Sphingosine Phosphate Lyase Regulates Murine Embryonic Stem Cell Proliferation and Pluripotency through an S1P2/STAT3 Signaling Pathway

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
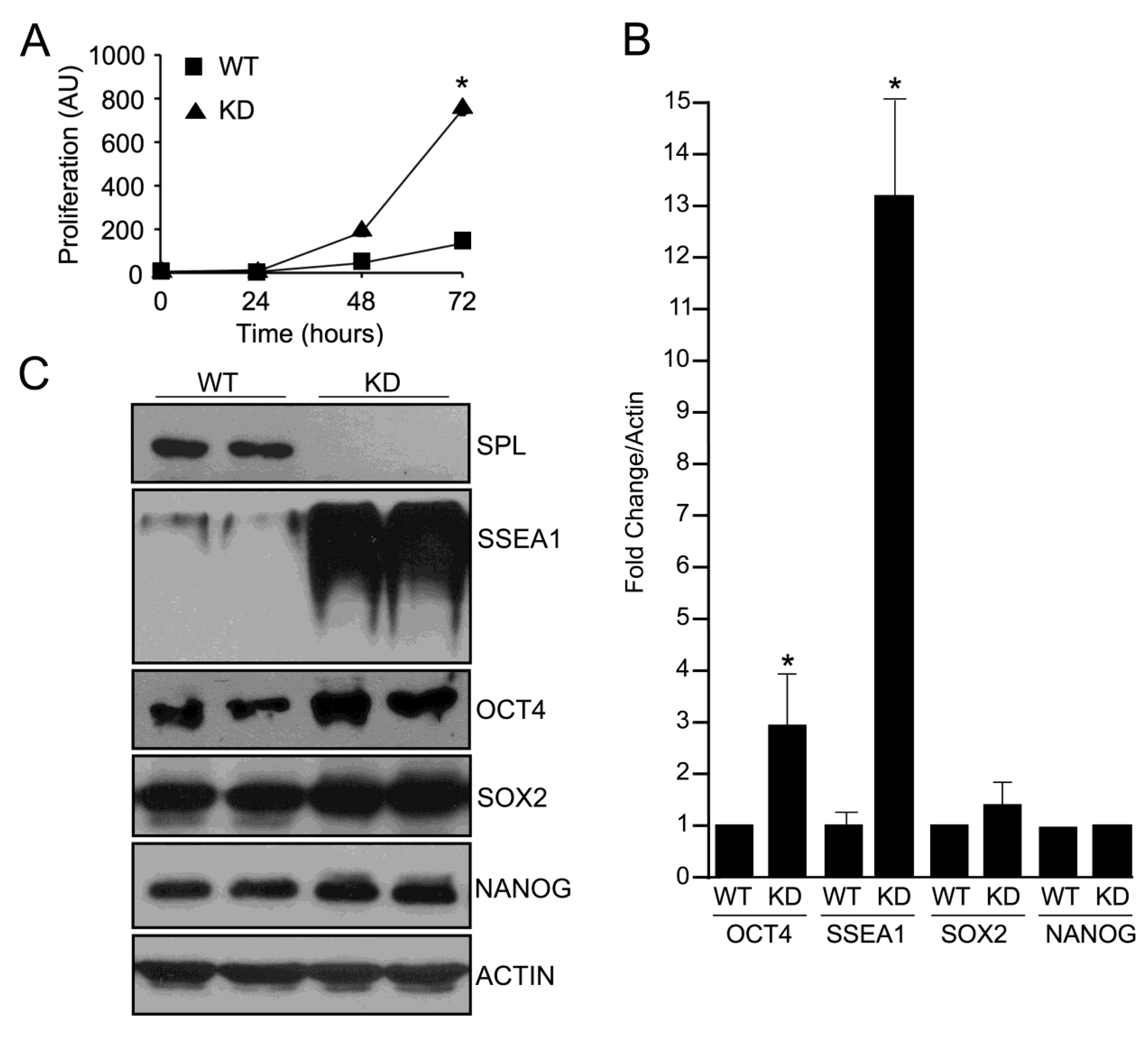
2.1. Silencing of SPL in mESCs
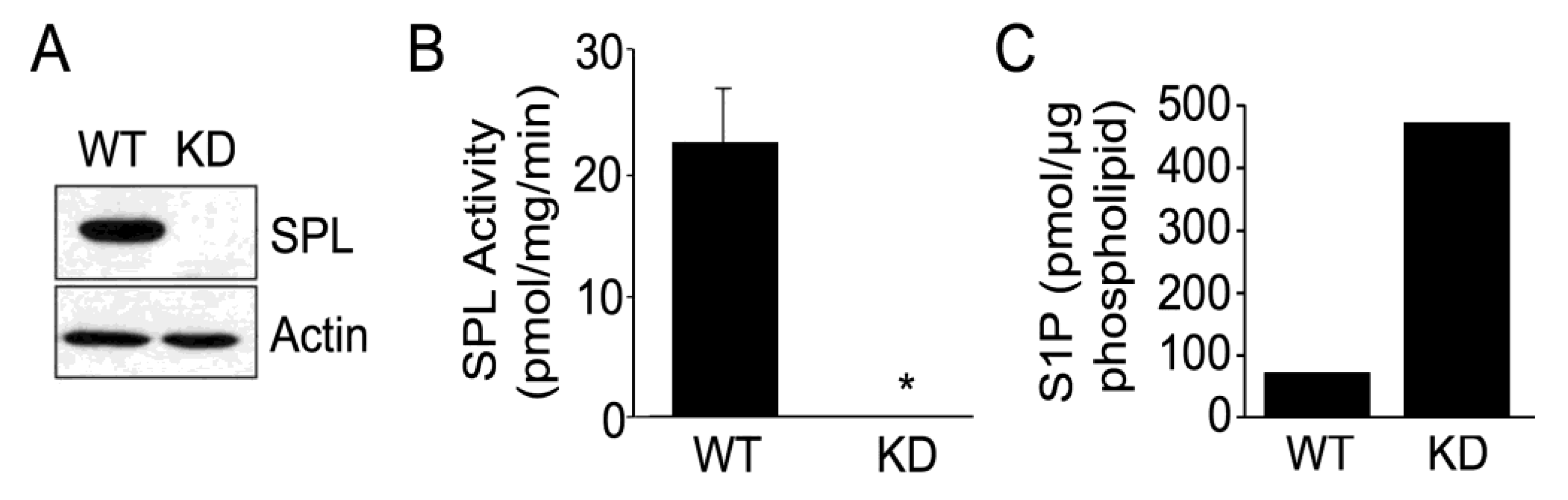
2.2. SPL Silencing Enhances mESC Proliferation and Pluripotency
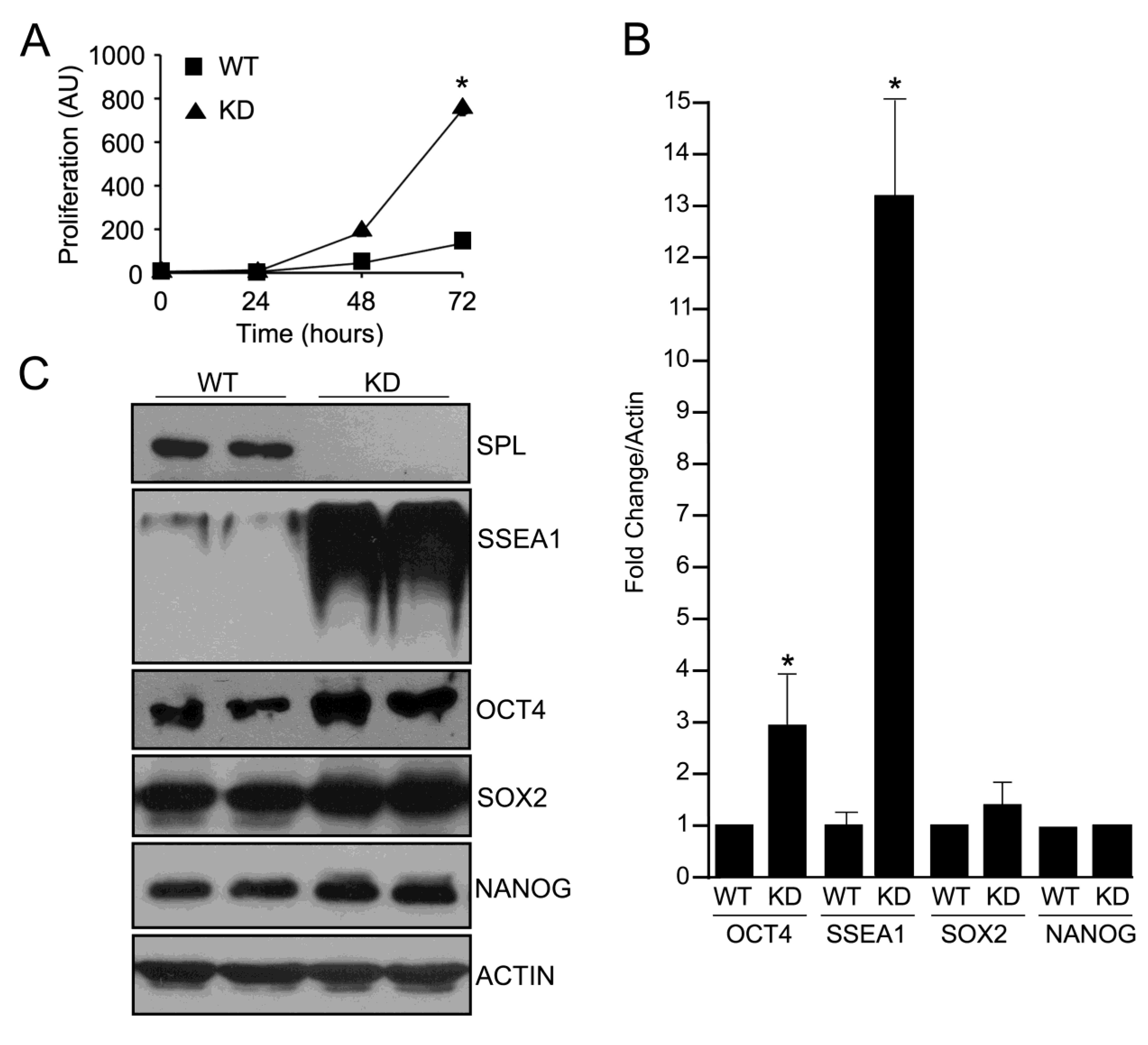
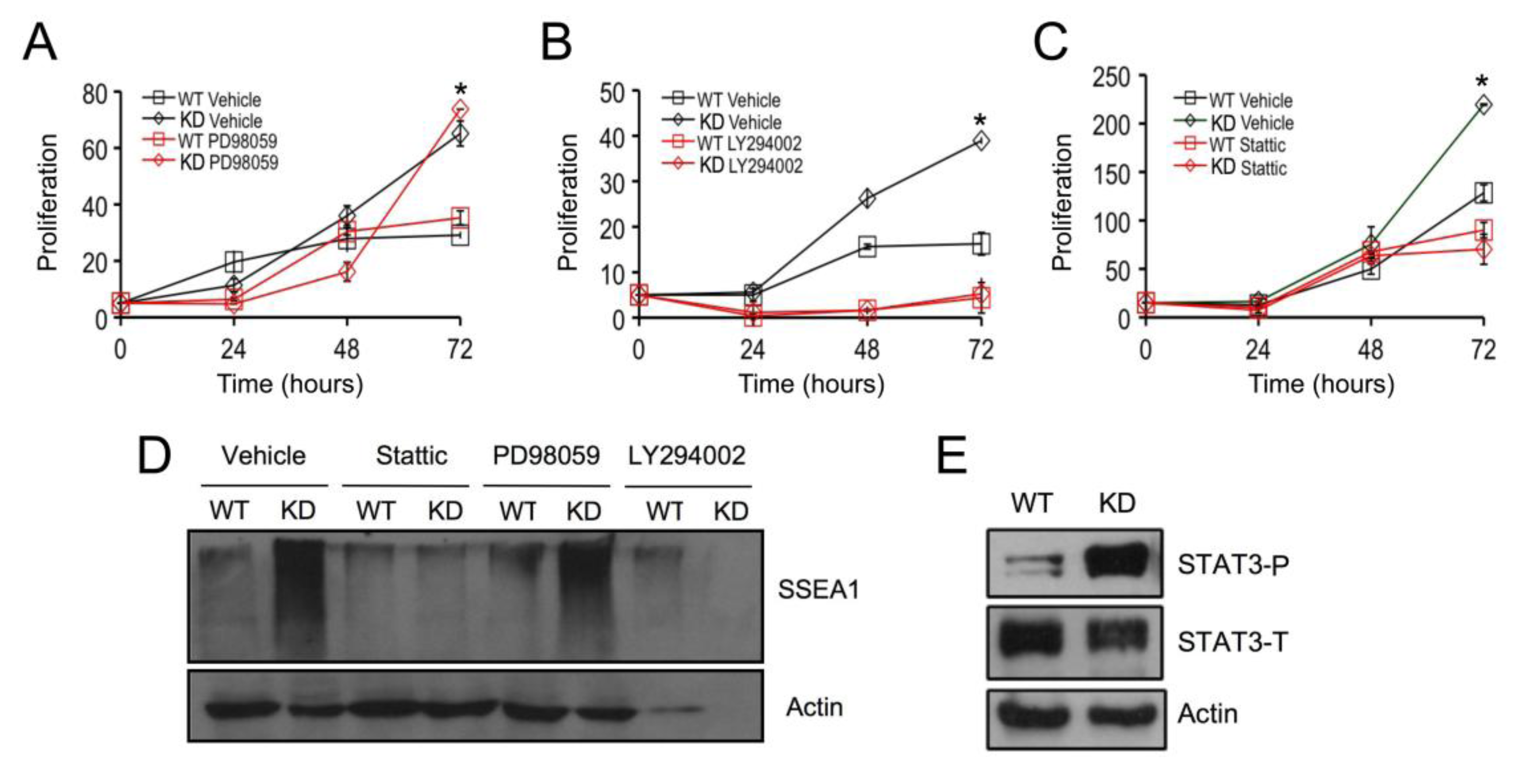
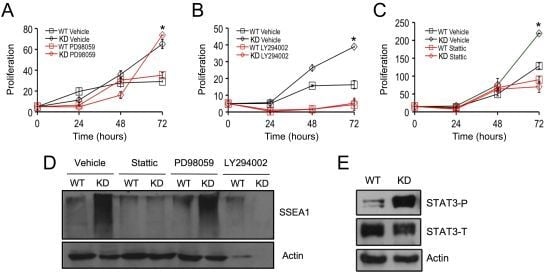
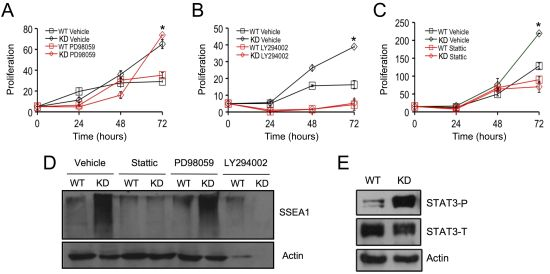
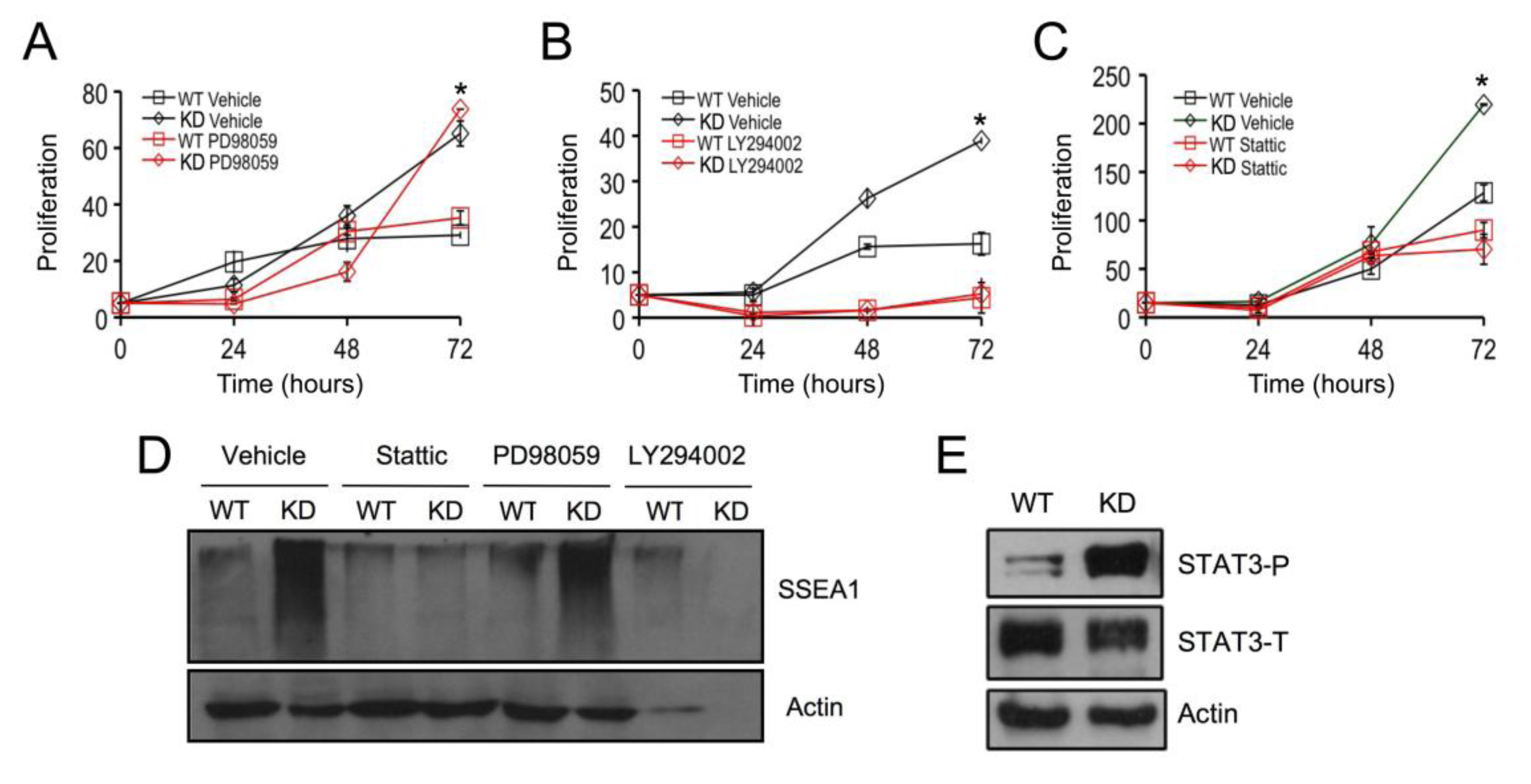
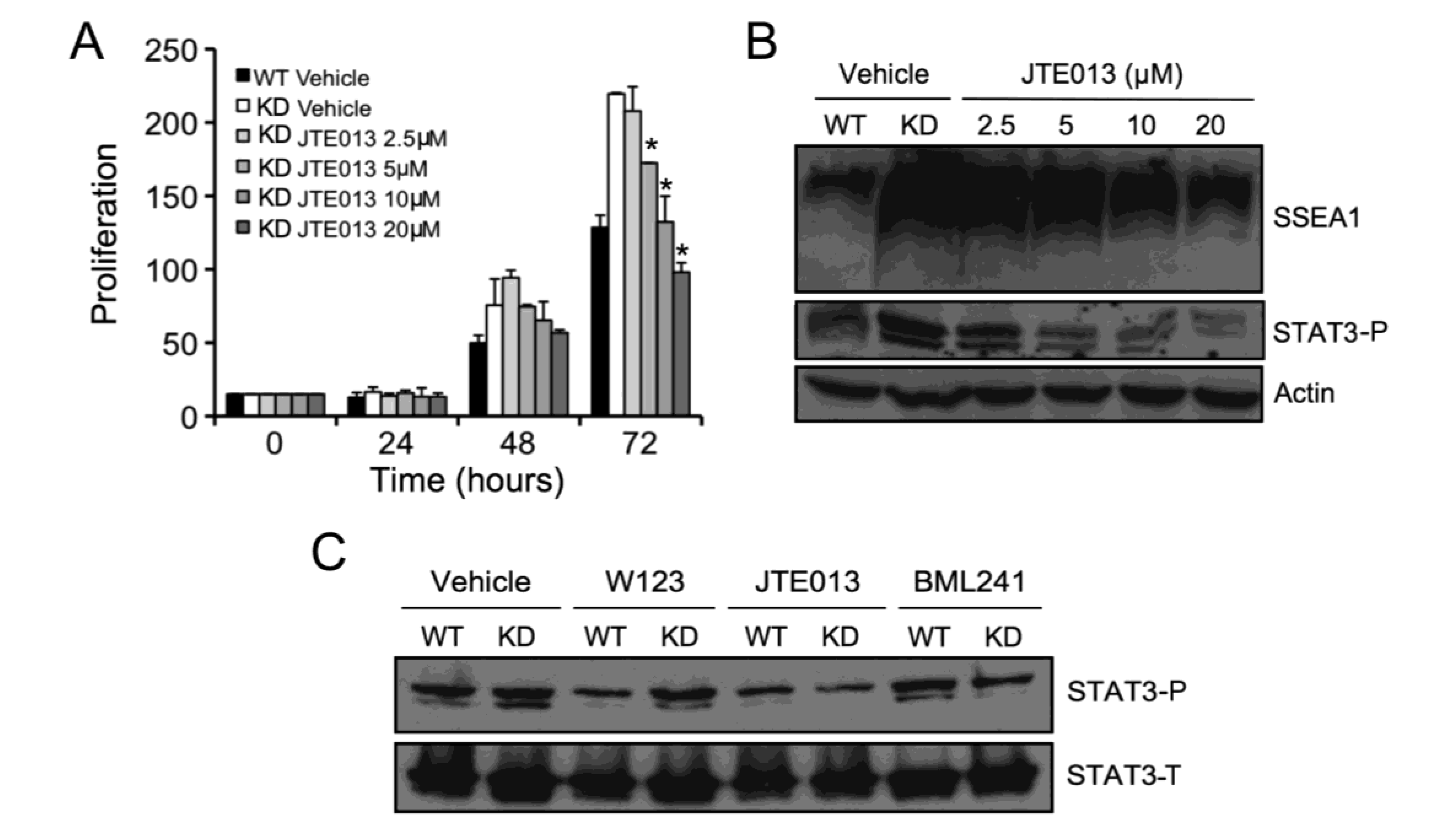
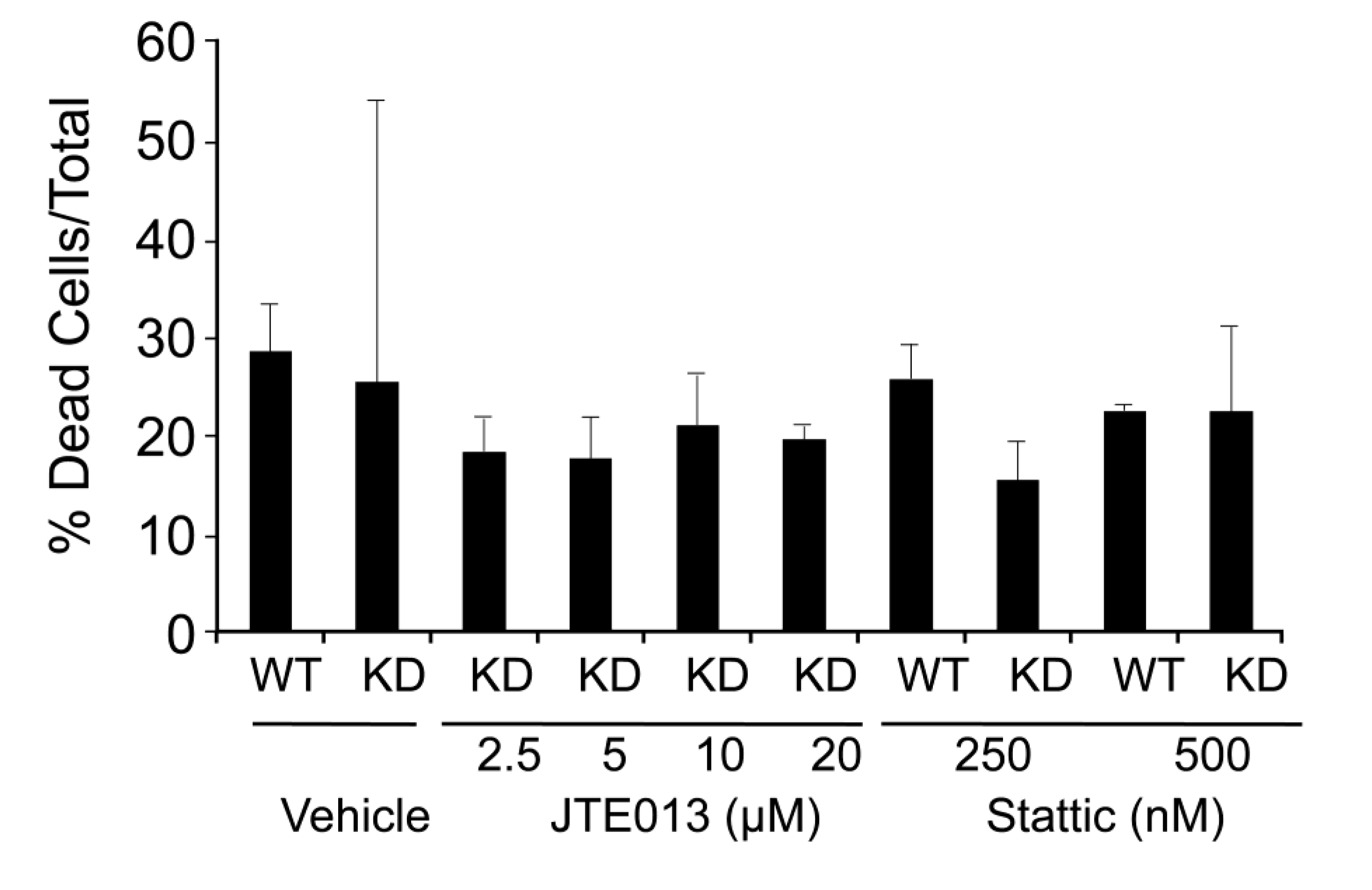
2.3. SPL Silencing Acts via STAT3 Signaling to Enhance mESC Proliferation and Pluripotency
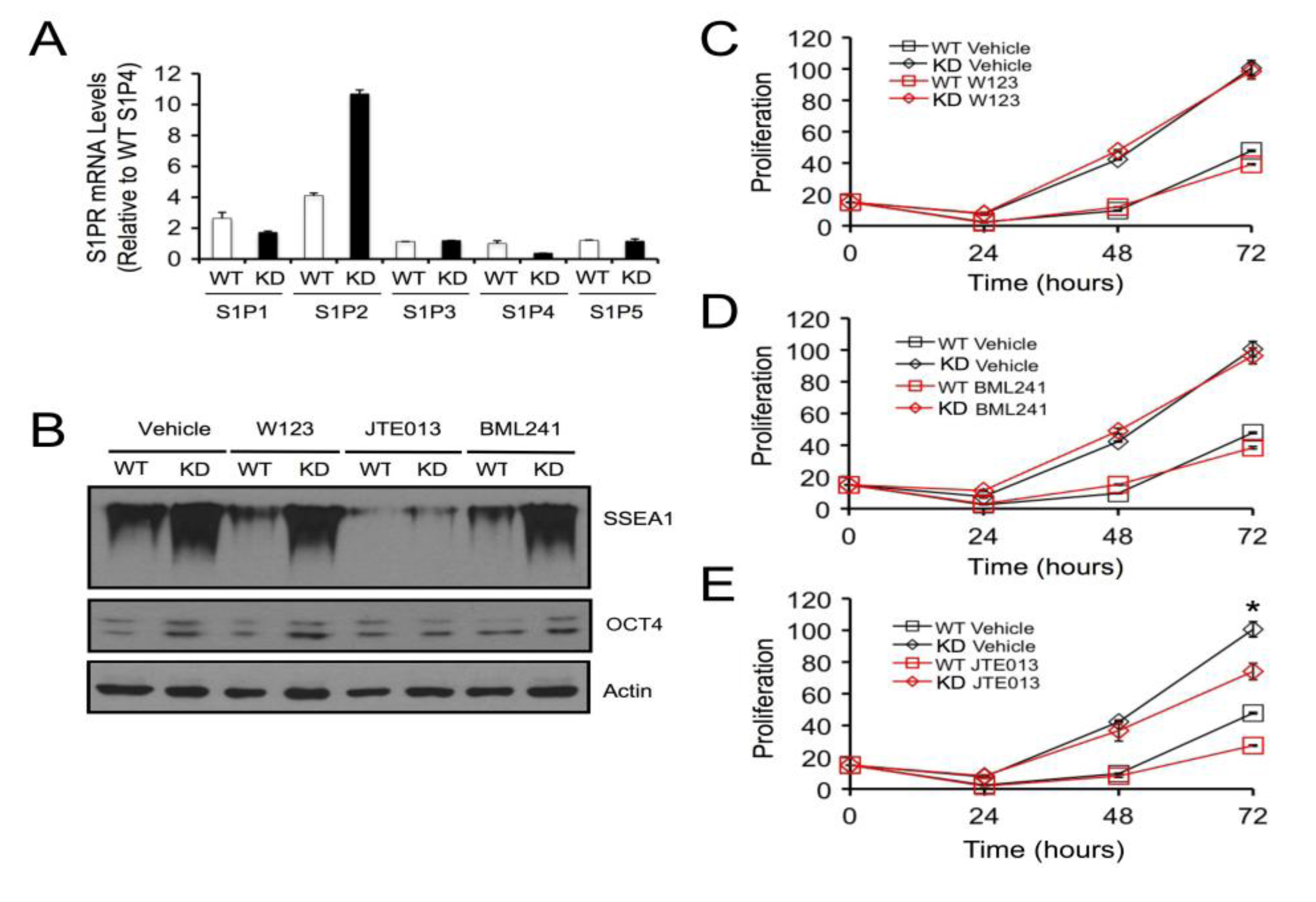
2.4. SPL Silencing Acts via S1P2 to Enhance mESC Proliferation and Pluripotency
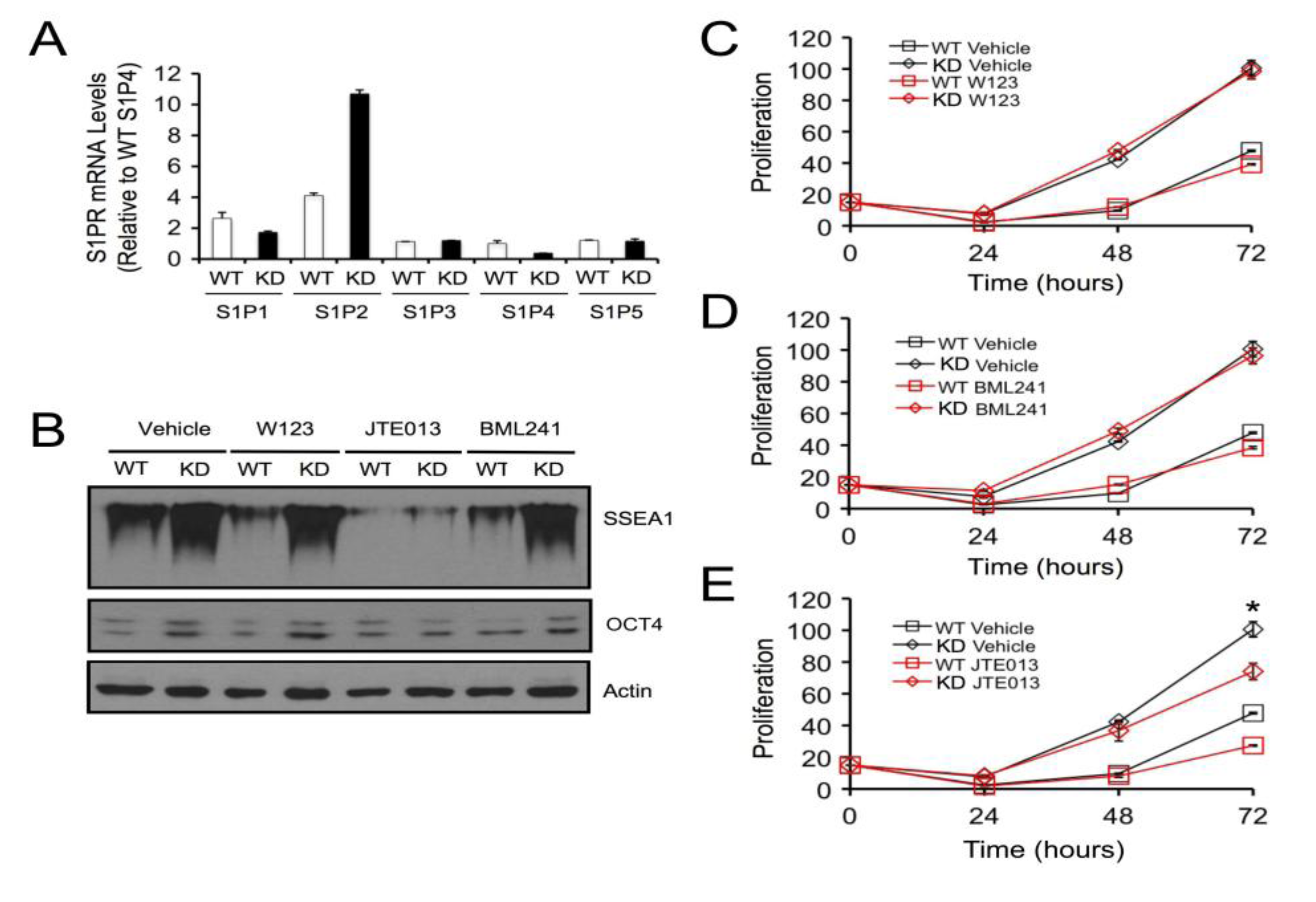
2.5. S1P2 Activates STAT3 Signaling in mESCs
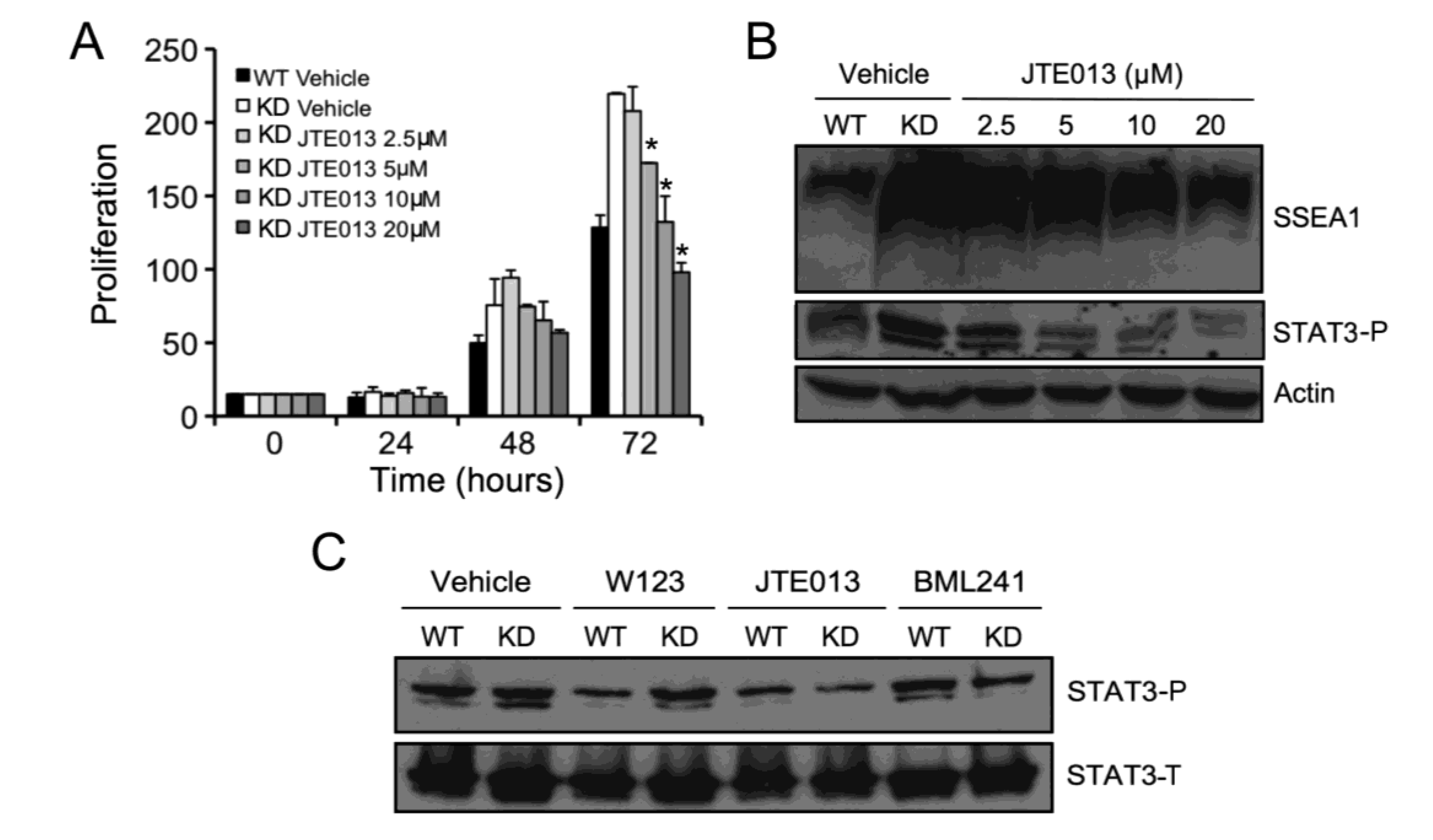
2.6. SPL-KD Cells are Capable of Generating Tissue Stem Cells
2.7. Discussion
3. Experimental Section
3.1. Embryonic Stem Cell Culture
3.2. SPL Knockdown
3.3. SPL Assay
3.4. Mass Spectrometry
3.5. Western Blotting
3.6. Cell Proliferation Assays
3.7. RT-PCR
3.8. Isolation of Satellite Cells from mESC Embryoid Bodies
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Williams, R.L.; Hilton, D.J.; Pease, S.; Willson, T.A.; Stewart, C.L.; Gearing, D.P.; Wagner, E.F.; Metcalf, D.; Nicola, N.A.; Gough, N.M. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 1988, 336, 684–687. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef]
- Kluk, M.J.; Hla, T. Role of the sphingosine 1-phosphate receptor edg-1 in vascular smooth muscle cell proliferation and migration. Circ. Res. 2001, 89, 496–502. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef]
- Lee, M.J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef]
- Nagata, Y.; Partridge, T.; Matsuda, R.; Zammit, P. Entry of muscle satellite cells into the cell cycle requires sphingolipid signaling. J. Cell Biol. 2006, 174, 245–253. [Google Scholar] [CrossRef]
- Seitz, G.; Boehmler, A.M.; Kanz, L.; Mohle, R. The role of sphingosine 1-phosphate receptors in the trafficking of hematopoietic progenitor cells. Ann. NY Acad. Sci. 2005, 1044, 84–89. [Google Scholar]
- Pebay, A.; Bonder, C.S.; Pitson, S.M. Stem cell regulation by lysophospholipids. Prostaglandins Other Lipid Mediat. 2007, 84, 83–97. [Google Scholar] [CrossRef]
- Pyne, N.J.; Long, J.S.; Lee, S.C.; Loveridge, C.; Gillies, L.; Pyne, S. New aspects of sphingosine 1-phosphate signaling in mammalian cells. Adv. Enzyme Regul. 2009, 49, 214–221. [Google Scholar] [CrossRef]
- Saba, J.D.; Nara, F.; Bielawska, A.; Garrett, S.; Hannun, Y.A. The bst1 gene of saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. J. Biol. Chem. 1997, 272, 26087–26090. [Google Scholar] [CrossRef]
- Rodgers, A.; Mormeneo, D.; Long, J.S.; Delgado, A.; Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate regulation of extracellular signal-regulated kinase-1/2 in embryonic stem cells. Stem Cells Dev. 2009, 18, 1319–1330. [Google Scholar] [CrossRef]
- Inniss, K.; Moore, H. Mediation of apoptosis and proliferation of human embryonic stem cells by sphingosine-1-phosphate. Stem Cells Dev. 2006, 15, 789–796. [Google Scholar] [CrossRef]
- Pebay, A.; Wong, R.C.; Pitson, S.M.; Wolvetang, E.J.; Peh, G.S.; Filipczyk, A.; Koh, K.L.; Tellis, I.; Nguyen, L.T.; Pera, M.F. Essential roles of sphingosine-1-phosphate and platelet-derived growth factor in the maintenance of human embryonic stem cells. Stem Cells 2005, 23, 1541–1548. [Google Scholar] [CrossRef]
- Harada, J.; Foley, M.; Moskowitz, M.; Waeber, C. Sphingosine-1-phosphate induces proliferation and morphological changes of neural progenitor cells. J. Neurochem. 2004, 88, 1026–1039. [Google Scholar] [CrossRef]
- Hurst, J.H.; Mumaw, J.; Machacek, D.W.; Sturkie, C.; Callihan, P.; Stice, S.L.; Hooks, S.B. Human neural progenitors express functional lysophospholipid receptors that regulate cell growth and morphology. BMC Neurosci. 2008, 9, 118. [Google Scholar] [CrossRef]
- He, X.; H'Ng S, C.; Leong, D.T.; Hutmacher, D.W.; Melendez, A.J. Sphingosine-1-phosphate mediates proliferation maintaining the multipotency of human adult bone marrow and adipose tissue-derived stem cells. J. Mol. Cell Biol. 2010, 2, 199–208. [Google Scholar] [CrossRef]
- Weinreb, M.; Shamir, D.; Machwate, M.; Rodan, G.A.; Harada, S.; Keila, S. Prostaglandin e2 (pge2) increases the number of rat bone marrow osteogenic stromal cells (bmsc) via binding the ep4 receptor, activating sphingosine kinase and inhibiting caspase activity. Prostaglandins Leukot. Essent. Fatty Acids 2006, 75, 81–90. [Google Scholar] [CrossRef]
- Wong, R.C.; Tellis, I.; Jamshidi, P.; Pera, M.; Pebay, A. Anti-apoptotic effect of sphingosine-1-phosphate and platelet-derived growth factor in human embryonic stem cells. Stem Cells Dev. 2007, 16, 989–1001. [Google Scholar] [CrossRef]
- Avery, K.; Avery, S.; Shepherd, J.; Heath, P.R.; Moore, H. Sphingosine-1-phosphate mediates transcriptional regulation of key targets associated with survival, proliferation, and pluripotency in human embryonic stem cells. Stem Cells Dev. 2008, 17, 1195–1205. [Google Scholar] [CrossRef]
- Burdon, T.; Stracey, C.; Chambers, I.; Nichols, J.; Smith, A. Suppression of shp-2 and erk signalling promotes self-renewal of mouse embryonic stem cells. Dev. Biol. 1999, 210, 30–43. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don't know jak-stat. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Boeuf, H.; Hauss, C.; Graeve, F.D.; Baran, N.; Kedinger, C. Leukemia inhibitory factor-dependent transcriptional activation in embryonic stem cells. J. Cell Biol. 1997, 138, 1207–1217. [Google Scholar] [CrossRef]
- Abe, K.; Hirai, M.; Mizuno, K.; Higashi, N.; Sekimoto, T.; Miki, T.; Hirano, T.; Nakajima, K. The yxxq motif in gp 130 is crucial for stat3 phosphorylation at ser727 through an h7-sensitive kinase pathway. Oncogene 2001, 20, 3464–3474. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. Stat3-induced s1pr1 expression is crucial for persistent stat3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of stat3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef]
- Kidder, B.L.; Yang, J.; Palmer, S. Stat3 and c-myc genome-wide promoter occupancy in embryonic stem cells. PLoS One 2008, 3, e3932. [Google Scholar] [CrossRef]
- Newbigging, S.; Zhang, M.; Saba, J.D. Immunohistochemical analysis of sphingosine phosphate lyase expression during murine development. Gene Expr. Patterns 2012, 13, 21–29. [Google Scholar] [CrossRef]
- Furusawa, T.; Ohkoshi, K.; Honda, C.; Takahashi, S.; Tokunaga, T. Embryonic stem cells expressing both platelet endothelial cell adhesion molecule-1 and stage-specific embryonic antigen-1 differentiate predominantly into epiblast cells in a chimeric embryo. Biol. Reprod. 2004, 70, 1452–1457. [Google Scholar] [CrossRef]
- Smith, K.P.; Luong, M.X.; Stein, G.S. Pluripotency: Toward a gold standard for human es and ips cells. J. Cell. Physiol. 2009, 220, 21–29. [Google Scholar] [CrossRef]
- Niwa, H.; Miyazaki, J.; Smith, A.G. Quantitative expression of oct-3/4 defines differentiation, dedifferentiation or self-renewal of es cells. Nat. Genet. 2000, 24, 372–376. [Google Scholar] [CrossRef]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. Gpr92 as a new g12/13- and gq-coupled lysophosphatidic acid receptor that increases camp, lpa5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef]
- Loh, K.C.; Leong, W.I.; Carlson, M.E.; Oskouian, B.; Kumar, A.; Fyrst, H.; Zhang, M.; Proia, R.L.; Hoffman, E.P.; Saba, J.D. Sphingosine-1-phosphate enhances satellite cell activation in dystrophic muscles through a s1pr2/stat3 signaling pathway. PLoS One 2012, 7, e37218. [Google Scholar] [CrossRef]
- Donati, C.; Cencetti, F.; Nincheri, P.; Bernacchioni, C.; Brunelli, S.; Clementi, E.; Cossu, G.; Bruni, P. Sphingosine 1-phosphate mediates proliferation and survival of mesoangioblasts. Stem Cells 2007, 25, 1713–1719. [Google Scholar] [CrossRef]
- Bandhuvula, P.; Fyrst, H.; Saba, J. A rapid fluorescent assay for sphingosine-1-phosphate lyase enzyme activity. J. Lipid Res. 2007, 48, 2769–2778. [Google Scholar] [CrossRef]
- Spelke, D.P.; Ortmann, D.; Khademhosseini, A.; Ferreira, L.; Karp, J.M. Methods for embryoid body formation: the microwell approach. Methods Mol. Biol. 2011, 690, 151–162. [Google Scholar] [CrossRef]
- Chang, H.; Yoshimoto, M.; Umeda, K.; Iwasa, T.; Mizuno, Y.; Fukada, S.; Yamamoto, H.; Motohashi, N.; Miyagoe-Suzuki, Y.; Takeda, S.; et al. Generation of transplantable, functional satellite-like cells from mouse embryonic stem cells. FASEB J. 2009, 23, 1907–1919. [Google Scholar] [CrossRef]
Supplementary File
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, G.S.; Kumar, A.; Saba, J.D. Sphingosine Phosphate Lyase Regulates Murine Embryonic Stem Cell Proliferation and Pluripotency through an S1P2/STAT3 Signaling Pathway. Biomolecules 2013, 3, 351-368. https://doi.org/10.3390/biom3030351
Smith GS, Kumar A, Saba JD. Sphingosine Phosphate Lyase Regulates Murine Embryonic Stem Cell Proliferation and Pluripotency through an S1P2/STAT3 Signaling Pathway. Biomolecules. 2013; 3(3):351-368. https://doi.org/10.3390/biom3030351
Chicago/Turabian StyleSmith, Gaelen S., Ashok Kumar, and Julie D. Saba. 2013. "Sphingosine Phosphate Lyase Regulates Murine Embryonic Stem Cell Proliferation and Pluripotency through an S1P2/STAT3 Signaling Pathway" Biomolecules 3, no. 3: 351-368. https://doi.org/10.3390/biom3030351