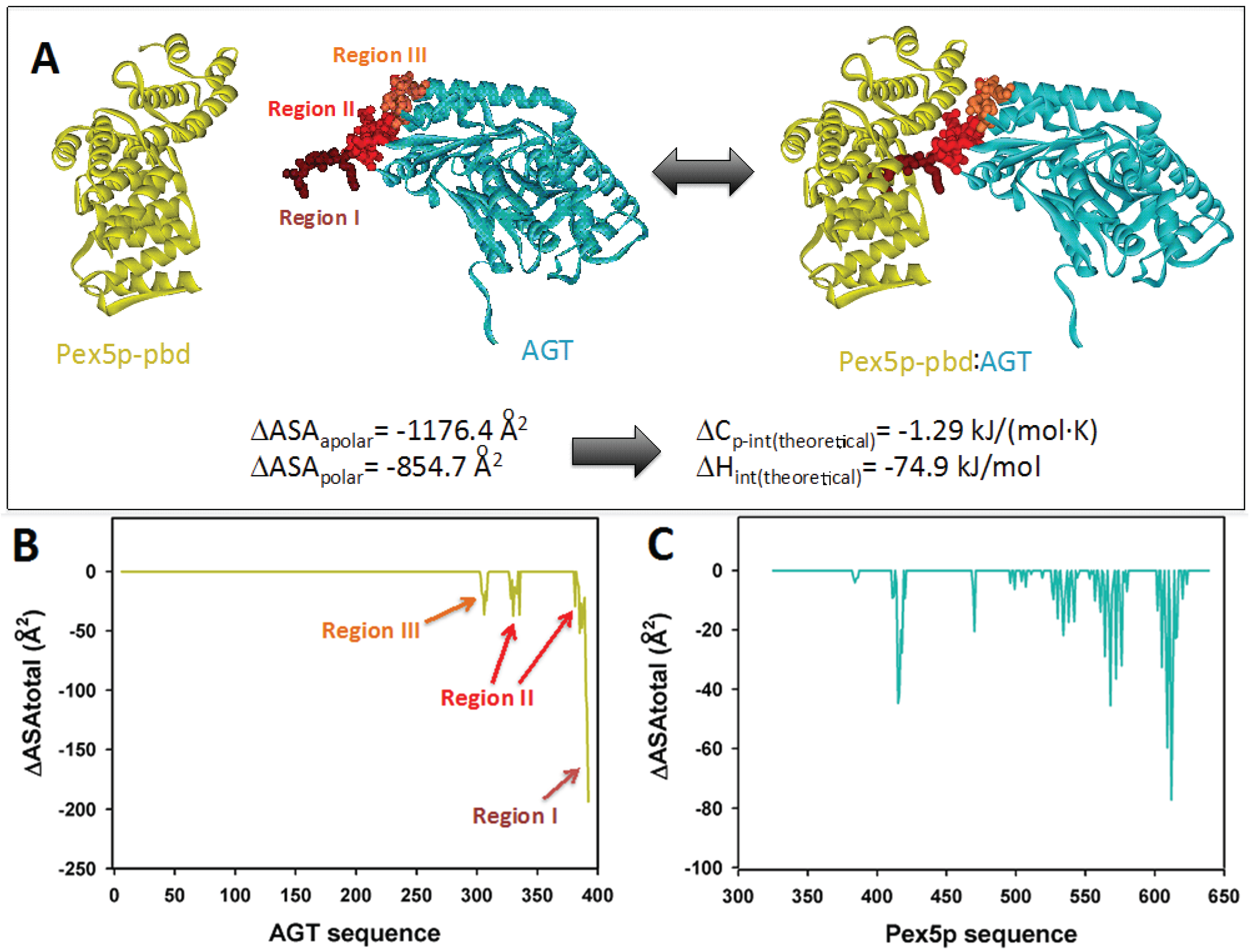
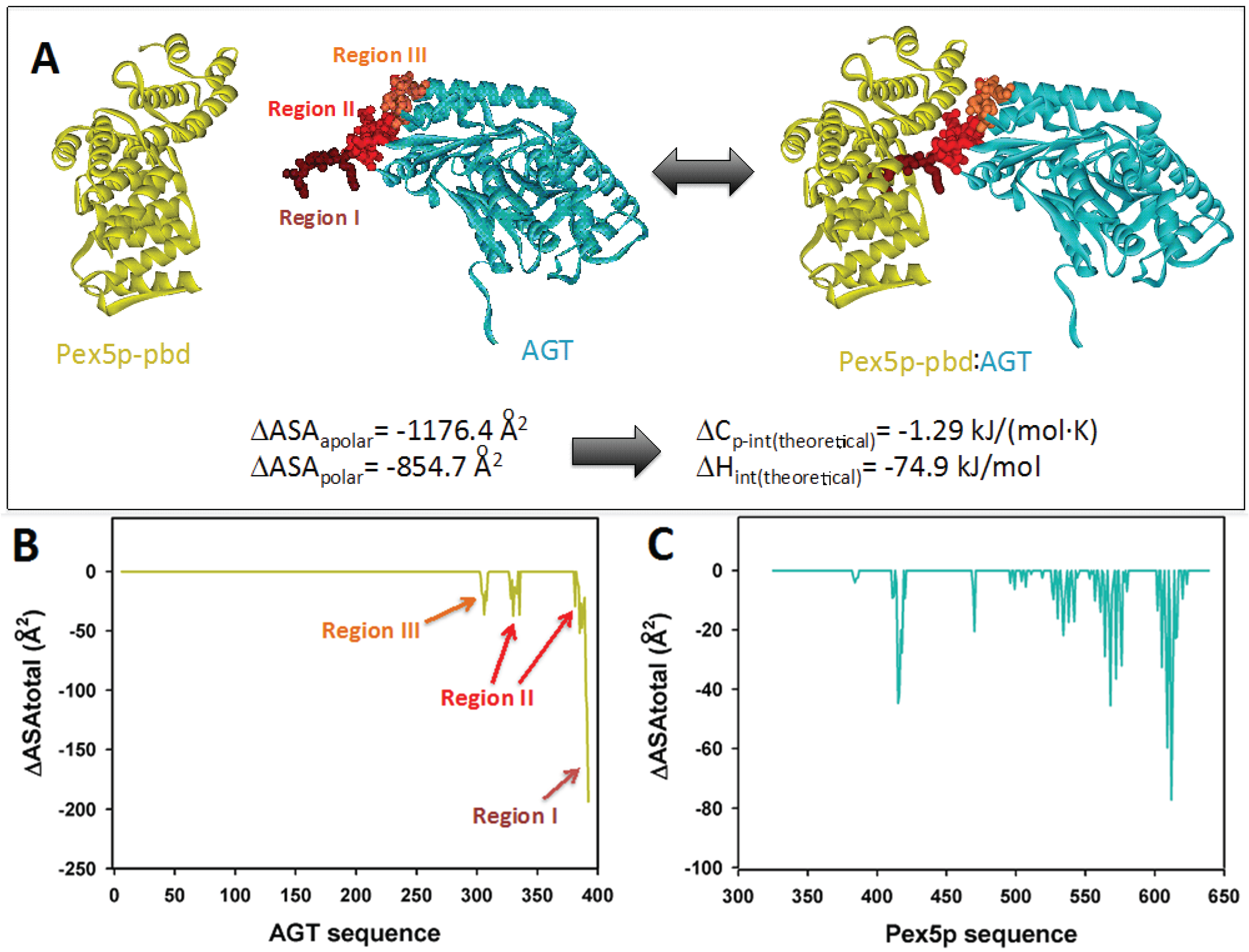
2.1. Modeling the Interaction of AGT and Pex5p-pbd
The interaction between AGT and the PTS1 binding domain of Pex5p (Pex5p-pbd) exclusively involves the C-terminal domain of AGT (residues 283–392) (PDB: 3R9A;
Figure 1A). While the overall conformation and activity of AGT are not significantly affected by the interaction, a conformational change is observed in the C-terminal bundle domain of Pex5p-pbd [
5]. The reported structure shows three topologically distinct regions of AGT interacting in the complex (
Figure 1A): (i) residues 389–392, which constitute the
minimal PTS1 sequence and interact with the central cavity of the TPR domains of Pex5p-pbd. This region forms an interface with the receptor of about 600 Å
2 [
5]; (ii) residues 381–388 (helix α13) and 327–330 (contained in the
ancillary targeting sequence or PTS1A; [
17]), and recently named the
extended PTS1 [
5]. Mutation of residues in this region show mild to moderate changes in affinity (about 2-fold lower affinity in Y330W and A328W, and about 5-fold in Y330A; [
5]); (iii) residues 303–307. Regions II and III form an additional surface of about 400 Å
2 [
5]. Changes in molecular surface upon complex formation at the residue level are shown in
Figure 1B,C.
Previous work with model peptides has shown that the recognition of PTS1 peptides by Pex5p-pbd resides to a large extent in the C-terminal tripeptide (positions −1 to −3; [
18]), even though residues upstream of this tripeptide may also contribute to the binding affinity by at least two orders of magnitude [
19]. However, these studies focused on the binding affinity (
i.e., binding free energy, Δ
G, which is related to the association constant
Ka by Δ
G = −R·T·ln
Ka), and did not investigate the possible effect on the enthalpic (Δ
H) and entropic (−
TΔ
S) contributions (Δ
G = Δ
H −
TΔ
S), which may suggest a link between changes in binding affinity with structural and dynamic differences in the complexes [
20,
21,
22]. Very recently, Fodor
et al. [
6] characterized the impact of specific residues of the human AGT PTS1 octapeptide sequence on the affinity for Pex5p-pbd by alanine scanning mutagenesis and isothermal titration calorimetry using full-length human AGT. Mutations at residues Q385, P388 and K389 (positions −8, −5 and −4 in AGT, respectively) had little effect on binding affinity (lower than 2-fold). Interestingly, the K389A mutant showed only 1.8-fold lower affinity but reduced the enthalpic penalization to binding by 10 kJ/mol. These findings are difficult to be solely explained by changes in molecular buried surface and alternatively may reflect significant structural reorganization in the complex and large enthalpy/entropy compensations. Mutations at the −3 and −2 positions have a much larger effect on binding affinity. The K390A
back-to-consensus mutation (-KKL to -AKL) increases the binding affinity for the Pex5p-pbd receptor, and this effect is mainly explained by a 4 kJ/mol decrease in the enthalpic penalization, likely due to the shrinkage of the binding site and optimization of short-range interactions reported by X-ray crystallography [
6]. Conversely, the K391A variant (that forms the very weak -KAL tripeptide) showed no detectable binding, which is in turn compensated in the double mutant K390A/K391A apparently due to structural optimization of specific interactions in the binding sites (further supported by the decrease in enthalpic penalization of 6 kJ/mol). The results from this elegant study suggest that the recognition of PTS1 cargo proteins by Pex5p is highly plastic from both structural and energetic viewpoints.
Figure 1.
(A) Molecular architecture of the AGT:Pex5-pbd complex (PDB code: 3R9A), indicating the three regions in AGT involved in the interaction surface. The amount of polar and apolar surface buried is determined from this complex and used to estimate theoretical values of intrinsic enthalpy and heat capacity of binding using Equations (1) and (2) (B) and (C) Changes in accessible surface at the residue level for AGT (B) and Pex5p-pbd (C) upon complex formation.
Figure 1.
(A) Molecular architecture of the AGT:Pex5-pbd complex (PDB code: 3R9A), indicating the three regions in AGT involved in the interaction surface. The amount of polar and apolar surface buried is determined from this complex and used to estimate theoretical values of intrinsic enthalpy and heat capacity of binding using Equations (1) and (2) (B) and (C) Changes in accessible surface at the residue level for AGT (B) and Pex5p-pbd (C) upon complex formation.
The availability of crystal structures for AGT: Pex5p-pbd complexes allow the estimation of some thermodynamic theoretical values corresponding to the intrinsic binding process (excluding conformational changes and ionization effects coupled to binding). Upon complex formation, total changes in polar and apolar surface (ΔASA
polar and ΔASA
apolar) are 855 Å
2 and 1176 Å
2 (
Figure 1A), therefore yielding theoretical values for the intrinsic Δ
Cp and Δ
H values upon binding of −1.29 kJ/(mol·K) and −74.9 kJ/mol based on Equations (1) and (2) (see also [
20]). These analyses set reference values to be contrasted with the calorimetric analyses performed in the present work, and to infer thermodynamic properties due to conformational changes. In the case that we use the complex AGT-WT-Pex5p-pbd to model different PTS1 sequences (
Table 1 and
Figure 2), we will obviously obtain a constant contribution from region III, and likely a variable contribution from regions I and II in the AGT (
Figure 1A and
Figure 2). Similarly, conformational changes in the C-terminal bundle of Pex5p-pbd are expected to remain constant across different modeled PTS1 sequences unless binding of AGT variants causes different long-range effects, while some differences in the TPR cavity are expected. To determine these intrinsic theoretical values for the different PTS1, we have modeled them using the available crystal structures, and the calculations of ΔASA
polar and ΔASA
apolar for the different PTS1 were carried out using the entire AGT protein and also for the 385–392 C-terminal octapeptide (compiled in
Table 2). It must be noted that the overall conformations of the three AGT-Pex5p-pbd crystallographic complexes available are very similar, and they mainly differ in the adaptation of Pex5p-pbd cavity to accommodate the reduced size of the mutated residues at positions −3 and −2 [
5,
6]. Thus, it is likely that the analyses of the pattern of interactions between AGT and Pex5p-pbd (
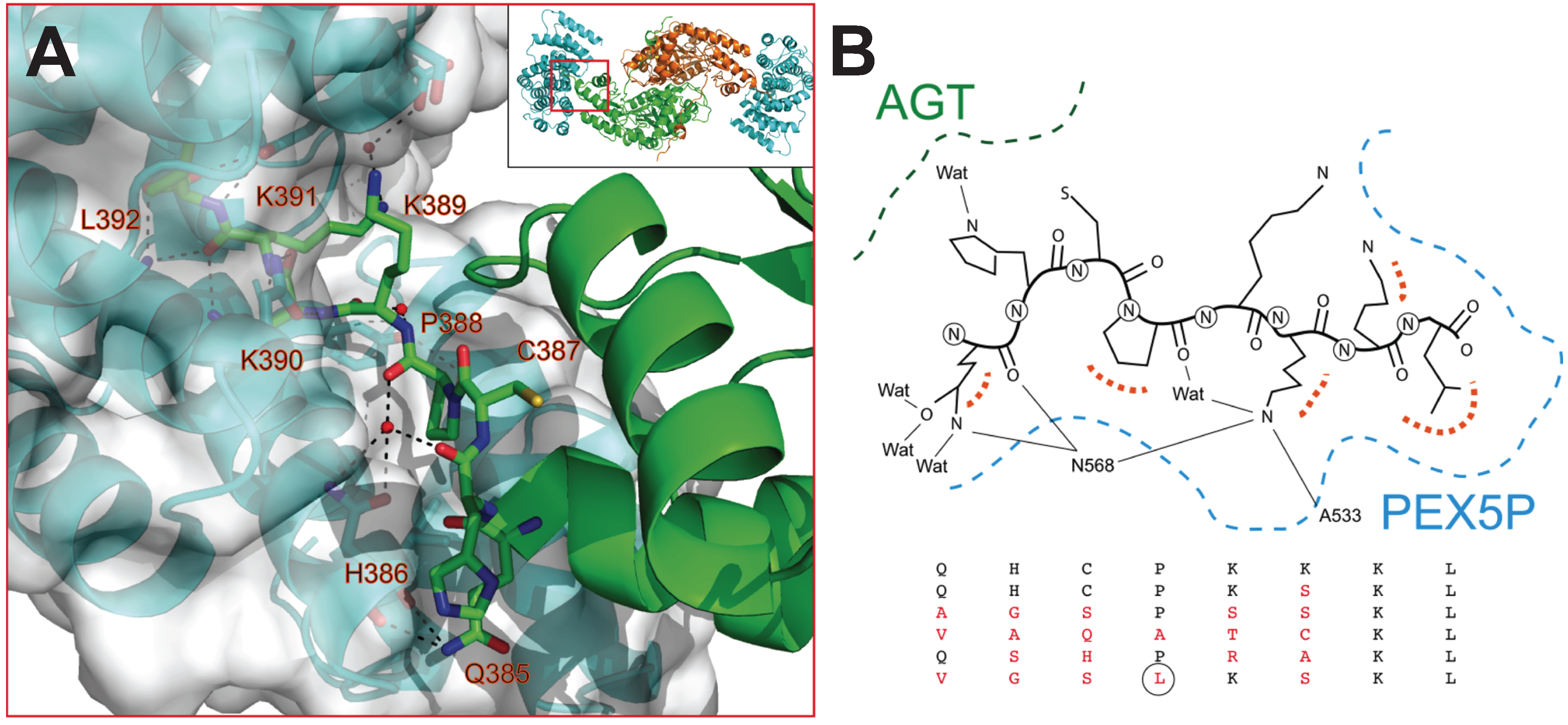
Figure 2B) can be used to explain our subsequent calorimetric analyses on the five AGT mutated versions to some extent. The C-terminal residues of AGT protrude into a central cavity of Pex5p-pbd forming both hydrogen bonds and hydrophobic interactions. Of them, residues K390, K391 and L392 are partially buried in a Pex5p-pbd central cavity while residues from Q385 to K389 are placed at the interface between AGT and the receptor. This suggests that mutations in the far C-terminal residues affect the structure of the receptor, while mutations in the rest of the tail may alter the relative position of the receptor with respect to AGT. To provide insights to this idea, we modeled the sequence of the five mutant proteins using as a template the structure of the AGT-Pex5p-pbd K390A mutant complex. Our models show that most mutations could be accommodated in the complex with minimal modifications of the original structure as none of them is totally buried in the complex interface. Conversely, the large and hydrophobic Leu side chain at 388 would require a structural reorganization (
Figure 2B).
Table 1.
PTS1 octapeptide sequences studied in this work. The protein sequence in which these octapeptides are naturally found, as well as their previously determined dissociation constant, are indicated [
7]. In red, we indicate the variations in sequence found when the peptides are compared with the PTS-1 sequence of human AGT. N.Det.: not determined.
Table 1.
PTS1 octapeptide sequences studied in this work. The protein sequence in which these octapeptides are naturally found, as well as their previously determined dissociation constant, are indicated [7]. In red, we indicate the variations in sequence found when the peptides are compared with the PTS-1 sequence of human AGT. N.Det.: not determined.
| PTS1 Sequence | Enzyme | Sequence | Kd (nM) |
|---|
| AGT | AGT | QHCPKKKL | 13490 |
| AGT-SKL | AGT + consensus tripeptide | QHCPKKL | N.Det. |
| BFE | L-Bifunctional enzyme | PKL | 1096 |
| HMG | 3-Hydroxy-3-methylglutaryl-CoA lyase | KL | 1995 |
| CRA | Carnitine acyl-transferase 1 | QPKL | 59 |
| ACO | Acyl-CoA oxidase 3 | KKL | 1.6 |
Figure 2.
(A) Structural details of the interaction between AGT and Pex5p-pbd (PDB: 3R9A) (B) Overview of the modeled structures of AGT with modified PTS1 sequences.
Figure 2.
(A) Structural details of the interaction between AGT and Pex5p-pbd (PDB: 3R9A) (B) Overview of the modeled structures of AGT with modified PTS1 sequences.
Table 2.
Changes in accessible surface (ΔASA) predicted for the AGT-Pex5p-pbd complexes studied here. Data correspond to the full length AGT with different PTS1 sequences.
Table 2.
Changes in accessible surface (ΔASA) predicted for the AGT-Pex5p-pbd complexes studied here. Data correspond to the full length AGT with different PTS1 sequences.
| PTS1 Sequence | ΔASA (for AGT) (Å2) | ΔASA (for Peptide) (Å2) |
|---|
| Apolar | Polar | Apolar | Polar |
|---|
| AGT | −1176 | −855 | −802 | −579 |
| AGT-SKL | −1075 | −812 | −718 | −599 |
| BFE | −1069 | −659 | −673 | −450 |
| HMG | −1100 | −703 | −715 | −493 |
| CRA | −1126 | −776 | −731 | −580 |
| ACO | −1168 | −713 | −783 | −519 |
2.2. PTS1 Sequence Dependence of the Interaction between AGT and Pex5p-pbd: Enthalpy-Entropy Compensations
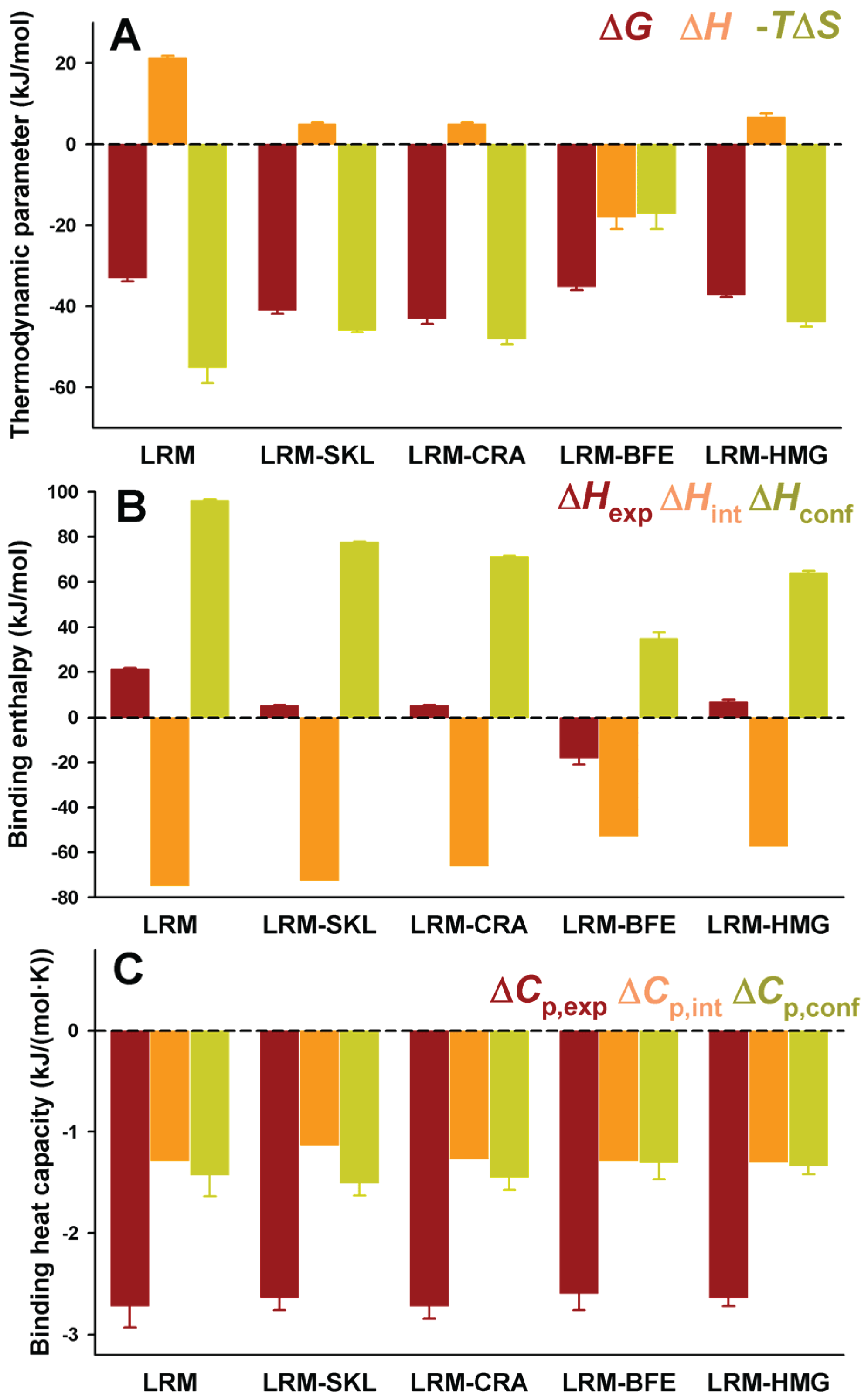
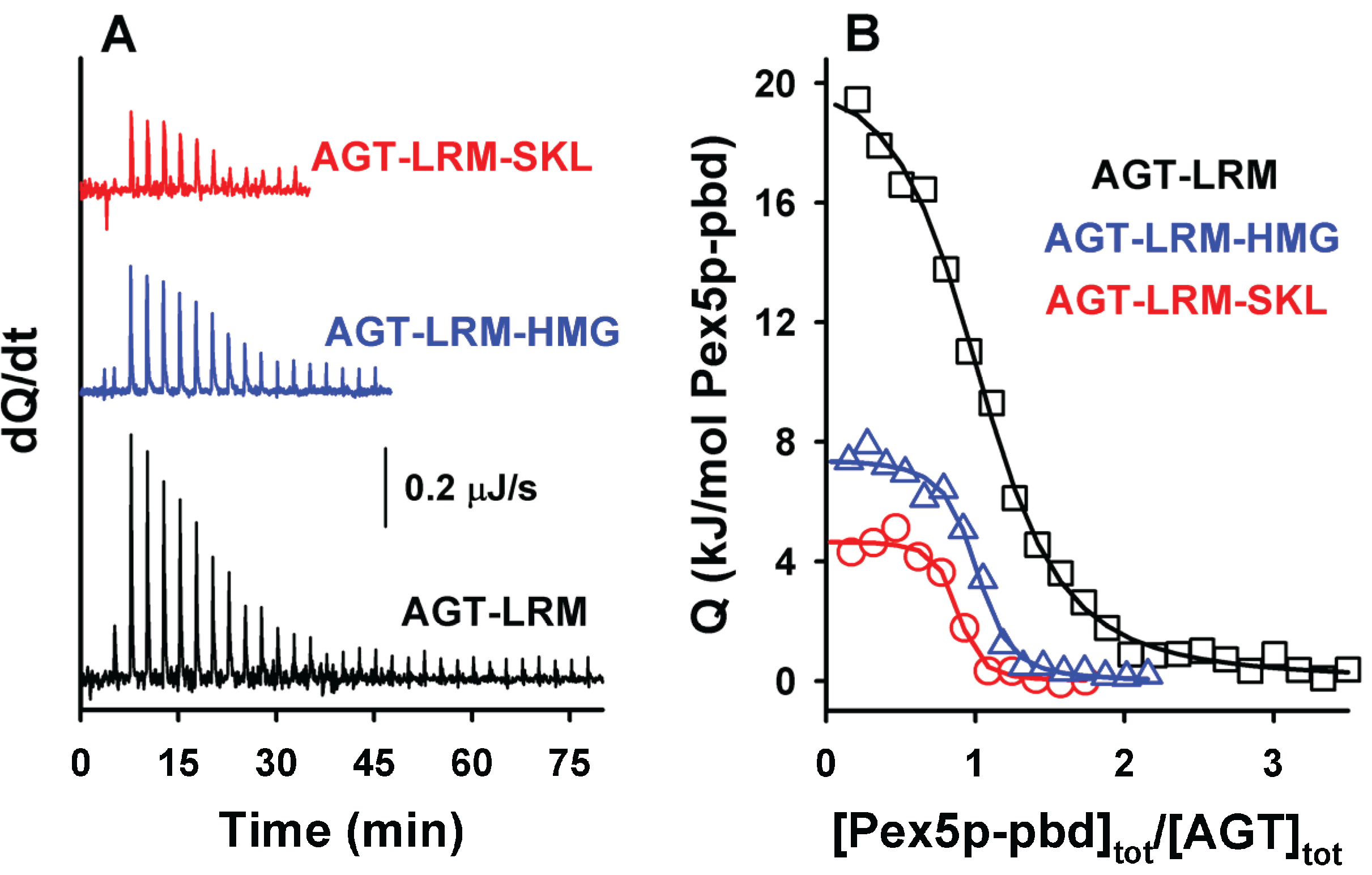
Binding of AGT to Pex5p-pbd show a stoichiometry of 1:1 (
Figure 3 and
Table 3, see also [
5,
12]). Interestingly, the wild-type (AGT) and disease-causing AGT-LRM mutant show virtually identical affinities as well as thermodynamic signatures (
Table 3). Introduction of the consensus -SKL tripeptide into either AGT or AGT-LRM variants produces a 50-fold increase in the affinity for the receptor, as well as a decrease in the enthalpic penalty of 16 kJ/mol. A similar behavior has been recently described for the -AKL tripeptide (mutation K390A of AGT; [
6]), and accordingly, may also reflect an optimization of the interactions between the PTS1 sequence of AGT and the Pex5-pbd in the consensus -SKL PTS1 sequence.
Figure 3.
ITC binding analysis of AGT-LRM and AGT-LRM-SKL proteins with Pex5p-pbd. Thermograms (A) and binding isotherms (B) at 25 °C for AGT-LRM, AGT-LRM-HMG and AGT-LRM-SKL; AGT-LRM (15 μM in subunit) was titrated using Pex5p-pbd 350 μM (29 injections of 1.2 μL) while AGT-LRM-HMG and AGT-LRM-SKL (20 μM in subunit) was titrated using Pex5p-pbd 250 μM (12–16 injections of 2–2.2 μL).
Figure 3.
ITC binding analysis of AGT-LRM and AGT-LRM-SKL proteins with Pex5p-pbd. Thermograms (A) and binding isotherms (B) at 25 °C for AGT-LRM, AGT-LRM-HMG and AGT-LRM-SKL; AGT-LRM (15 μM in subunit) was titrated using Pex5p-pbd 350 μM (29 injections of 1.2 μL) while AGT-LRM-HMG and AGT-LRM-SKL (20 μM in subunit) was titrated using Pex5p-pbd 250 μM (12–16 injections of 2–2.2 μL).
Table 3.
Thermodynamic binding parameters for the interaction of Pex5p-pbd with AGT and AGT-LRM variants at pH 7.4 and 25 °C. Data are mean ± s.d. of three independent titrations.
Table 3.
Thermodynamic binding parameters for the interaction of Pex5p-pbd with AGT and AGT-LRM variants at pH 7.4 and 25 °C. Data are mean ± s.d. of three independent titrations.
| AGT Variant | N | Kd | ΔG (kJ/mol) | ΔH (kJ/mol) | -TΔS (kJ/mol) | ΔCpa (kJ/mol·K) |
|---|
| AGT | 1.0 ± 0.1 | 1.4 ± 0.2 μM | −33.5 ± 0.4 | 21.7 ± 3.7 | −55.2 ± 1.2 | −2.5 ± 0.4 |
| AGT-LRM | 1.1 ± 0.1 | 1.6 ± 0.4 μM | −33.0 ± 0.8 | 21.3 ± 0.4 | −55.3 ± 3.6 | −2.7 ± 0.2 |
| AGT-SKL | 0.8 ± 0.1 | 44 ± 10 nM | −43.1 ± 0.9 | 6.3 ± 0.8 | −49.4 ± 1.7 | −2.7 ± 0.1 |
| AGT-LRM-SKL | 0.9 ± 0.1 | 65 ± 27 nM | −41.0 ± 0.7 | 5.0 ± 0.3 | −46.0 ± 0.5 | −2.6 ± 0.1 |
As noted above, residues upstream from the C-terminal tripeptide of the PTS1 sequence may strongly modulate binding affinity [
18,
19]. Therefore, we have introduced PTS1 octapeptides previously described by [
7] and displaying widely different binding affinities for Pex5p-pbd (
Table 1). As expected from this previous report, we observed an increase in binding affinity compared to the natural PTS1 sequence of AGT (
Figure 4A and
Table 4). On the AGT-LRM background, several PTS1 sequences (AGT-SKL, CRA and HMG,
Table 1) increased the affinity with a significant reduction of the enthalpic penalties (about 16 kJ/mol), while the BFE sequence (showing only a 2-fold increase in affinity) shows much favorable enthalpic contribution (about 35 kJ/mol) and much less favorable entropic contribution to binding (
Figure 4A). These changes in the enthalpic contributions to binding could be at least partly explained by structural optimization of the PTS1 sequence at the Pex5p-pbd binding site similar to that proposed for the AGT–SKL variant (
Table 3) and found for the K390A AGT mutant [
6]. We must also note that the changes in binding affinity found are relatively modest in most of the cases due to significant enthalpy/entropy compensations (
Figure 4A and [
6]), highlighting the complex sequence/energetic dependence of the molecular recognition of PTS1 cargo proteins by Pex5p-pbd, which goes beyond the –SKL consensus tripeptide.
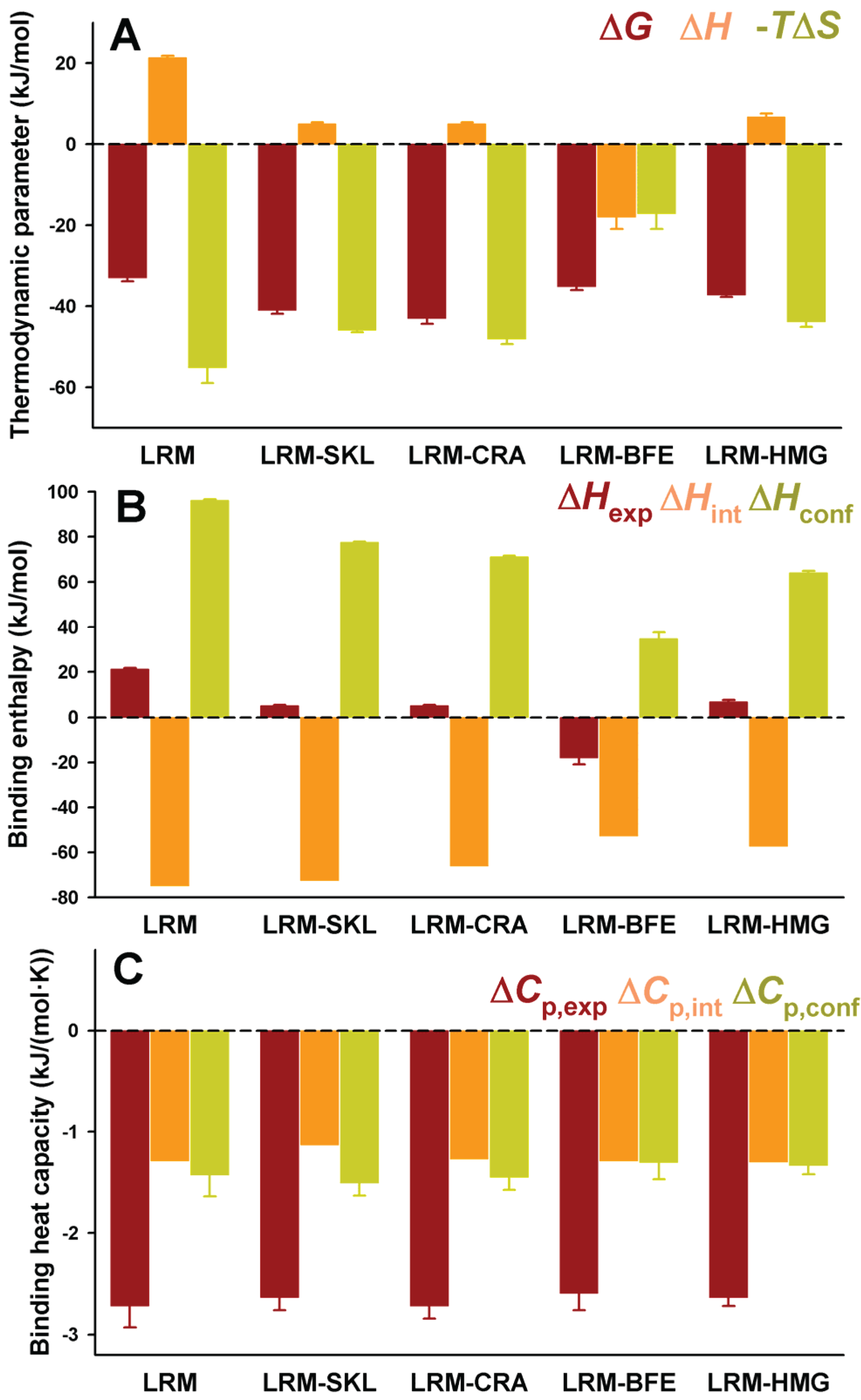
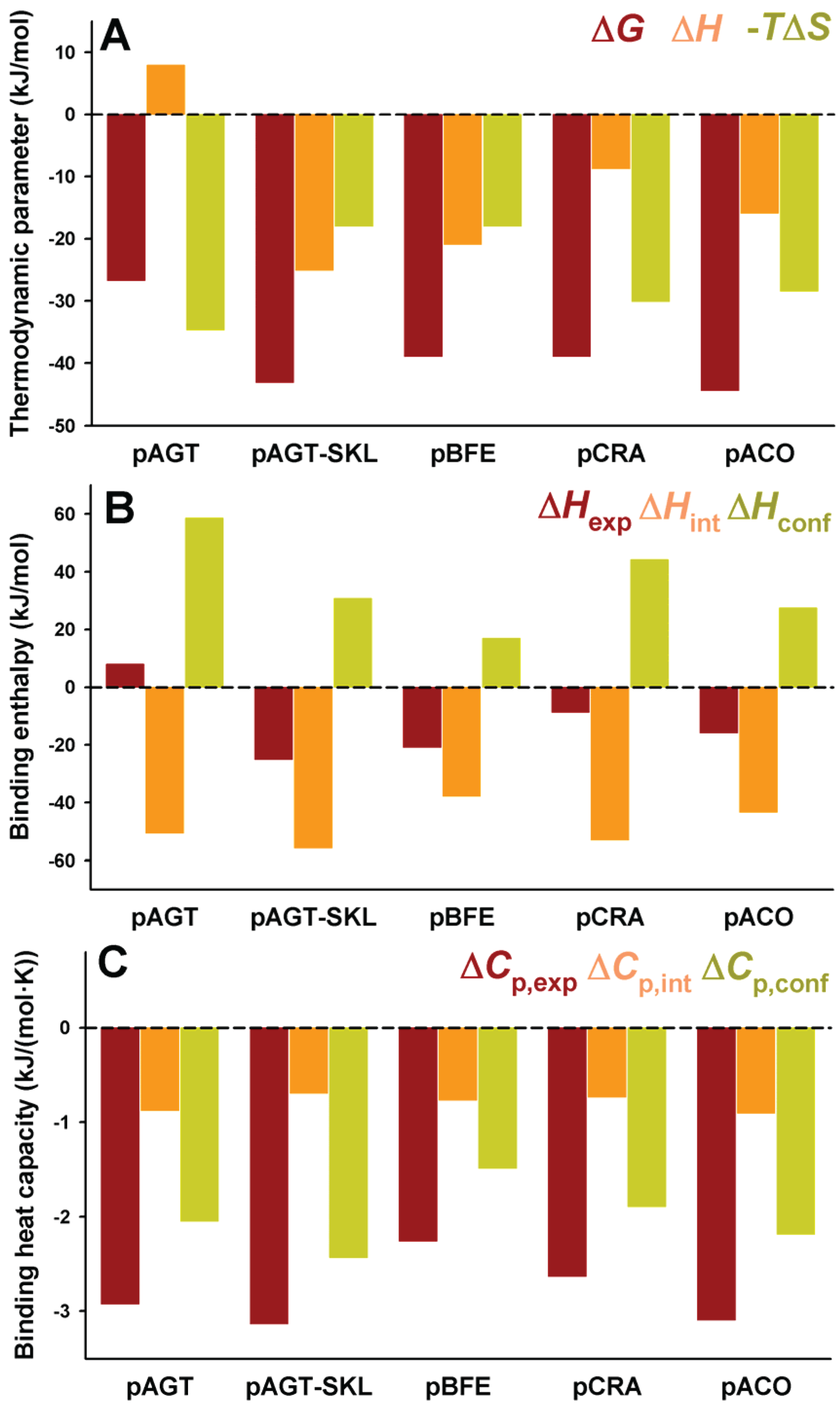
Figure 4.
Thermodynamic binding properties for the interaction of protein variants on the AGT-LRM background with Pex5p-pbd. (
A) Free energy (Δ
G), enthalpy (Δ
H) and entropy (−
TΔ
S) contributions to the binding reaction; (
B,
C) Thermodynamic dissection of binding enthalpies (
B) and heat capacity changes (Δ
Cp,
C) into their intrinsic contribution (estimated from polar and apolar ΔASA using the crystal structure for the complex;
Figure 1) and the contribution from conformational changes. In (
A,B), data are mean ± s.d. for three independent experiments at 25 °C.
Figure 4.
Thermodynamic binding properties for the interaction of protein variants on the AGT-LRM background with Pex5p-pbd. (
A) Free energy (Δ
G), enthalpy (Δ
H) and entropy (−
TΔ
S) contributions to the binding reaction; (
B,
C) Thermodynamic dissection of binding enthalpies (
B) and heat capacity changes (Δ
Cp,
C) into their intrinsic contribution (estimated from polar and apolar ΔASA using the crystal structure for the complex;
Figure 1) and the contribution from conformational changes. In (
A,B), data are mean ± s.d. for three independent experiments at 25 °C.
Table 4.
Thermodynamic binding parameters for the interaction of PTS1 peptides and AGT-LRM variants with Pex5p-pbd at pH 7.4 and 25 °C determined by ITC. N.Det. not determined.
Table 4.
Thermodynamic binding parameters for the interaction of PTS1 peptides and AGT-LRM variants with Pex5p-pbd at pH 7.4 and 25 °C determined by ITC. N.Det. not determined.
| AGT a | Kd | Peptide b | Kd |
|---|
| AGT-LRM | 1.6 ± 0.4 μM | pAGT | 19 μM |
| AGT-LRM-SKL | 65 ± 27 nM | pAGT-SKL | 29 nM |
| AGT-LRM-CRA | 40 ± 25 nM | pCRA | 157 nM |
| AGT-LRM-BFE | 0.74 ± 0.25 μM | pBFE | 145 nM |
| AGT-LRM-HMG | 0.28 ± 0.04 μM | pHMG | N.Det. |
| AGT-LRM-ACO | N.Det. | pACO | 14 nM |
The experimental enthalpic contributions to binding may be structurally rationalized using well-known structure/energetic relationships [
20]. Since the binding enthalpies are not dependent on the chosen buffer (giving similar values in HEPES and phosphate; data not shown [
23]), contributions from ionization events upon binding should be ruled out. Therefore, the experimental binding enthalpies can be dissected in two main terms: (i) Δ
Hint, arising from the surface area (polar and apolar) buried upon binding; this term can be calculated from the crystal structures (
Figure 1) and models provided in this work (
Figure 2 and
Table 2); (ii) Δ
Hconf, which arises from conformational changes in the cargo protein and Pex5p-pbd receptor upon binding. This term is evaluated from the difference between the experimental and intrinsic binding enthalpies. We must note that this energetic dissection is aimed to detect large differences between experimental and structure-derived theoretical values, thus allowing propose the existence of different conformational changes upon cargo binding, but not provide a detailed structural-energetic description of complex formation that could be a difficult task even if high-resolution crystal structures would be available for the complexes. These two terms (Δ
Hint and Δ
Hconf) have been estimated for the five variants on the AGT-LRM background experimentally studied (
Figure 4B). Δ
Hint seems to moderately vary among these five variants, from −57 kJ/mol to −75 kJ/mol (mean ± s.d. = −65 ± 10 kJ/mol), being always favorable to binding. However, Δ
Hconf largely varies among these AGT proteins, with values ranging from 35 to 96 kJ/mol (mean ± s.d. = 69 ± 22 kJ/mol), and is thus always unfavorable to binding. We must finally note that the contributions from Δ
Hconf and Δ
Hint show little or no correlation with the binding affinity, supporting complex enthalpy/entropy compensations ultimately leading to the given binding affinity for Pex5p-pbd-AGT complex formation.
A similar dissection has been performed for the binding heat capacities, for which the experimental values are virtually identical for the five AGT-LRM variants studied (
Figure 4C). In this case, both contributions are of the same sign and contribute similarly to the experimental value (mean ± s.d. of −1.26 ± 0.07 and −1.40 ± 0.08 kJ/mol, for the intrinsic and conformational components, respectively), and thus, very small changes in each contribution among the AGT proteins studied. This likely reflects the different contributions of polar and apolar surface burial to the binding enthalpies and heat capacities (see Equations (1) and (2)).
2.3. Interaction between PTS1 Nonapeptides and Pex5p-pbd Provide Insight into the Contribution from Ancillary Regions to Binding Energetics
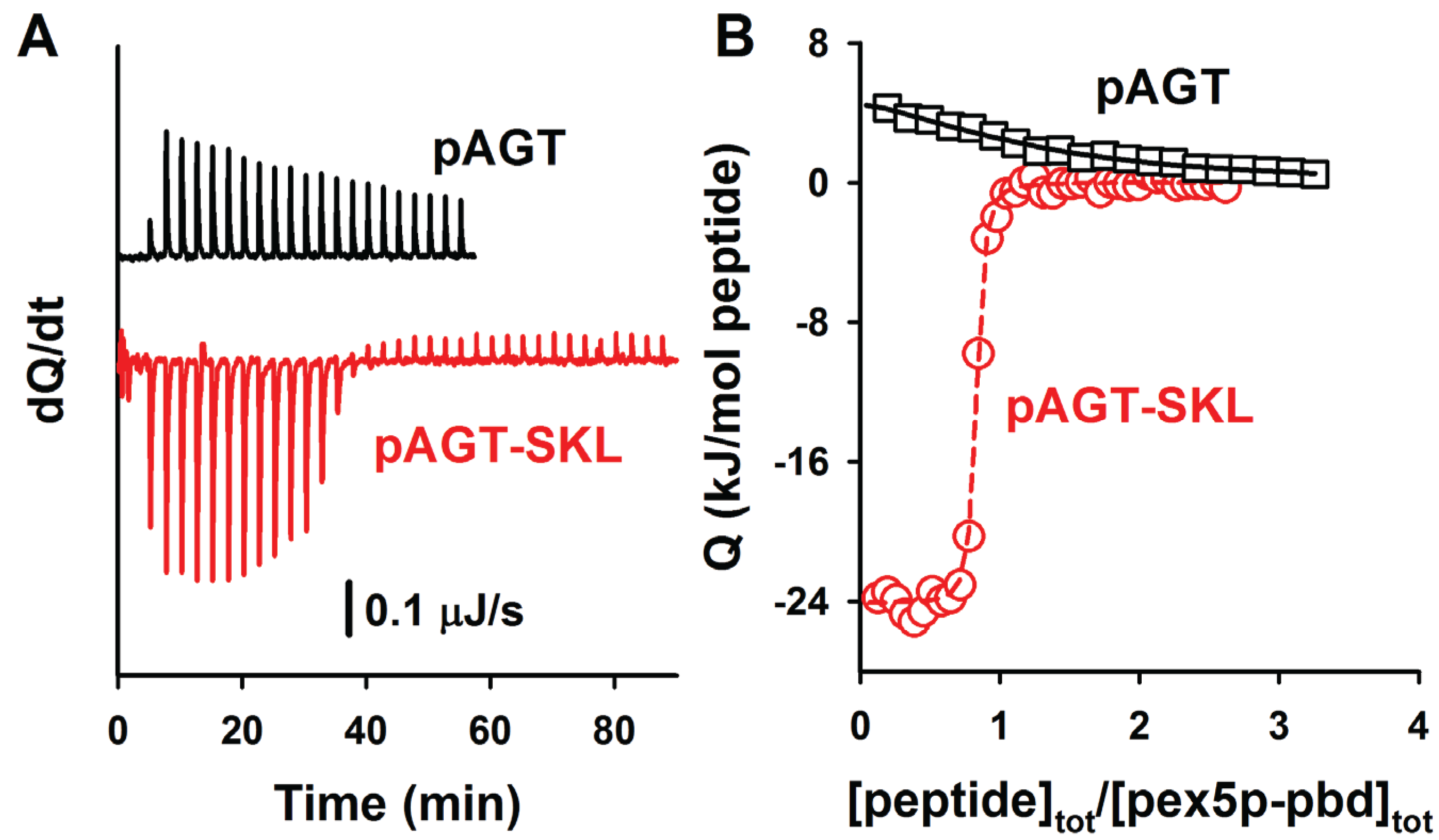
Representative ITC binding analyses of nonapeptides (containing PTS1 octapeptides
plus a N-terminal tyrosine residue) to Pex5p-pbd are shown in
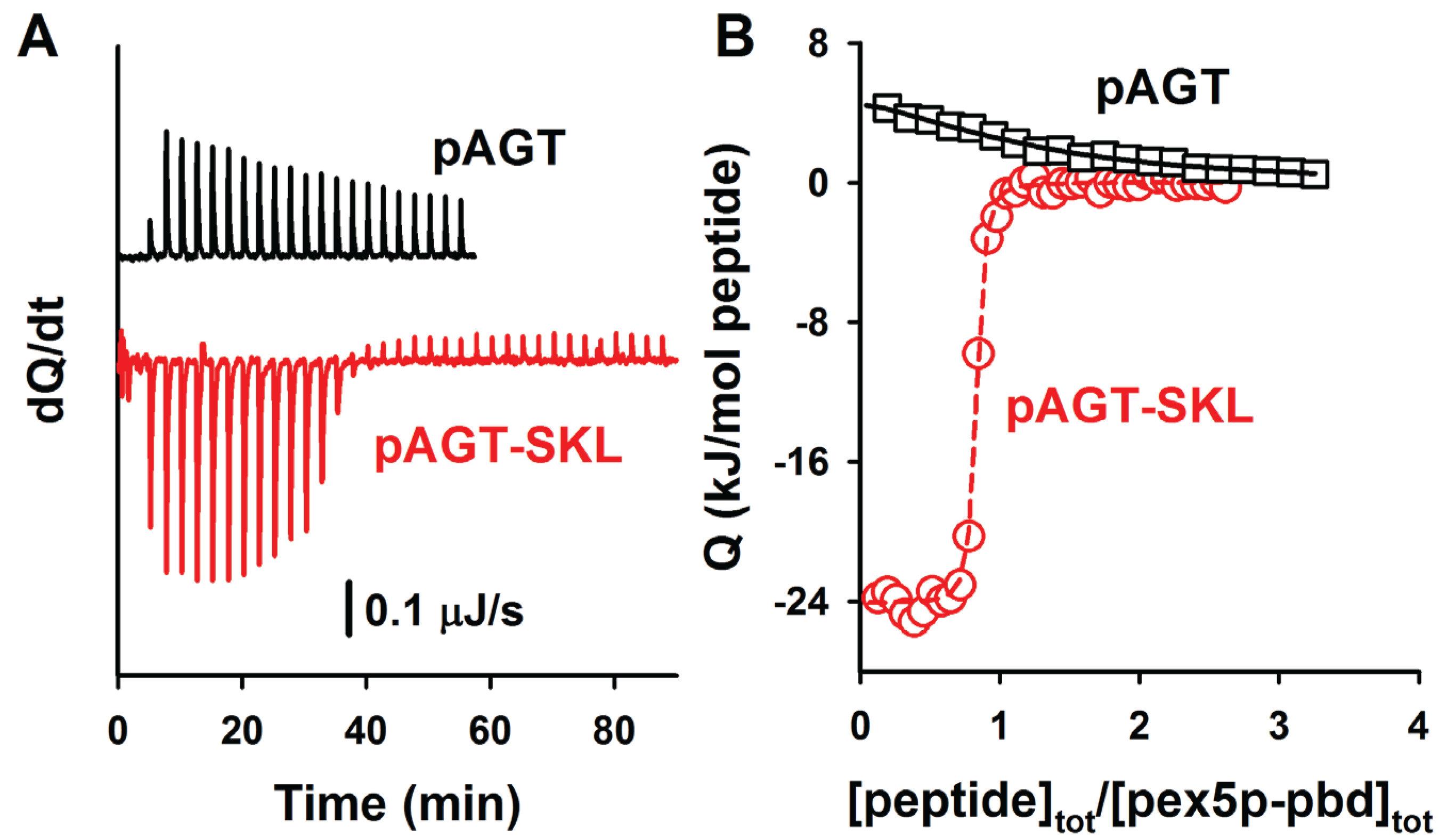
Figure 5. A comparison of the binding affinities of these PTS1 nonapeptides with their corresponding full-length AGT counterparts reveals about one order of magnitude lower affinity for the peptides (
Table 4; with the exception of AGT-SKL, that is close to the detection limit of ITC for a direct titration), in reasonable agreement with previous reports for AGT and SCP2 proteins [
5,
6,
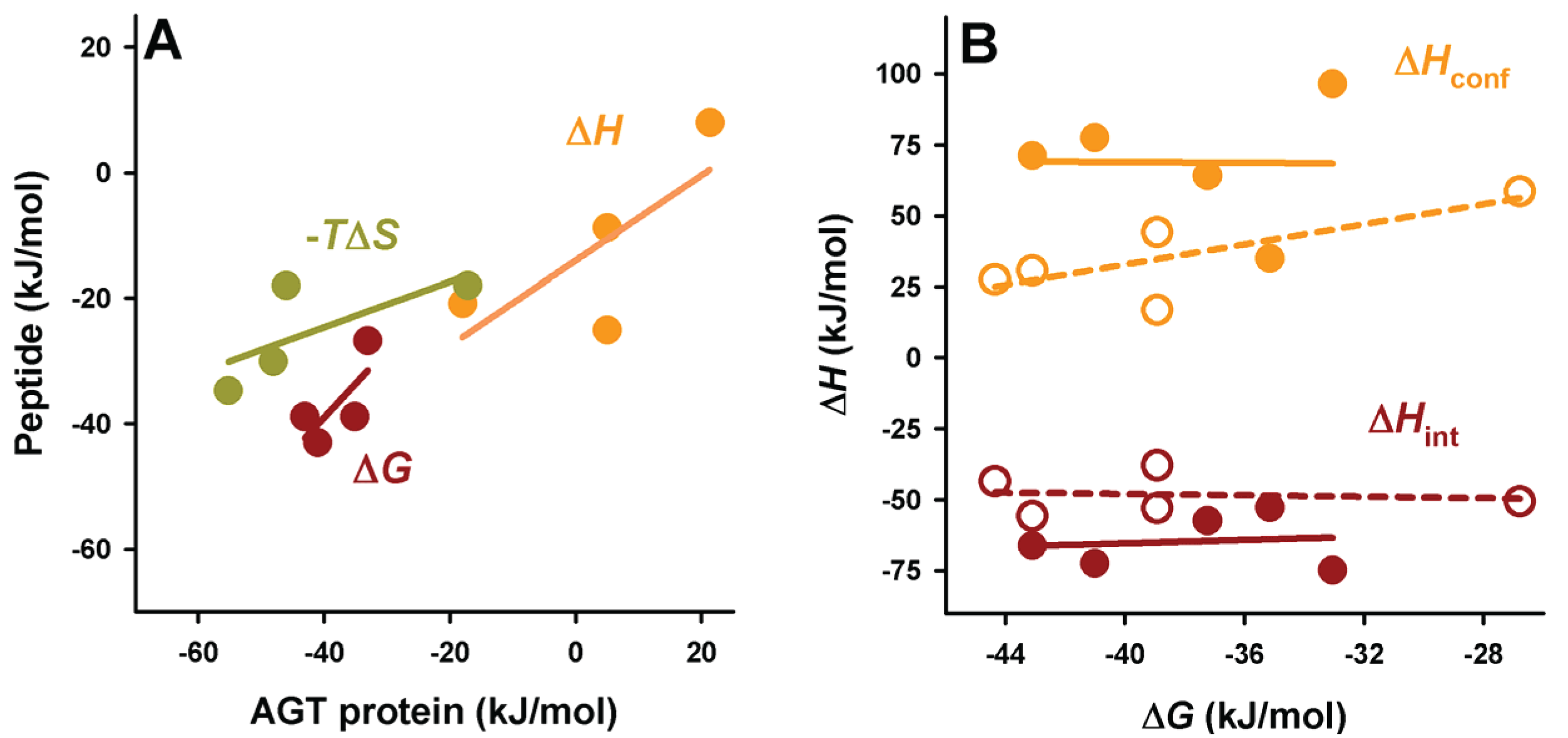
7]. The lower affinity in the peptides likely reflects the contributions from ancillary regions in the full-length cargo protein to complex formation, even though other minor contributions (such as electrostatic effects due to the presence of a charged N-terminal amine group in the peptides) should not be ruled out. Nevertheless, the difference in affinity between AGT proteins and PTS1 nonapeptides shows a reasonable correlation in terms of binding enthalpies and entropies (
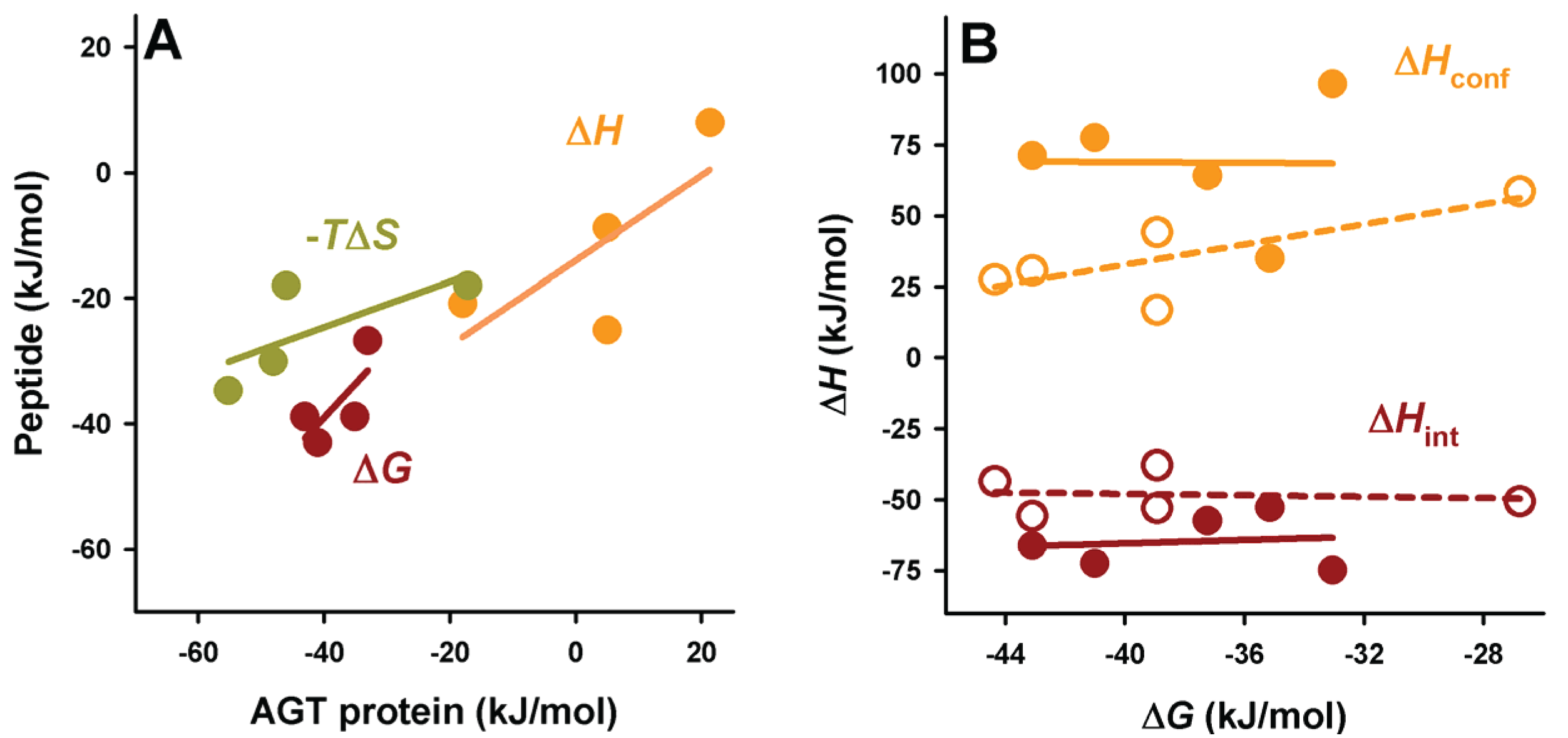
Figure 6A), suggesting the conservation of this context dependent cargo recognition (full-length protein
vs. peptide) to some extent. This supports the idea that, overall, the binding mode of all AGT proteins to Pex5p-pbd is similar and mainly encoded in the C-terminal octapeptide sequence, while other ancillary regions in the AGT protein roughly add one order of magnitude to the binding affinity.
Figure 5.
ITC analysis of the binding of the peptides pAGT and pAGT-SKL to Pex5p-pbd. Thermograms (A) and binding isotherms (B) at 25 °C for pAGT (squares) and pAGT-SKL (circles); Pex5p 20 μM was titrated using 0.5 mM of pAGT and pAGT-SKL peptides (25–30 injections of 1.2–1.5 μL).
Figure 5.
ITC analysis of the binding of the peptides pAGT and pAGT-SKL to Pex5p-pbd. Thermograms (A) and binding isotherms (B) at 25 °C for pAGT (squares) and pAGT-SKL (circles); Pex5p 20 μM was titrated using 0.5 mM of pAGT and pAGT-SKL peptides (25–30 injections of 1.2–1.5 μL).
A comparative analyses of the intrinsic and conformational contributions to the experimental Δ
H and Δ
Cp value may provide some insight into this gain in affinity between full-length cargo protein and the nonapeptides (
Figure 6 and
Figure 7). In general, the peptides show a lower enthalpic penalization to binding, which indicates that a significant fraction of this penalization in the full-length protein arises from ancillary regions, and is somewhat entropically compensated. While the contributions to the enthalpy show similar nature in the full-length AGT and the peptides, the average favorable contribution from the intrinsic component is 17 kJ/mol lower in the peptide, while the conformational component reduces its penalization by about 33 kJ/mol, thus supporting the notion that the ancillary regions contribute favorably in terms of solvent exposure but unfavorably in terms of conformational changes (which is the main source of the enthalpic penalization to binding in the full-length AGT). As found for full-length proteins, the correlation between intrinsic and conformational components to the binding affinity is weak (
Figure 6B). In the case of binding heat capacities, the behavior of the peptides is similar to that of the full-length protein, but as expected, the contribution of the intrinsic component is lower due to the smaller changes in solvent accessibility compared to the full-length protein.
Figure 6.
Context dependence of binding thermodynamics to Pex5p-pbd. (A) correlation of binding thermodynamic parameters between AGT proteins and PTS1 nonapeptides; (B) Binding free energies show little or no correlation with the different contributions (conformational or intrinsic) to binding enthalpies. In panel b, closed symbols are for AGT proteins and open symbols for PTS1 nonapeptides.
Figure 6.
Context dependence of binding thermodynamics to Pex5p-pbd. (A) correlation of binding thermodynamic parameters between AGT proteins and PTS1 nonapeptides; (B) Binding free energies show little or no correlation with the different contributions (conformational or intrinsic) to binding enthalpies. In panel b, closed symbols are for AGT proteins and open symbols for PTS1 nonapeptides.
Figure 7.
Thermodynamic binding properties for the interaction of PTS1 nonapeptides with Pex5p-pbd. (
A) Free energy (Δ
G), enthalpy (Δ
H) and entropy (−
TΔ
S) contributions to the binding reaction; (
B,
C) Thermodynamic dissection of binding enthalpies (
B) and heat capacity changes (Δ
Cp,
C) into their intrinsic contribution (estimated from polar and apolar ΔASA using the crystal structure for the complex;
Figure 1) and the contribution from conformational changes. In (
A,B), data are means for two independent experiments at 25.
Figure 7.
Thermodynamic binding properties for the interaction of PTS1 nonapeptides with Pex5p-pbd. (
A) Free energy (Δ
G), enthalpy (Δ
H) and entropy (−
TΔ
S) contributions to the binding reaction; (
B,
C) Thermodynamic dissection of binding enthalpies (
B) and heat capacity changes (Δ
Cp,
C) into their intrinsic contribution (estimated from polar and apolar ΔASA using the crystal structure for the complex;
Figure 1) and the contribution from conformational changes. In (
A,B), data are means for two independent experiments at 25.
2.4. In Vitro Stabilization of Pex5-pbd Upon Peptide Binding Correlate with Binding Affinity
Protein-protein interactions may lead to intracellular stabilization of the protein partners, and alterations in binding and subsequent stabilization may contribute to human disease [
24,
25]. Stabilization (at least
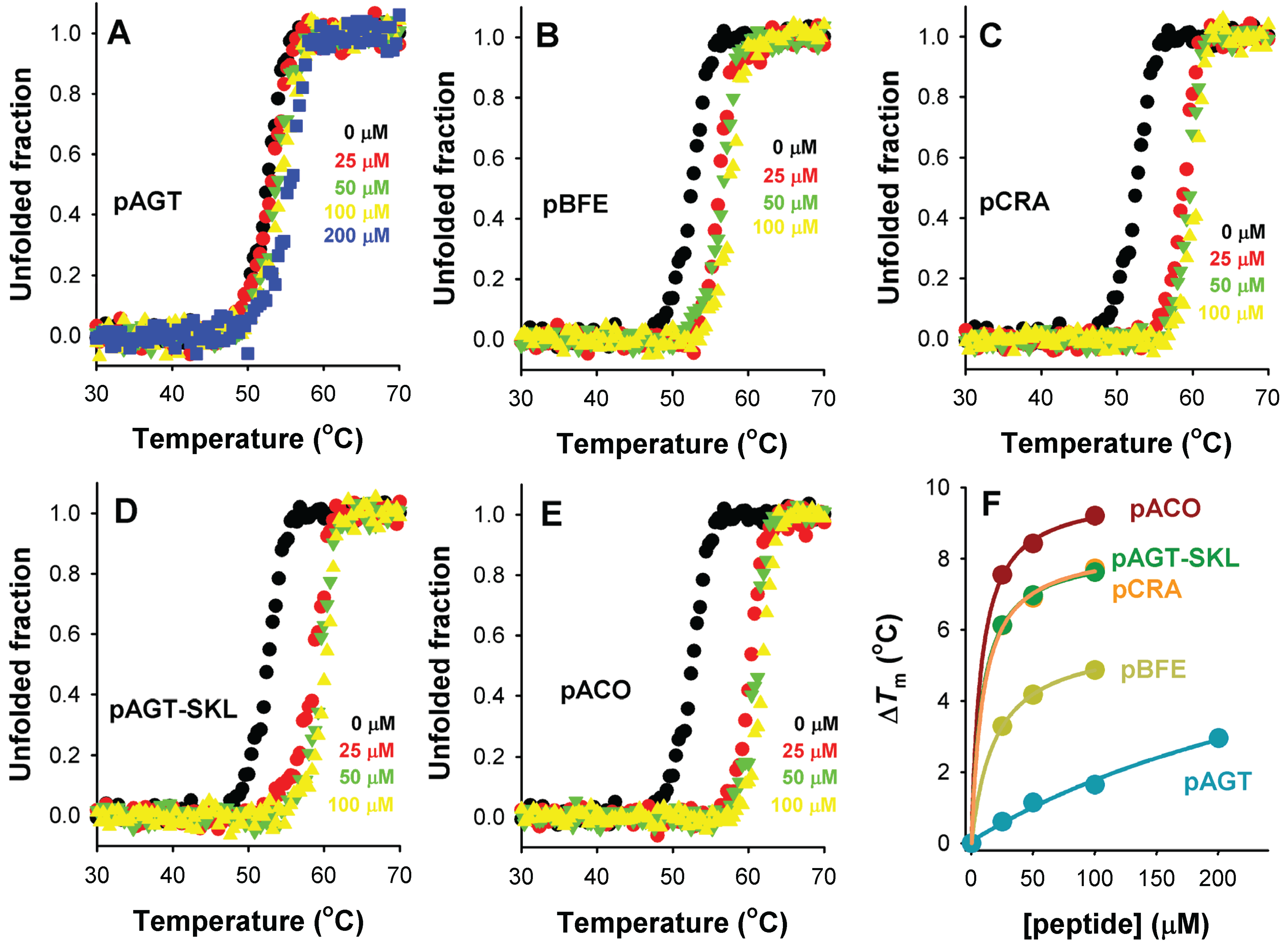
in vitro) is often interpreted as the preferential binding of the partners as folded conformations rather than as non-native conformations, and in principle, the degree of stabilization may be related to binding affinity. We have thus tested whether binding of PTS1 sequences may stabilize Pex5p-pbd using our set of peptides, instead of AGT proteins because the latter show extremely high thermal and kinetic stabilities
in vitro [
12,
26]. To this end, we have used two complementary approaches: thermal denaturation experiments monitored by circular dichroism (
Figure 8), and proteolysis kinetics under native conditions (
Figure 9).
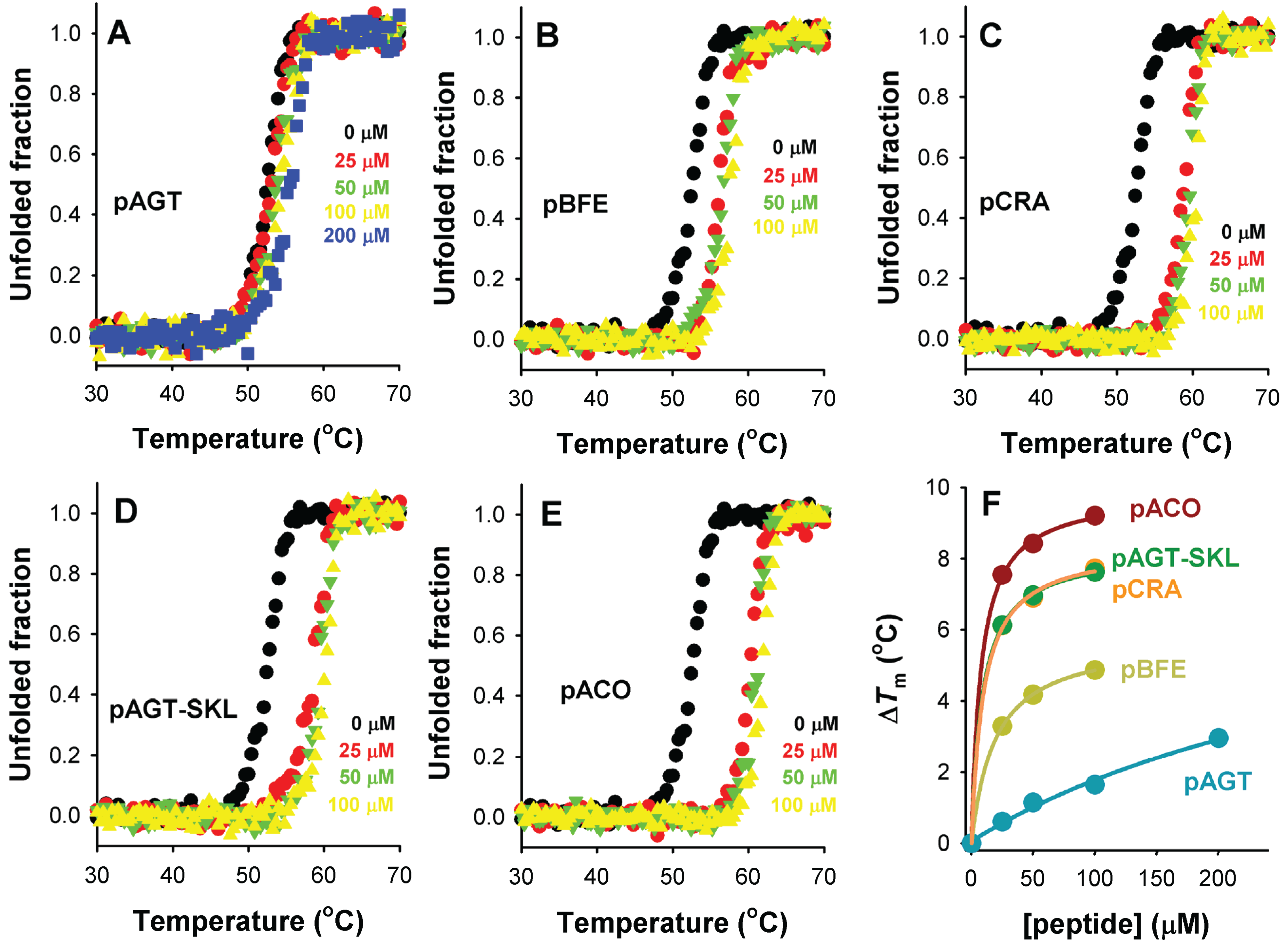
Figure 8.
Thermal stabilization of Pex5p-pbd upon PTS1 nonapeptide binding. (A–E) show thermal denaturation profiles in the absence or presence of different peptide concentrations (as the colors indicated); (F) shows the thermal up-shift (ΔTm) of Pex5p-pbd as a function of peptide concentration.
Figure 8.
Thermal stabilization of Pex5p-pbd upon PTS1 nonapeptide binding. (A–E) show thermal denaturation profiles in the absence or presence of different peptide concentrations (as the colors indicated); (F) shows the thermal up-shift (ΔTm) of Pex5p-pbd as a function of peptide concentration.
Thermal denaturation of Pex5p-pbd is irreversible and kinetically controlled (data not shown [
23]), yielding a single denaturation transition with a
Tm of ~51 °C (
Figure 8). Addition of increasing concentrations of PTS1 nonapeptides leads to a gradual stabilization of Pex5p-pbd (
Figure 8A–E). Interestingly, the peptide concentration dependence of thermal stabilization correlates well with their corresponding binding affinities (
Figure 8F), which is compatible with a scenario in which the Pex5p-pbd peptide complex is kinetically protected against thermal denaturation, and thus, that the amount of free Pex5p-pbd protein determines to a large extent the rate of protein denaturation. Therefore, we hypothesize that cargo binding to Pex5-pbd may stabilize both the receptor and the cargo protein intracellularly, and this stabilization is plausibly linked to the binding affinity and the levels of Pex5p-pbd and cargo proteins.
Proteolysis is a useful tool to examine the effect of ligand binding on protein stability and dynamics [
27,
28,
29]. We have thus examined changes in Pex5-pbd dynamics upon peptide binding by determining the proteolysis kinetic pattern in the presence of thermolysin (
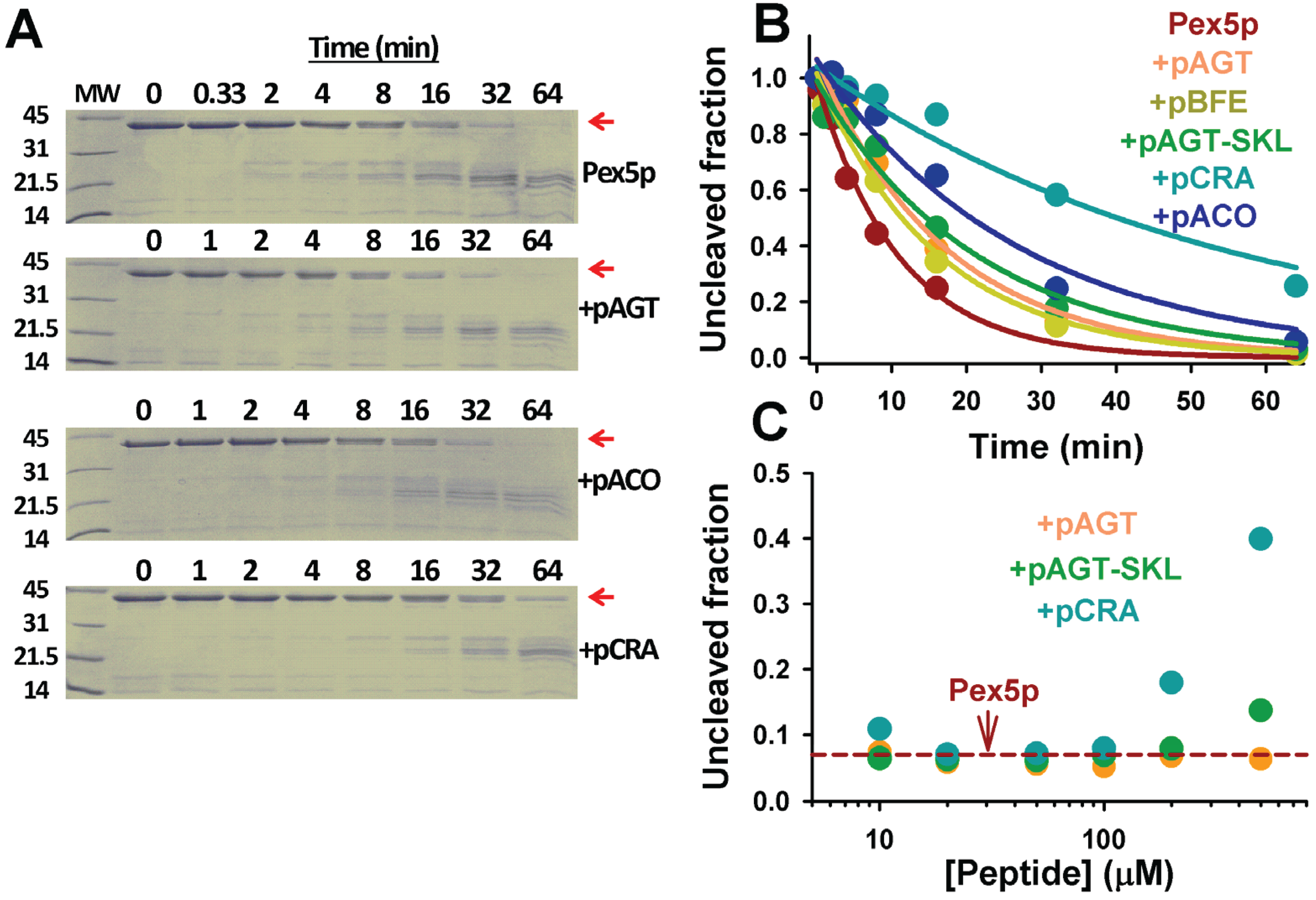
Figure 9). Pex5p-pbd is very sensitive to proteolytic attack by thermolysin, and displays a half-life of about 8 min in the presence of a 1:2000 protease:protein ratio. Addition of PTS1 nonapeptides protect towards proteolysis without changing the partial proteolysis pattern (
Figure 9A), from a mild 1.5-fold with p.AGT and p.BFE to a 5-fold in p.CRA (
Figure 9B). This protective effect is concentration-dependent (
Figure 9C) and correlates fairly well with their corresponding affinities, as also observed for thermal denaturation of Pex5-pbd. This suggests that proteolysis kinetics are reflecting peptide binding effects on Pex5p-pbd dynamics and/or conformational stability.
Figure 9.
Proteolysis of Pex5p-pbd in the absence and presence of PTS1 nonapeptides. (A) SDS-PAGE gels of proteolysis kinetics of Pex5p-pbd in the absence or presence of peptides. MW indicates molecular weight markers in kDa, and the native band of Pex5p-pbd is indicated by a red arrow. Gels were stained with Coomassie blue brilliant; (B) Proteolysis kinetics of the native band of Pex5p-pbd. Lines are best-fits to a single exponential function; (C) Fraction of native Pex5p-pbd after 32 min proteolysis in the absence (horizontal dashed red line) and presence of different peptide concentrations. Thermolysin and Pex5p-pbd concentrations were 10 nM and 20 μM, respectively. In (A) and (B) peptide concentration was 500 μM.
Figure 9.
Proteolysis of Pex5p-pbd in the absence and presence of PTS1 nonapeptides. (A) SDS-PAGE gels of proteolysis kinetics of Pex5p-pbd in the absence or presence of peptides. MW indicates molecular weight markers in kDa, and the native band of Pex5p-pbd is indicated by a red arrow. Gels were stained with Coomassie blue brilliant; (B) Proteolysis kinetics of the native band of Pex5p-pbd. Lines are best-fits to a single exponential function; (C) Fraction of native Pex5p-pbd after 32 min proteolysis in the absence (horizontal dashed red line) and presence of different peptide concentrations. Thermolysin and Pex5p-pbd concentrations were 10 nM and 20 μM, respectively. In (A) and (B) peptide concentration was 500 μM.
2.5. Partial Correction of the Mistargeting Phenotype in a Disease-Causing AGT Variant by Enhancing PTS1 Binding Affinity
Folding, misfolding and mistargeting of human AGT inside cells is a remarkably complex process [
12]. Wild-type AGT is known to transiently interact with molecular chaperones (Hsp70 and Hsp90) on its way to reach the functional and peroxisomal competent dimeric state [
26]. However, upon synthesis, PH1 disease-causing variants are known to interact more strongly with Hsp70, Hsp90 and Hsp60 chaperones [
12,
13,
26,
30], and this behavior is either observed for variants classically associated to mitochondrial mistargeting or to peroxisomal aggregation. Similarly, PH1 variants associated to mistargeting or aggregation are often thermally and kinetically unstable, especially in their apo-form [
12,
26]. In some cases, aggregation has also been linked to altered conformational dynamics of the N-terminal of human AGT [
31]. On the basis of all these results, we have recently proposed that several steps of the intracellular folding and assembly of human AGT would be affected in PH1 variants, and thus, may be potentially targeted for pharmacological treatment of PH1. We must note that it is possible that mitochondrial mistargeting and/or misfolding leading aggregation may kinetically trap the human AGT protein along its folding/misfolding/mistargeting processes, and therefore simply targeting steps from an equilibrium perspective (for instance, using native-state ligands) may not be efficient as new therapeutic approaches. Nevertheless, the fact that a given PH1 variant may lead to either mitochondrial mistargeting or enhanced aggregation depending on the expression conditions in eukaryotic cells [
11,
12] suggests that human AGT is not kinetically trapped along its folding/misfolding/mistargeting pathways and its final fate is amenable to modulation by environmental conditions (temperature, the presence of osmolytes, ligands, and so on).
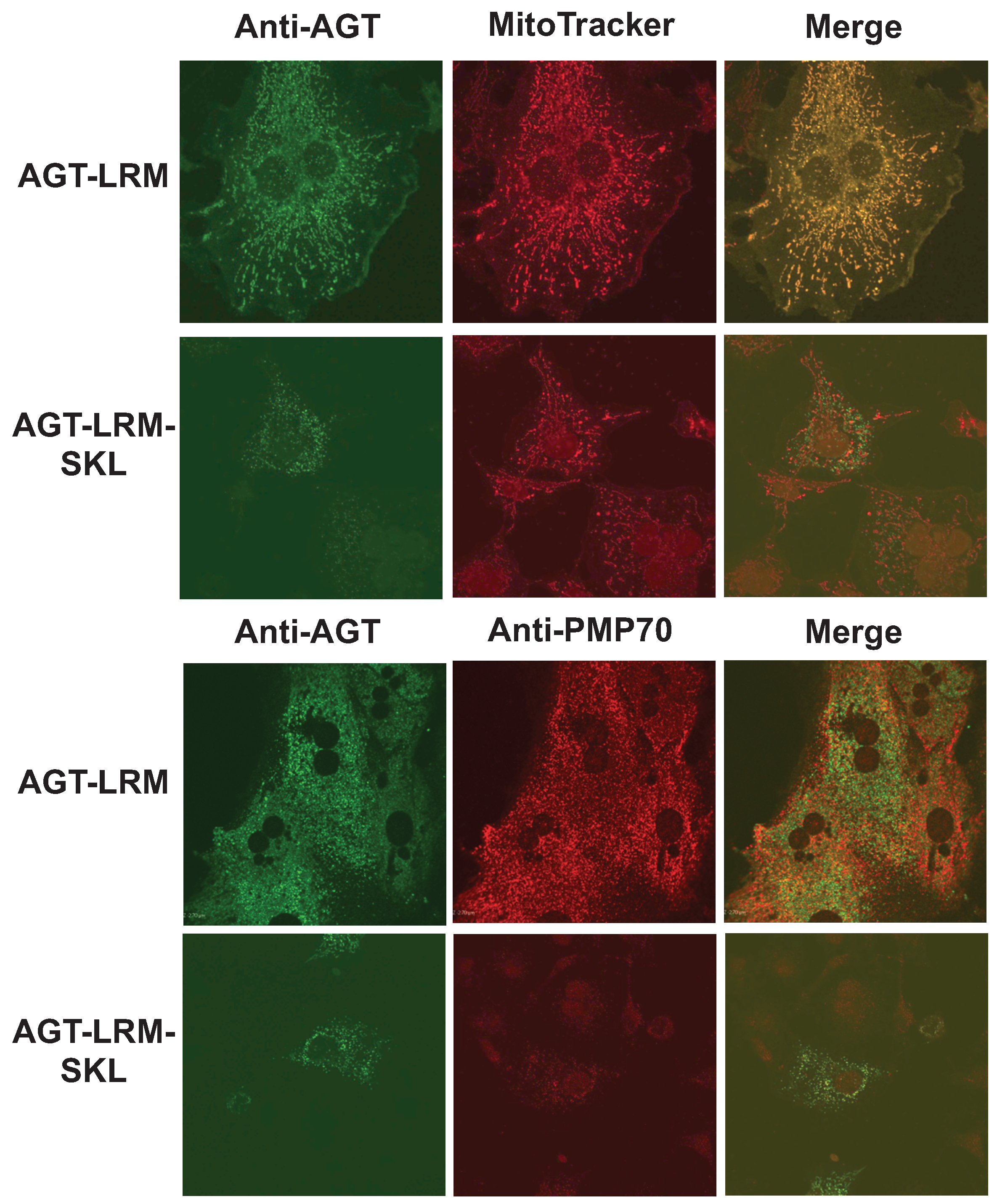
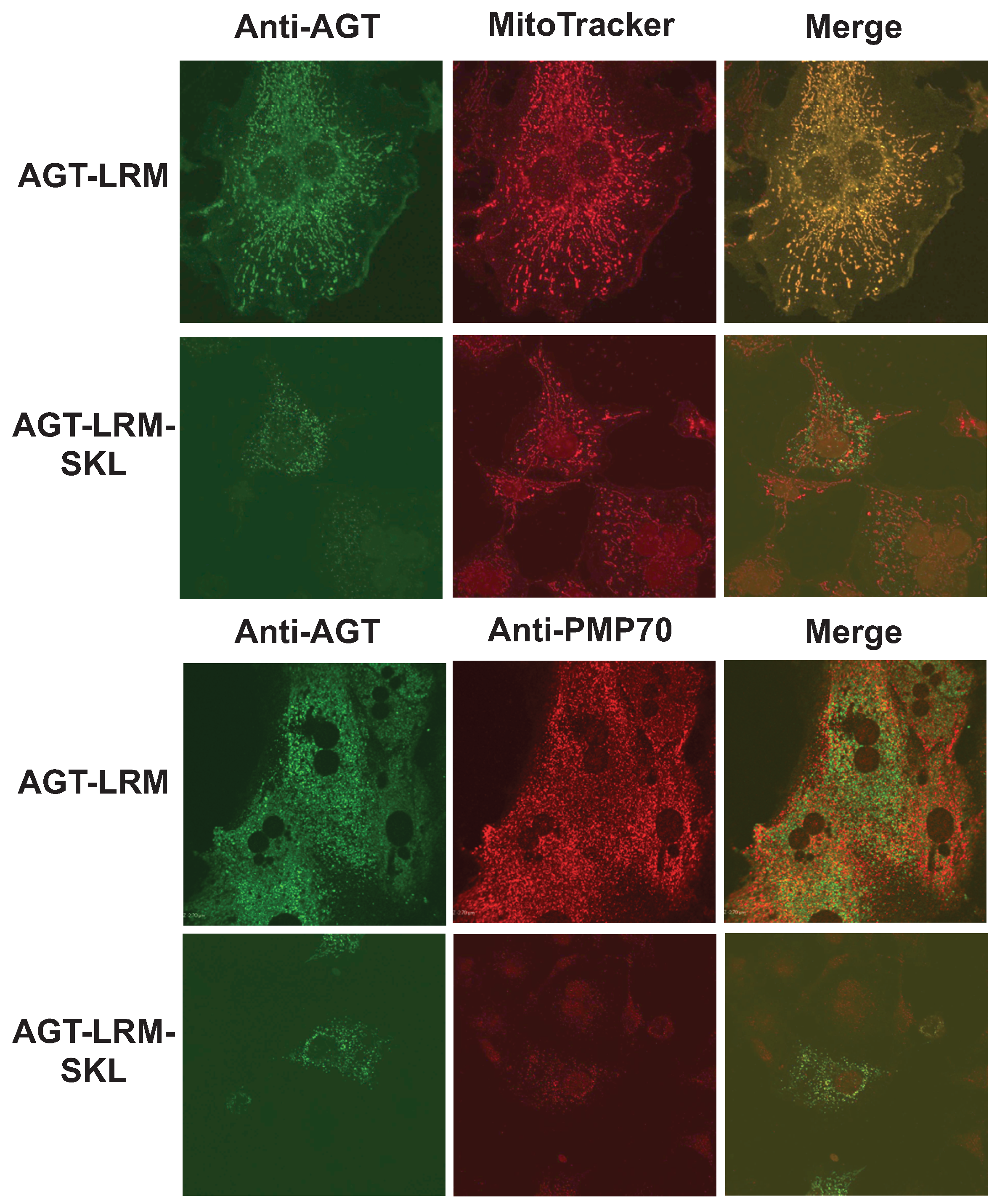
We have thus studied whether an increase in binding affinity of the human AGT native state for Pex5p could shift the equilibrium of human AGT along is folding/mistargeting pathways inside cells. To do so, we transiently transfected CHO cells with plasmids expressing the AGT-LRM variant, which is classically associated with mitochondrial mistargeting, and this variant with the PTS1 sequence mutated to the high affinity consensus –SKL C-terminal tripeptide (AGT-LRM-SKL). Confocal immunofluorescence images of a total of 443 cells were analyzed for the expression of AGT-LRM and AGT-LRM-SKL proteins and its colocalization with either mitochondria or peroxisomal markers is given in
Table 5. As we show in
Figure 10, the AGT-LRM variant is mostly found in mitochondria (R
r = 0.90, with 92% colocalization). However, in AGT-LRM-SKL, we observe a significant fraction of the protein in peroxisomes (84%), with a significant decrease in the colocalization with mitotracker in the mitochondria (R
r = 0.47, with 40% colocalization). The 95% confidence intervals for the percentage of mitochondrial localization in the two tested situations are disparate: in AGT-LRM 0.66 <
m < 1.16
vs. AGT-LRM-SKL: 0.24 <
m < 0.56. Similarly, the amount of AGT-LRM localized in peroxisomes (17%) appears significantly lower than AGT-LRM-SKL (84%). Both colocalization parameters correlate well with their respective Pearson’s coefficients: 0.39 in the AGT-LRM
vs. 0.79 in AGT-LRM-SKL. The 95% confidence intervals are disparate in this case too: 0.15 <
m < 0.44
vs. 0.68 <
m < 1. However, the standard error for the study of AGT-LRM colocalization with peroxisomal marker PMP70 is relatively high, making the confidence interval wide.
Table 5.
Descriptive statistics of intensity correlation coefficient-based (ICCB) parameters in CHO cells. Mean Pearson’s correlation coefficient (
r) and the mean Manders’ percentage of colocalization (
m) were determined as described previously [
32], in the 4 experimental situations for: 1. AGT-LRM transfected cells in mitochondria; 2. AGT-LRM transfected cells in peroxisomes; 3. AGT-LRM-SKL transfected cells in mitochondria and 4. AGT-LRM-SKL transfected cells in peroxisomes. Abbreviations: AGT = alanine: glyoxylate aminotransferase, Mt = mitochondria, Px = peroxisomes. As a % of colocalization, we used Manders’ overlap R in situations 1 and 4, and split Manders’ M
1 in situations 2 and 3.
Table 5.
Descriptive statistics of intensity correlation coefficient-based (ICCB) parameters in CHO cells. Mean Pearson’s correlation coefficient (r) and the mean Manders’ percentage of colocalization (m) were determined as described previously [32], in the 4 experimental situations for: 1. AGT-LRM transfected cells in mitochondria; 2. AGT-LRM transfected cells in peroxisomes; 3. AGT-LRM-SKL transfected cells in mitochondria and 4. AGT-LRM-SKL transfected cells in peroxisomes. Abbreviations: AGT = alanine: glyoxylate aminotransferase, Mt = mitochondria, Px = peroxisomes. As a % of colocalization, we used Manders’ overlap R in situations 1 and 4, and split Manders’ M1 in situations 2 and 3.
| | Experimental situations |
|---|
| ICCB Parameters | 1. AGT-LRM
Mt: AGT + Mitotracker | 2 AGT-LRM
Px:AGT + PMP70 | 3. AGT-SKL
Mt:AGT + mitotracker | 4. AGT-SKL
Px:AGT + PMP70 |
|---|
| Pearson Rr | % Coloc | Pearson Rr | % Coloc | Pearson Rr | % Coloc | Pearson Rr | % Coloc |
|---|
| # cells | 75 | 50 | 169 | 149 |
| r/m ± S.E. | 0.90 ± 0.12 | 0.92 ± 0.12 | 0.39 ± 0.15 | 0.17 ± 0.15 | 0.47 ± 0.08 | 0.40 ± 0.08 | 0.79 ± 0.08 | 0.84 ± 0.08 |
Figure 10.
The consensus AGT-SKL sequence partially corrects mitochondrial mistargeting of AGT-LRM. Immunolocalization of AGT-LRM and AGT-LRM-SKL with mitochondrial (mitotracker) and peroxisomal (PMP-70) markers.
Figure 10.
The consensus AGT-SKL sequence partially corrects mitochondrial mistargeting of AGT-LRM. Immunolocalization of AGT-LRM and AGT-LRM-SKL with mitochondrial (mitotracker) and peroxisomal (PMP-70) markers.
This interpretation is also coherent with recent findings [
33] that blocking mitochondrial import machinery partly resolves the mistargeting of AGT-LRM. Thus, the final localization of AGT-LRM seems to depend on the balance between the opposing forces of mitochondrial and peroxisomal import machineries, mediated by interactions with cellular chaperones and Pex5p, respectively.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}