Regulation of Mammalian Gene Dosage by Long Noncoding RNAs

RNA biology program, Sanford-Burnham Medical Research Institute, La Jolla, CA 92037, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomolecules 2013, 3(1), 124-142; https://doi.org/10.3390/biom3010124
Submission received: 21 December 2012
/
Revised: 23 January 2013
/
Accepted: 25 January 2013
/
Published: 4 February 2013
Abstract
:Recent transcriptome studies suggest that long noncoding RNAs (lncRNAs) are key components of the mammalian genome, and their study has become a new frontier in biomedical research. In fact, lncRNAs in the mammalian genome were identified and studied at particular epigenetic loci, including imprinted loci and X-chromosome inactivation center, at least two decades ago—long before development of high throughput sequencing technology. Since then, researchers have found that lncRNAs play essential roles in various biological processes, mostly during development. Since much of our understanding of lncRNAs originates from our knowledge of these well-established lncRNAs, in this review we will focus on lncRNAs from the X-chromosome inactivation center and the Dlk1-Dio3 imprinted cluster as examples of lncRNA mechanisms functioning in the epigenetic regulation of mammalian genes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Long non-coding RNAs are defined as RNAs over 200 nt in length that do not encode proteins. These RNAs were previously considered as transcriptional “noise” or by-products until the development of technologies allowing unbiased high throughput sequencing of transcripts in cells. In 2002, following large-scale sequencing of mouse cDNA libraries, Okazaki et al. revealed that a huge proportion of the mammalian transcriptome does not code for proteins and defined lncRNAs as a significant transcript class [1]. The recent ENCODE (Encyclopedia of DNA Elements) study reported over 9640 lncRNA loci in the human genome, roughly half the number of protein-coding genes [2]. These studies have changed our view of the mammalian genome and underscored the importance of understanding lncRNA function at a mechanistic level. Insight into lncRNA function comes mainly from the study of individual lncRNAs, particularly those identified decades ago from specific genomic loci using traditional gene mapping approaches. The most studied lncRNAs are perhaps those transcribed from the X-chromosome inactivation center and from imprinted loci.
2. Xist lncRNA and X-Chromosome Inactivation
Female mammals have two X-chromosomes and therefore potentially a two-fold excess of X-linked genes relative to XY males. Such an imbalance in X-linked genes would lead to female early embryonic lethality. To overcome this problem, female mammals silence one (Xi) of the two X chromosomes to equalize X-linked gene dosages between the sexes during early development, a process known as X-chromosome inactivation (XCI) [3,4]. A large body of literature exists relevant to different types of XCI (imprinted vs. random), how XCI evolved, and the involvement of XCI in cancer development. Those topics are reviewed elsewhere [5,6,7,8,9]; here, we focus on the role played by lncRNAs in XCI.
2.1. The X Inactivation Center and Xist lncRNA
Early investigators discovered an X-linked locus, the X chromosome inactivation center (Xic), required to trigger X chromosome inactivation [10,11,12]. Amazingly, this center is enriched with lncRNAs, most of them functioning in XCI (Figure 1A). The best-studied and most important of these is the 17Kb X-inactivation specific transcript, known as Xist, which is expressed exclusively from the inactivated X-chromosome and essential for the establishment of XCI in early development [13,14,15]. XCI is arbitrarily divided into three stages. At the pre-XCI stage, Xist is expressed at low levels from two active X-chromosomes. During the initiation stage of XCI, Xist is upregulated and begins to “spread” along one of the X-chromosomes and converts that allele into heterochromatin characterized by: (1) genome-wide loss of euchromatic marks; (2) gain of heterochromatic marks; (3) increased DNA methylation; and (4) silencing of gene expression. This very dynamic stage requires Xist. Upon establishment of XCI, the inactive X remains silenced throughout subsequent cell divisions. During this maintenance stage, Xist remains highly expressed from the inactive X. However, deletion of Xist RNA does not result in chromosome-wide gene reactivation [16,17,18]. Therefore, Xist RNA is not required at this stage of XCI [19].
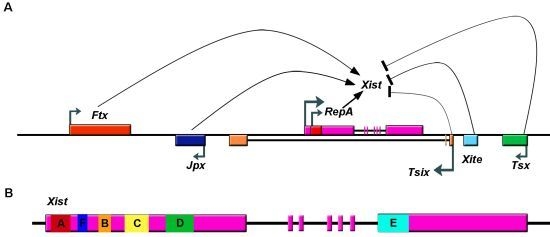
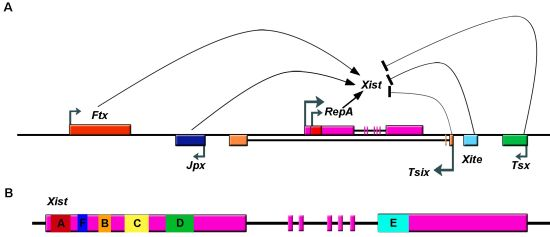
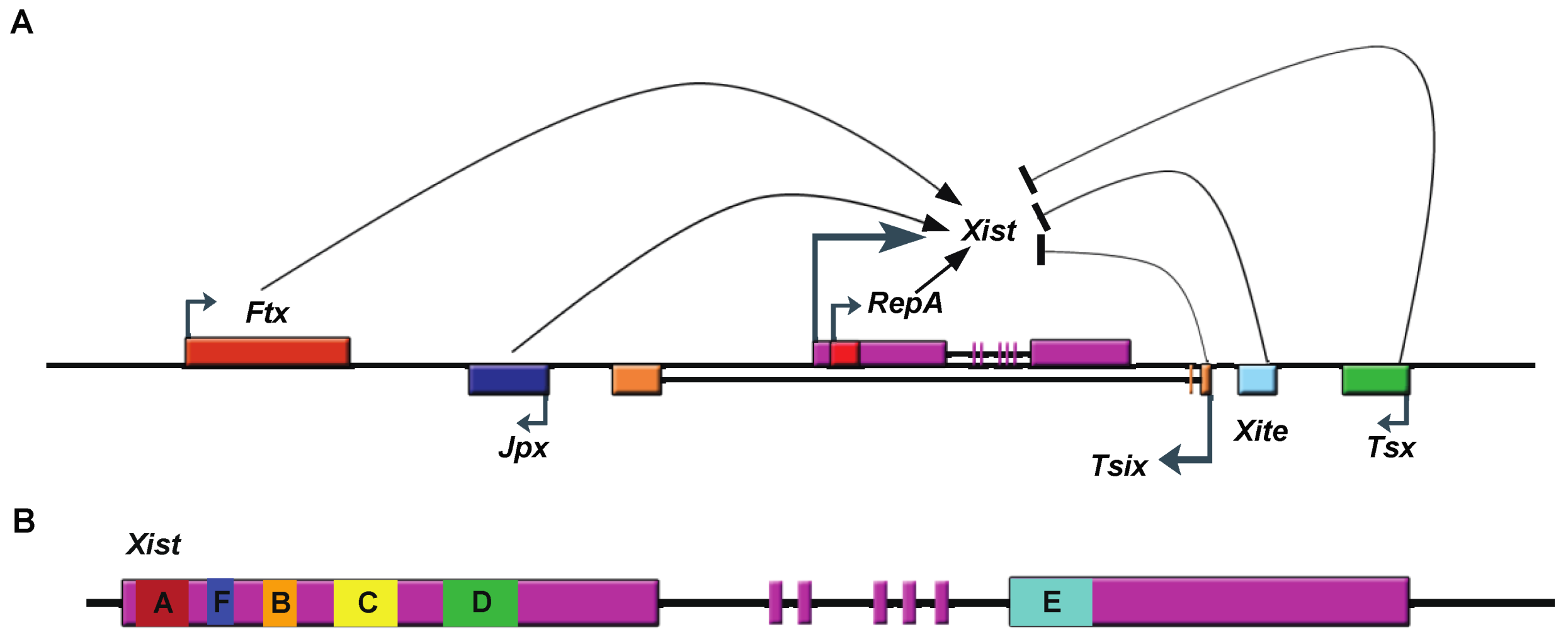
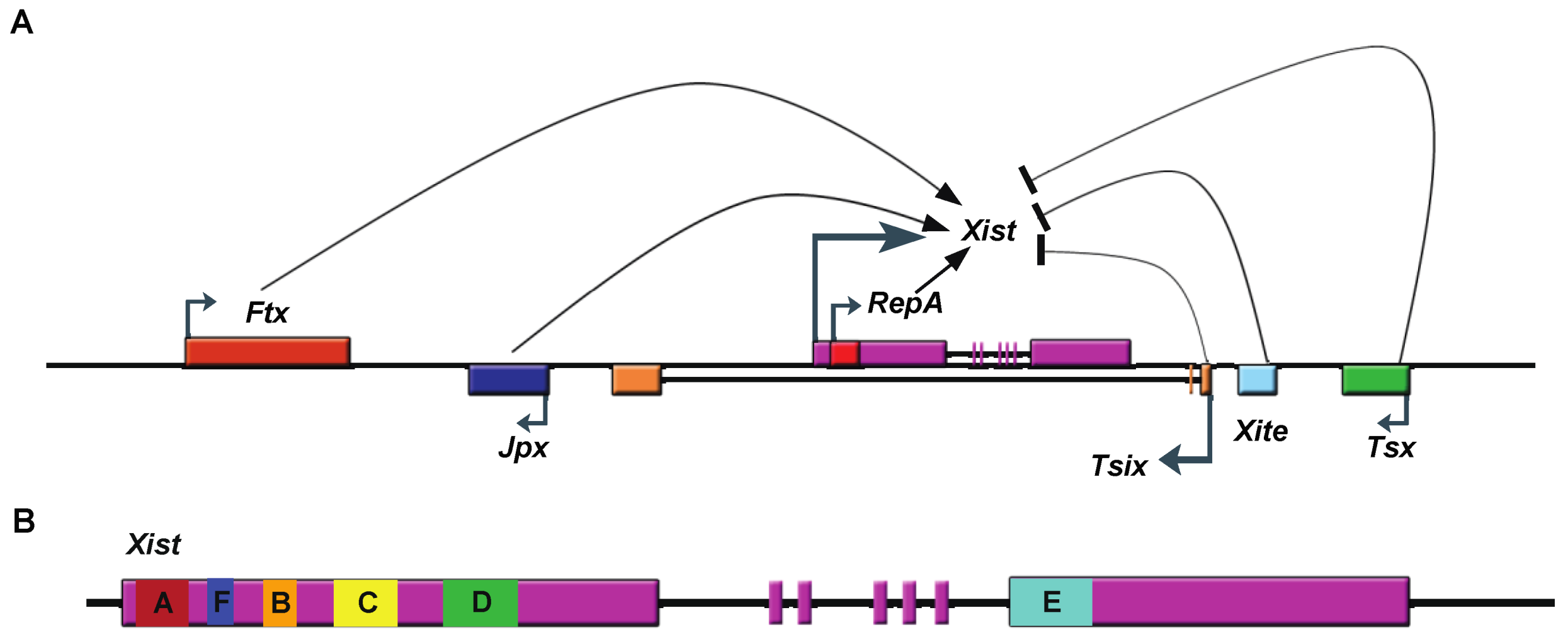
Figure 1.
Schematic drawing of the Xic and of Xist. A. lncRNAs transcribed from Xic, a region of the X-chromosome necessary and sufficient to trigger XCI. These lncRNAs include Xist (X-inactivation specific transcript), Tsix (antisense of Xist), Tsx (testis-specific X-linked gene), Xite (X-inactivation intergenic transcription element), RepA RNA, Jpx (also known as Enox (Expressed Neighbor of Xist)), and Ftx (Five prime to Xist). Ftx, JPX, and RepA lncRNAs promote Xist transcription, while Tsix, Xite and Tsx inhibit it. B. Gene structure of Xist RNA showing location of tandem repeats (A-F). Exons are represented as boxes.
Figure 1.
Schematic drawing of the Xic and of Xist. A. lncRNAs transcribed from Xic, a region of the X-chromosome necessary and sufficient to trigger XCI. These lncRNAs include Xist (X-inactivation specific transcript), Tsix (antisense of Xist), Tsx (testis-specific X-linked gene), Xite (X-inactivation intergenic transcription element), RepA RNA, Jpx (also known as Enox (Expressed Neighbor of Xist)), and Ftx (Five prime to Xist). Ftx, JPX, and RepA lncRNAs promote Xist transcription, while Tsix, Xite and Tsx inhibit it. B. Gene structure of Xist RNA showing location of tandem repeats (A-F). Exons are represented as boxes.
2.2. Xist RNA Contains Multiple Functional Domains
Like proteins, lncRNA also exhibits functional domains. Xist RNA contains six different repeat regions (A to F) (Figure 1B). The most well studied is the repeat A region located at the Xist RNA 5’-end [20]. Transgene studies suggest that Xist lacking the repeat A region cannot initiate gene silencing [20]. The function of that region was subsequently investigated in vivo using a gene targeting approach. Surprisingly, deletion of the A-repeats altered regulation of the mutant Xist allele such that it was not expressed [21], making analysis of that deficiency difficult in vivo. Ex vivo studies using mouse embryonic stem cells (mESCs) revealed at least two independent mechanisms for the silencing function of the A-repeats. One study suggests that the region is required for proper splicing of Xist RNA by directly interacting with splicing factor ASF/SF2 [22]. In another study, we discovered that a shorter transcript within the Xist locus is transcribed through the RepA region in mESCs and we named it RepA RNA [23]. Knockdown of that RepA transcript drastically decreased Xist levels, similar to the phenotype observed following in vivo deletion of the repeat A region of Xist, suggesting that RepA RNA is an important regulator of Xist expression. We found that both Xist and RepA RNA can bind the chromatin repressor Polycomb repressive complex 2 (PRC2) through repeat A region and such interactions are required for the establishment of the chromosome-wide H3K27-3me heterochromatic mark during the initiation of XCI.
Several laboratories have analyzed the structure of the repeat A domain in order to understand the molecular basis for RNA-protein interaction. The A-repeat region contains 7.5–8.5 tandem repeats (variable among different species) of a conserved ~26-mer sequence. Computational analysis indicates that each repeat has a double stem loop structure [20]. However, NMR studies of an in vitro transcribed 26-mer showed that only the first predicted hairpin is formed, while the second predicted hairpin mediates duplex formation among different repeats [24,25]. One limitation of NMR studies is that only a short sequence, such as one of the A repeats, can be analyzed. By carrying out a FRET experiment using chemical and enzymatic probes, another study examined the structure of the whole domain and found that the A region contains two long stem-loop structures, each including four repeats [26]. These studies highlighted the importance of RNA’s structure for its function.
2.3. Upregulation of Xist RNA during XCI Initiation
Since Xist upregulation is crucial for XCI, much effort has been made to elucidate cis-elements and trans-acting factors that control its expression. The following sections describe how lncRNAs and pluripotency factors regulate Xist expression.
2.3.1. LncRNAs
In addition to Xist, the XIC harbors several lncRNAs (Figure 1A). One of them, Tsix, is transcribed antisense to Xist and covers the entire Xist locus [30,31,32,33,34]. At pre-XCI stage, Tsix expression exceeds that of Xist. When Tsix is deleted, Xist is non-randomly upregulated from the disrupted allele, suggesting that Tsix represses Xist expression. Several mechanisms have been proposed to explain this activity. One is that Tsix modulates the chromatin state of the Xist promoter. Evidence supporting this hypothesis is that the Xist promoter exists in different chromatin states in wildtype cells versus cells expressing Tsix truncation constructs [35,36,37], and that Tsix lacking the last exon, which overlaps with the Xist promoter region, fails to regulate Xist [38]. Another possible mechanism is that Tsix forms an RNA duplex with Xist and regulates it through the RNAi pathway [39]. Although Dicer and Tsix reportedly regulate Xist synergistically in mESCs [39], no microRNA has yet been identified from the Xist/Tsix locus. We found that Tsix RNA can also bind the PRC2 complex and have proposed that Tsix controls Xist activity by titrating away Xist-interacting proteins [23]. Furthermore, Tsix couples with several proteins acting as pluripotency factors in early embryos and ESCs to maintain low expression of Xist [40]. (This mechanism is discussed below.) In addition to Tsix, other studies have shown that lncRNAs Xite (X-Inactivation Intergenic Transcription Element) [41] and Tsx (Testis Specific X-linked Gene) [42] repress Xist expression through Tsix activation, while Jpx (also known as Enox (Expressed Neighbor of Xist)) [43] and Ftx (Five prime to Xist) [44] activates Xist RNA through unknown mechanisms.
2.3.2. Pluripotency Factors
Since undifferentiated female ESCs have two active X chromosomes and express low levels of Xist from both, it has long been thought that X-inactivation is coupled to the pluripotency state of mESCs. Recent studies support this hypothesis by showing that pluripotent factors play a repressive role in Xist regulation. Conditional knockout of Oct4 (also known as Pou5f1) or Nanog in male mESCs, where Xist is expressed at low levels and then silenced after differentiation, results in unexpected Xist up-regulation during differentiation [45]. Furthermore, coating of both X chromosomes with Xist is seen in female differentiating ES cells following Oct4 knockdown [46]. The first intron of the Xist locus exhibits binding sites for Oct4, Nanog, and Sox2 [45,47,48]. However, deletion of intron1 results in a small increase in Xist expression, suggesting these factors may alter Xist function by binding other genetic elements [49]. Indeed, Oct4 also directly binds and activates Tsix and Xite to repress Xist [46]. In addition to the pluripotency factors noted above, a recent study found the protein encoded by the newly identified pluripotency gene Prdm14 also represses Xist RNA expression [47].
Interestingly, while one set of pluripotency factors represses Xist, a different set of factors activates Tsix. For example, expression of Klf4, c-Myc, and Rex1 promotes Tsix expression [40], and Rex1 is required for efficient Tsix elongation. Factors such as RNF12 (Ring finger protein 12), an X-linked E3 ubiquitin ligase that targets Rex1 for degradation, can activate Xist expression even in male mESCs [50] and is essential for XCI initiation [51]. Therefore, Tsix and pluripotency factors act synergistically to repress Xist in undifferentiated ESCs.
2.4. Loading and Spreading of Xist on the Inactivated X Chromosome
The most extraordinary feature of Xist is its ability to “coat” almost an entire X-chromosome. How Xist RNAs coat and spread to inactivate the X chromosome remains an open question. Interestingly, naturally occurring or induced X:autosome translocations show different degrees of XCI spread from the X into the autosomal DNA segment, suggesting that specific sequences facilitate spreading [52,53]. These observations prompted investigators to look for potential differences in X-chromosome and autosome sequences. LINE (long interspersed elements) are candidates for mediating this effect due to their higher density on the X chromosome compared to autosomes [54]. The theory proposed by Mary Lyon stated that interspersed repetitive LINE elements act as booster elements to promote spread of Xist RNA [54]. A transgenic study in mESCs showed that chromosome regions with higher LINE density are inactivated more efficiently by a Xist transgene [53]. A recent study suggests that silenced LINE elements contribute to formation of a heterochromatic compartment initiated by Xist RNA and that a particular type of active LINEs may participate in local propagation of the XCI into regions that would otherwise escape [55]. Nonetheless, the exact function of LINE elements in XCI remains to be studied.
In terms of trans-acting factors hnRNPU (also known as SAF-A), a known nuclear scaffold protein, is known to be enriched on the inactive X chromosome [56] and to function in Xist RNA loading [57,58]. A recent study showed that YY1, a RNA/DNA binding protein, tethers Xist RNA to the inactive X [27]. However, since YY1 binding sites are highly abundant throughout the mammalian genome, it is unclear where the specificity of YY1-specific guidance of Xist onto the X-chromosome rather than autosomes lies. Factors mediating this activity remain to be identified.
2.5. Xist Promotes Formation of a Heterochromatic Xi
Xist RNA accumulation on Xi leads to chromatin changes, such as DNA hypermethylation, enrichment of macroH2A, loss of the euchromatic mark H3K4-3me and chromosome-wide establishment of heterochromatic mark H3K27-3me and mono ubiquinated H2A (H2AK119u1) [19,59,60,61,62,63]. How can Xist lead to these global changes? Since Xist coats an entire X, it has long been postulated that Xist RNA binds and carries silencing factors, which are deposited as it coats regions during XCI. Thus, much effort has been applied to identifying Xist-interacting proteins.
The best-studied Xist-interacting trans-factors are Polycomb group proteins. These proteins are highly enriched on the inactive X-chromosome during XCI establishment [60,61,64]. Deletion of some Polycomb proteins, such as Eed, leads to reactivation of the silenced X in extra-embryonic tissues, highlighting their essential role in XCI [65]. However, Polycomb proteins do not seem to affect random X-inactivation in embryos and mESCs, suggesting the existence of a functionally redundant mechanism regulating XCI [66]. Polycomb proteins form two major complexes. One of those, Polycomb Repressive Complex 2 or PRC2, is a histone methyltransferase that trimethylates H3 lysine 27 [67]. Upon recognizing H3K27-3me, another complex, PRC1, is recruited to specific genomic loci to establish ubiquinated histone H2A (H2AK119u1) and compact chromatin for gene silencing. Studies using inducible transgenes showed that PRC2 and PRC1 are localized to Xi in a Xist RNA-dependent manner [68,69]. As discussed earlier, it has been suggested that PRC2 is recruited onto Xi by directly interacting with Xist/RepA lncRNAs via repeat A region [23,26,70]. Furthermore, phosphorylation of Ezh2, the catalytic subunit of PRC2, increases PRC2’s RNA binding ability [71]. Notably, a Xist mutant lacking the A-repeats retains the ability to recruit PRC2 and PRC1 to Xi [60,69], indicating that other Xist sequences can recruit Polycomb proteins directly or indirectly. Interestingly, in cells with the depletion of a key component of PRC2, some PRC1 proteins can localize to induced Xist RNA while others cannot, suggesting Xist recruits PRC1 by both PRC2 dependent and independent mechanisms [69]. The PRC2 independent mechanism is supported by a study showing PRC1 can find its genomic targets in PRC2-deficient cells through the interaction with RYBP protein [72]. Surprisingly, trithorax proteins, such as Ash2L, which typically antagonize Polycomb function and are primarily linked to gene activation, also bind Xist, suggesting that either Trithorax proteins also function in gene repression or that Xist RNA can activate specific genes [58].
2.6. Perspective
Studies of Xist RNA in the past two decades have greatly advanced our understanding of how lncRNAs regulate gene expression epigenetically. However, many questions remain unanswered. For example, it is unclear how Xist specifically spreads along the future Xi but not along the activated X. It is also not yet understood how Xist RNA interacts with so many different proteins and what determines the specificity of those interactions. And since Polycomb proteins do not function in establishment of XCI in embryos, the mechanisms used to establish the repressive state of the Xi are not yet identified.
3. Imprinted lncRNA Gtl2
Genomic imprinting is another dosage regulation mechanism used by mammalian cells. While most autosomal genes are expressed from two parental alleles, some genes are expressed monoallelically in a parent-of-origin manner. This epigenetic phenomenon is termed imprinting. To date, over 100 imprinted genes have been validated in mouse (see the Mouse Imprinting Catalog: http://www.mousebook.org/catalog.php?catalog=imprinting).
Like X-inactivation, genomic imprinting is tightly controlled by epigenetic mechanisms such as DNA methylation and chromatin modification [73]. Interestingly, within a cluster in which imprinted genes are co-regulated, at least one gene encodes a lncRNA, suggesting the importance of lncRNA in regulating this process [74]. Indeed, some imprinted lncRNAs, such as Air and Kcnq1OT1, have been shown to interact with chromatin modifiers to silence reciprocally imprinted protein coding genes [75,76,77]. Here, we will use the imprinted Dlk1-Dio3 locus as an example to assess the functional role of lncRNAs in control of imprinting and discuss possible mechanisms used by Gtl2 (also named Meg3), the maternally expressed lncRNA at the Dlk1-Dio3 locus, to regulate imprinting at this locus.
3.1. Discovery of Gtl2
Gtl2 was discovered following an insertional mutagenesis gene trap screen to isolate genes differentially regulated during mouse embryonic development [78]. The gene trap insertion at a region 2.3 kb upstream from Gtl2 promoter caused dwarfism in mouse offspring only through paternal inheritance, not via maternal transmission [78,79]. Subsequent isolation of the Gtl2 gene suggested that its product was a non-coding RNA (ncRNA) based on the observation that the Gtl2 contained no significant open reading frames [79]. Gtl2 was mapped to a region of mouse chromosome 12 (12qF1) thought to contain imprinted genes, due to the observation that in that region uniparental disomy led to embryonic lethality [80]. The paternal-specific growth defect emerging from the insertional mutation plus observation of decreased Gtl2 expression levels in parthenogenetic embryos led to the hypothesis that Gtl2 is paternally imprinted [79]. However, soon after, several independent studies demonstrated that Gtl2 is in fact expressed exclusively from the maternal allele [81,82,83]. The growth phenotype observed following the insertional mutagenesis screen was later suggested to result from perturbations in imprinting of the whole locus, particularly the down regulation of several paternally expressed protein coding genes [84,85].
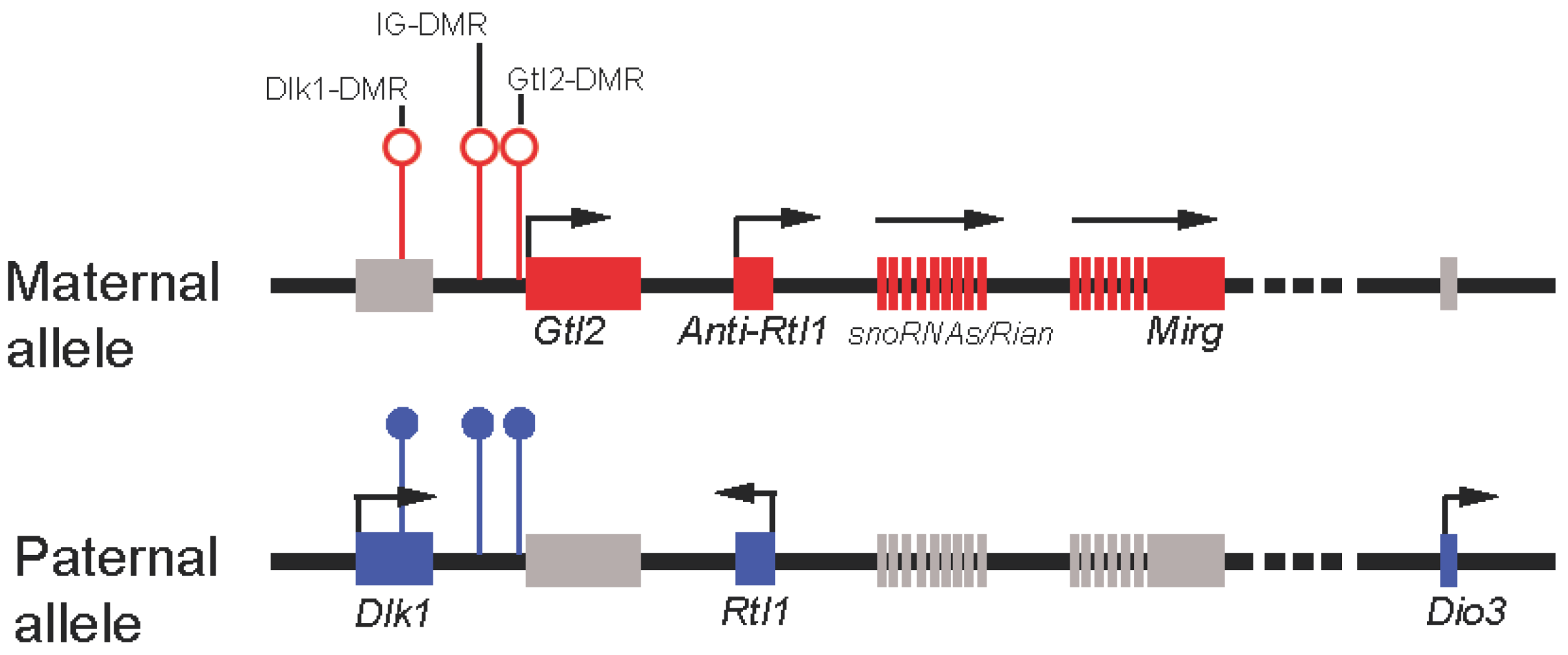
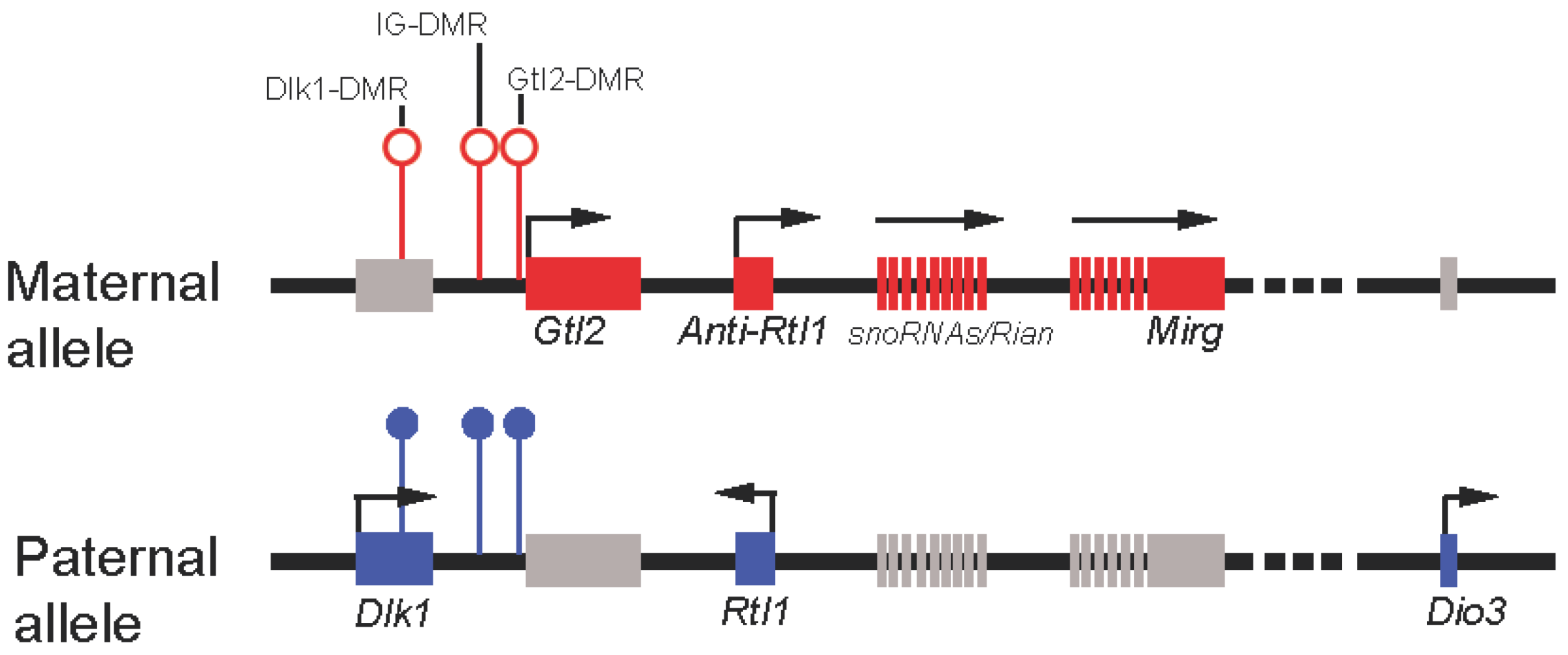
Not long after the discovery of Gtl2, other imprinted genes that form a cluster and are co-regulated with Gtl2 were also identified [81,83,86,87,88,89]. Interestingly, genes preferentially expressed from the paternally-inherited chromosome are all protein-coding genes, including Dlk1, Rtl1, and Dio3 (Figure 2), whereas genes expressed maternally all encode ncRNAs, namely, Gtl2, Anti-Rtl1, Rian, Mirg, and a large cluster of multiple snoRNAs/microRNAs (Figure 2). All maternally-expressed ncRNAs in this locus are transcribed in the same orientation. This combined with tissue-specific expression of those ncRNAs suggested coordinated expression of the maternally-expressed genes at this locus [90]. It has also been suggested that all of those ncRNAs, with the exception of Gtl2, may come from a long polycistronic transcript [90]. Imprinting is typically regulated by a cis-element called the differentially methylated region (DMR). Three DMRs, located between Dlk1 and Gtl2 (hence intergenic DMR or IG-DMR), at the Gtl2 promoter, and downstream of Dlk1 have been identified [91]. While IG-DMR has been shown to regulate the imprinting status of all genes at this locus, the function of the other DMRs remains poorly understood.
Figure 2.
Schematic representation of the mouse imprinted locus Dlk1-Dio3. Red rectangles represent maternally-expressed genes, and blue represent paternally-expressed genes. Gray rectangles represent repressed genes. Open circles represent unmethylated DMRs, and filled represent methylated DMRs.
Figure 2.
Schematic representation of the mouse imprinted locus Dlk1-Dio3. Red rectangles represent maternally-expressed genes, and blue represent paternally-expressed genes. Gray rectangles represent repressed genes. Open circles represent unmethylated DMRs, and filled represent methylated DMRs.
3.2. Gtl2 Function
3.2.1. Gtl2 Regulates Genomic Imprinting
Genetic evidence from Gtl2 knockout mice suggests that Gtl2 regulates genomic imprinting. One study showed that mice with maternal deletion (300 bp of the promoter and exons 1–5) of the Gtl2 gene show bidirectional loss of imprinting of all genes in the Dlk1-Dio3 locus, whereas no effect was detected when the deletion was transmitted paternally, suggesting that Gtl2 controls genomic imprinting at this locus [92]. Gtl2 knockout mice phenocopy IG-DMR knockout mice [92,93], implying that Gtl2 (and/or its downstream ncRNAs) potentially regulates Dlk1-Dio3 imprinting. Indeed, the same study showed that maternal deletion of Gtl2 resulted in increased IG-DMR methylation on the maternal allele [92]. Thus, Gtl2 and/or its downstream ncRNAs may function to maintain the unmethylated status of the maternal IG-DMR through an unknown mechanism. Interestingly, an earlier study deleting ~4Kb of the Gtl2 promoter plus exons 1–5 yielded significantly different results [94]. The authors did not observe a change in DNA methylation or the imprinted status of any genes within this region when the deletion was transmitted maternally. However, the aberrant imprinted status of Dlk1, Rian, and Mirg was detected when deletion mutation was transmitted paternally. The difference between these two studies could lie in the variable size of the deletion of the Gtl2 promoter (300 bp vs.~4 Kb). Since the region flanking the Gtl2 promoter is a well-established DMR, these discrepant results suggest a functional role for the Gtl2 DMR and underscore the importance of studying Gtl2 function using approaches that minimally perturb the DMR, such as RNAi.
3.2.2. Gtl2 Regulates Cellular Reprogramming
A recent study showed that mouse induced Pluripotent Stem Cells (iPSCs) in which Gtl2 and its downstream ncRNAs were aberrantly silenced contributed poorly to chimaeras and failed to generate mice in which every cell is contributed by an iPSC (all-iPSC mice) [95]. In contrast, iPSCs showing normal expression of lncRNAs contributed to high-grade chimaeras and generated viable all-iPSC mice [95]. Further investigation of possible mechanisms underlying repression of maternally imprinted genes revealed that in the iPSCs showing aberrant silencing of lncRNAs from this locus, the maternal IG-DMR was hypermethylated and active histone marks such as H3/H4 acetylation and H3K4 methylation were absent at the Gtl2 promoter [95]. In a subsequent publication, these authors reported that ascorbic acid facilitates cellular reprogramming by preventing silencing of Gtl2 and downstream lncRNAs [96]. These findings highlight a role for Gtl2 in preserving the developmental potential of iPSCs during reprogramming.
3.2.3. Gtl2 Functions in Tumorigenesis
Loss of GTL2/MEG3 is seen in many tumor types, including human pituitary adenoma, brain tumors, and liver tumors [97,98,99]. GTL2/MEG3 overexpression inhibits tumor cell proliferation in vitro [98,99,100,101,102]. Interestingly, comparison of gene expression profiles in brain between Gtl2 knockout and wild-type mice revealed an upregulation of genes correlated with angiogenesis in knockout mouse [103], suggesting Gtl2 may function as a tumor suppressor in part by inhibiting angiogenesis. Furthermore, it was suggested that a postulated tumor-suppressing function of Gtl2 might stem from its ability to induce p53 accumulation, thereby activating expression of some p53 target genes [99,100,101].
3.3. Molecular Mechanisms Potentially Underlying Gtl2 Regulation of the Dlk1-Dio3 Locus
lncRNAs employ diverse mechanisms to regulate gene expression at both the transcriptional and post-transcriptional level (reviewed in [104]). Here, we assess possible molecular mechanisms used by Gtl2 to regulate genes within the Dlk1-Dio3 locus. The modes of regulation considered are not mutually exclusive: Gtl2 function might be exerted through combinatorial mechanisms or carried out differentially in response to specific environmental cues.
3.3.1. Gtl2 Maintains Dlk1-Dio3 Imprinting By Protecting the Maternal IG-DMR from Methylation
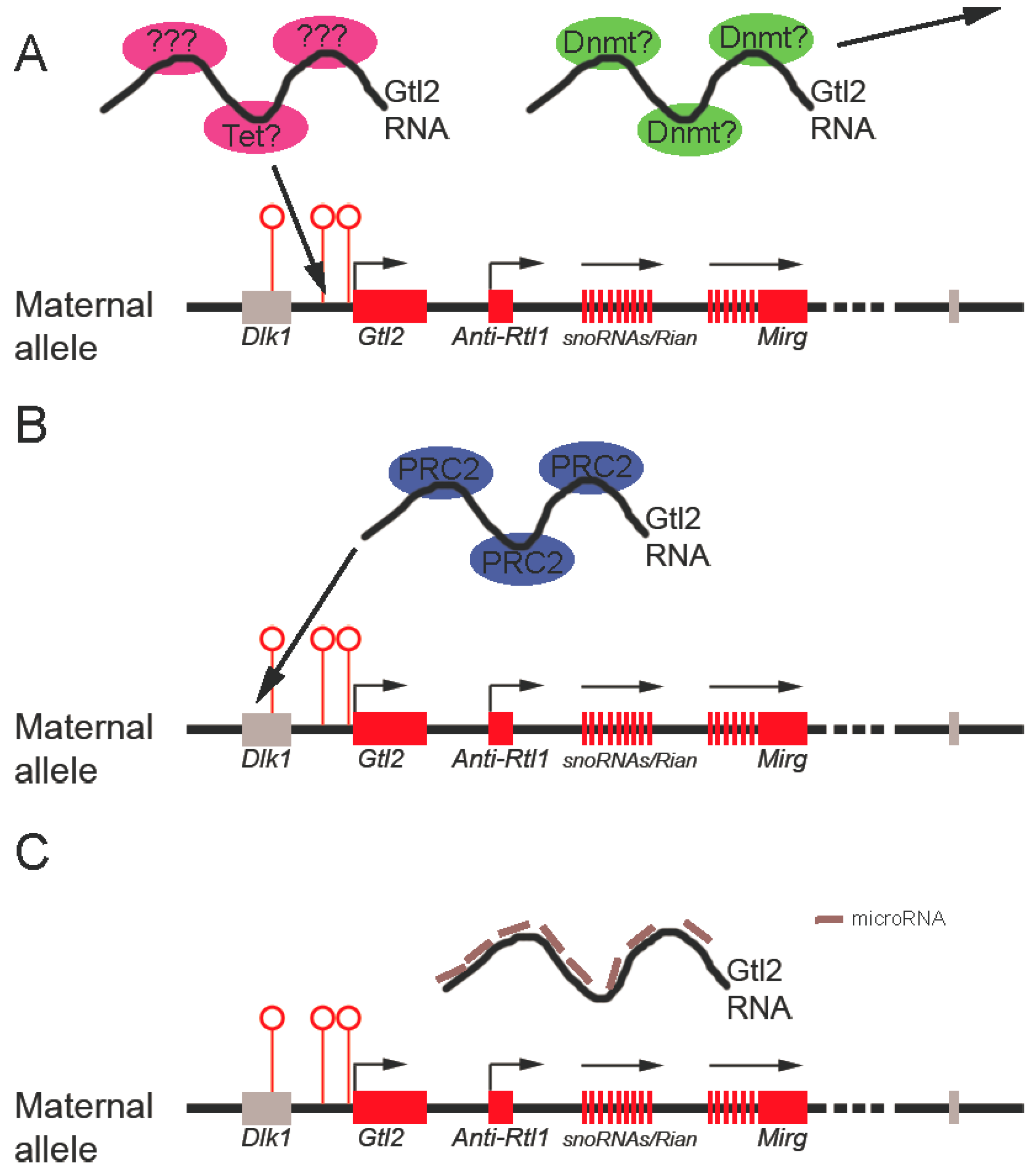
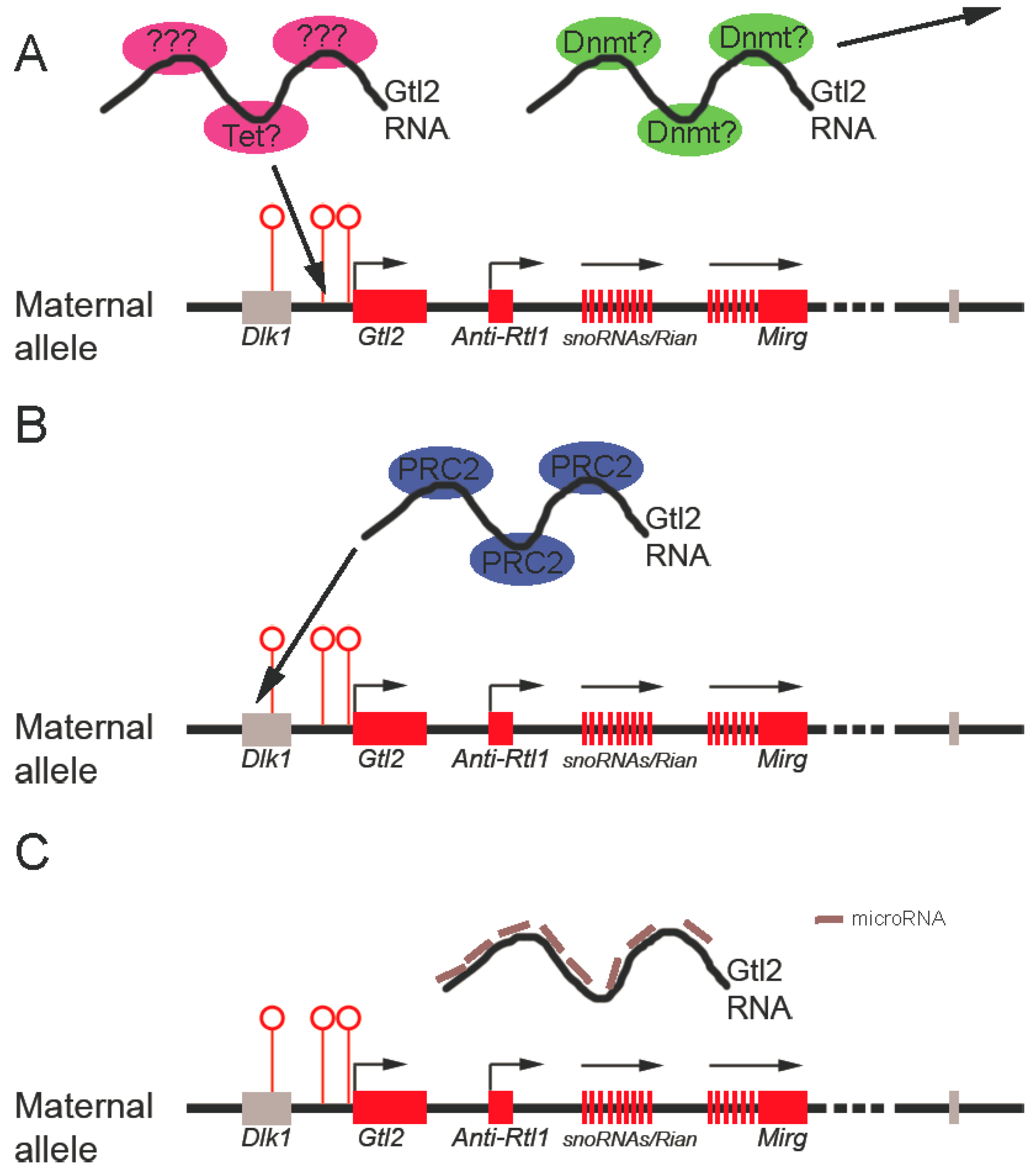
Maintaining the proper differential methylation status of the germline DMR is crucial for mono-allelic expression within an imprinting cluster. The observation that targeted deletion of the IG-DMR from the mouse Dlk1-Dio3 locus caused loss of imprinting when the deletion occurred at the maternal rather than the paternal allele demonstrated that the maternally-inherited unmethylated DMR is essential for the imprinting status of the locus [93]. That Gtl2 knockdown results in hypermethylation of the maternal IG-DMR suggests that Gtl2 is required to maintain its unmethylated status. Potential mechanisms include Gtl2 RNA interaction with and sequestration of positive DNA methylation regulators (such as DNA methyltransferases) or by Gtl2 actively recruiting negative regulators of DNA demethylation (such as Tet proteins) to keep IG-DMR unmethylated (Figure 3A). These mechanisms are not yet known.
Figure 3.
Possible mechanisms underlying Gtl2 regulation of the imprinted Dlk1-Dio3 locus. A. Gtl2 may function to maintain the unmethylated status of the IG-DMR by titrating away DNA methyltransferases (Dnmt in the figure) and/or by recruiting DNA demethylation machinery (such as Tet or unknown proteins). B. Gtl2 could silence Dlk1 expression by recruiting PRC2 complexes to the Dlk1 genomic region. C. Finally, Gtl2 may serve as competitive endogenous RNA (ceRNA) to keep maternally-expressed genes active by titrating away inhibitory microRNAs targeting those genes. Red rectangles represent maternally-expressed genes. Gray rectangles represent repressed genes. Open circles represent unmethylated DMRs.
Figure 3.
Possible mechanisms underlying Gtl2 regulation of the imprinted Dlk1-Dio3 locus. A. Gtl2 may function to maintain the unmethylated status of the IG-DMR by titrating away DNA methyltransferases (Dnmt in the figure) and/or by recruiting DNA demethylation machinery (such as Tet or unknown proteins). B. Gtl2 could silence Dlk1 expression by recruiting PRC2 complexes to the Dlk1 genomic region. C. Finally, Gtl2 may serve as competitive endogenous RNA (ceRNA) to keep maternally-expressed genes active by titrating away inhibitory microRNAs targeting those genes. Red rectangles represent maternally-expressed genes. Gray rectangles represent repressed genes. Open circles represent unmethylated DMRs.
3.3.2. Gtl2 silences Gene Expression through Recruitment of Chromatin Modifying Machinery
The role of histone modification in regulating imprinting of the Dlk1-Dio3 locus is less studied than that of DNA methylation. Nevertheless, one study showed that in midgestation mouse embryos, differential histone acetylation occurred between the maternal and paternal alleles of the Gtl2-DMR, but not of the IG-DMR [105]. The Gtl2-DMR of the active maternal allele was hyperacetylated on H3 and H4, while the silent paternal allele showed a low level of acetylation of both histones. A more recent study that characterized the allele-specific epigenetic profile of the Gtl2-DMR in greater detail confirmed the allele-specific acetylation pattern and revealed enrichment of the active histone methylation mark (H3K4 methylation) in the maternal allele in midgestation mouse embryos [106]. It is not clear whether this differential histone modification is instructive for imprinting, or simply reflects the transcriptional state of the genes.
Our recent studies demonstrate that Gtl2 directly binds PRC2, and that Gtl2 knockdown in mouse ESCs results in a reduced Ezh2 occupancy at the Dlk1 promoter and increased Dlk1 expression [107]. This data suggests that Gtl2 can silence gene expression from the maternal allele of the Dlk1-Dio3 locus through chromatin modification (Figure 3B). It is currently unclear that how Gtl2 recruits Ezh2 to chromatin and whether Gtl2 uses the same mechanism to silence genes other than Dlk1 within the same imprinted locus.
3.3.3. Gtl2 Acts as a ceRNA to Maintain Expression of Maternally-Expressed ncRNAs
The observation that Gtl2 deletion abolishes expression of downstream lncRNAs and that expression of these RNAs is coordinated suggests that Gtl2 functions to maintain expression of other maternally-expressed ncRNAs from the Dlk1-Dio3 locus [90,93]. The recently proposed “competitive endogenous RNA” (ceRNA) theory proposes that RNA transcripts that contain sequences similar to microRNA response elements (MREs) may compete with that pool of microRNAs and alter their availability in cells, thus modulating the level of transcripts targeted by those microRNAs [108]. It has also been suggested that ncRNAs may serve as more efficient ceRNAs than protein-coding RNAs because the binding of microRNA to lncRNAs are not interfered by translation [109]. Indeed, a recent study showed that a muscle-specific lincRNA, linc-MD1, can act as a sponge to titrate away miR-133 and regulate the expression of key transcription factors governing muscle differentiation, suggesting the feasibility of this model [110]. Therefore, Gtl2 may serve as a ceRNA to titrate away microRNAs targeting other maternally-expressed transcripts at the same locus (Figure 3C). However, as yet, no study has analyzed potential MREs shared between Gtl2 and those maternally-expressed transcripts. It will be of interest to determine whether lncRNAs regulate each other through this novel mechanism.
4. Conclusions
Since the completion of the Human Genome Project, our perception of the mammalian genome has undergone a dramatic shift. The number of non protein-coding transcripts identified over the past decade has increased exponentially. We now realize that lncRNA is far from the “dark matter” of the genome, but instead plays a critical function in gene regulation. Since XCI and genomic imprinting are two well-defined molecular activities, lncRNAs from XIC or imprinted loci provide ideal systems to understand epigenetic control of how genes are regulated. The mechanisms identified will likely be applicable to newly discovered lncRNAs.
Acknowledgements
This work is supported by an AACR-Aflac, Inc. Career Development Award for Pediatric Cancer Research (J.C.Z.) and a CIRM Training Grant (Y.W.).
References
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; Kondo, S.; Nikaido, I.; Osato, N.; Saito, R.; Suzuki, H.; et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cdnas. Nature 2002, 420, 563–573. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar]
- Barr, M.L.; Bertram, E.G. A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 1949, 163, 676. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene action in the x-chromosome of the mouse (mus musculus l). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Barakat, T.S.; Gribnau, J. X chromosome inactivation in the cycle of life. Development 2012, 139, 2085–2089. [Google Scholar] [CrossRef]
- Gribnau, J.; Grootegoed, J.A. Origin and evolution of x chromosome inactivation. Curr. Opin. Cell Biol. 2012, 24, 397–404. [Google Scholar] [CrossRef]
- Lee, J.T. Gracefully ageing at 50, x-chromosome inactivation becomes a paradigm for rna and chromatin control. Nat. Rev. Mol. Cell Biol. 2011, 12, 815–826. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Heard, E. Fifty years of x-inactivation research. Development 2011, 138, 5049–5055. [Google Scholar] [CrossRef]
- Agrelo, R.; Wutz, A. Cancer progenitors and epigenetic contexts: An xisting connection. Epigenetics: Official J. DNA Methyl. Soc. 2009, 4, 568–570. [Google Scholar] [CrossRef]
- Rastan, S. Non-random x-chromosome inactivation in mouse x-autosome translocation embryos--location of the inactivation centre. J. Embryol. Exp. Morphol. 1983, 78, 1–22. [Google Scholar]
- Rastan, S.; Robertson, E.J. X-chromosome deletions in embryo-derived (ek) cell lines associated with lack of x-chromosome inactivation. J. Embryol. Exp. Morphol. 1985, 90, 379–388. [Google Scholar]
- Brown, C.J.; Lafreniere, R.G.; Powers, V.E.; Sebastio, G.; Ballabio, A.; Pettigrew, A.L.; Ledbetter, D.H.; Levy, E.; Craig, I.W.; Willard, H.F. Localization of the x inactivation centre on the human x chromosome in xq13. Nature 1991, 349, 82–84. [Google Scholar]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human x inactivation centre is expressed exclusively from the inactive x chromosome. Nature 1991, 349, 38–44. [Google Scholar]
- Borsani, G.; Tonlorenzi, R.; Simmler, M.C.; Dandolo, L.; Arnaud, D.; Capra, V.; Grompe, M.; Pizzuti, A.; Muzny, D.; Lawrence, C.; et al. Characterization of a murine gene expressed from the inactive x chromosome. Nature 1991, 351, 325–329. [Google Scholar]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; Cooper, P.; Smith, S.; McCabe, V.M.; Norris, D.P.; Penny, G.D.; Patel, D.; Rastan, S. Conservation of position and exclusive expression of mouse xist from the inactive x chromosome. Nature 1991, 351, 329–331. [Google Scholar]
- Brown, C.J.; Willard, H.F. The human x-inactivation centre is not required for maintenance of x-chromosome inactivation. Nature 1994, 368, 154–156. [Google Scholar]
- Csankovszki, G.; Panning, B.; Bates, B.; Pehrson, J.R.; Jaenisch, R. Conditional deletion of xist disrupts histone macroh2a localization but not maintenance of x inactivation. Nat. Genet. 1999, 22, 323–324. [Google Scholar] [CrossRef]
- Wutz, A.; Jaenisch, R. A shift from reversible to irreversible x inactivation is triggered during es cell differentiation. Mol. Cell 2000, 5, 695–705. [Google Scholar]
- Wutz, A. Gene silencing in x-chromosome inactivation: Advances in understanding facultative heterochromatin formation. Nat. Rev. Genet. 2011, 12, 542–553. [Google Scholar] [CrossRef]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of xist rna. Nat. Genet. 2002, 30, 167–174. [Google Scholar]
- Hoki, Y.; Kimura, N.; Kanbayashi, M.; Amakawa, Y.; Ohhata, T.; Sasaki, H.; Sado, T. A proximal conserved repeat in the xist gene is essential as a genomic element for x-inactivation in mouse. Development 2009, 136, 139–146. [Google Scholar]
- Royce-Tolland, M.E.; Andersen, A.A.; Koyfman, H.R.; Talbot, D.J.; Wutz, A.; Tonks, I.D.; Kay, G.F.; Panning, B. The a-repeat links asf/sf2-dependent xist rna processing with random choice during x inactivation. Nat. Struct. Mol. Biol. 2010, 17, 948–954. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat rna to the mouse x chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef]
- Duszczyk, M.M.; Zanier, K.; Sattler, M. A nmr strategy to unambiguously distinguish nucleic acid hairpin and duplex conformations applied to a xist rna a-repeat. NucleicAcids Res. 2008, 36, 7068–7077. [Google Scholar] [CrossRef]
- Duszczyk, M.M.; Sattler, M. (1)h, (1)(3)c, (1)(5)n and (3)(1)p chemical shift assignments of a human xist rna a-repeat tetraloop hairpin essential for x-chromosome inactivation. Biomolecular NMRAssign. 2012, 6, 75–77. [Google Scholar] [CrossRef]
- Maenner, S.; Blaud, M.; Fouillen, L.; Savoye, A.; Marchand, V.; Dubois, A.; Sanglier-Cianferani, S.; Van Dorsselaer, A.; Clerc, P.; Avner, P.; et al. 2-d structure of the a region of xist rna and its implication for prc2 association. PLoS Biol. 2010, 8, e1000276. [Google Scholar] [CrossRef]
- Jeon, Y.; Lee, J.T. Yy1 tethers xist rna to the inactive x nucleation center. Cell 2011, 146, 119–133. [Google Scholar] [CrossRef]
- Beletskii, A.; Hong, Y.K.; Pehrson, J.; Egholm, M.; Strauss, W.M. Pna interference mapping demonstrates functional domains in the noncoding rna xist. Proc. Natl. Acad. Sci. USA 2001, 98, 9215–9220. [Google Scholar] [CrossRef]
- Sarma, K.; Levasseur, P.; Aristarkhov, A.; Lee, J.T. Locked nucleic acids (lnas) reveal sequence requirements and kinetics of xist rna localization to the x chromosome. Proc. Natl. Acad. Sci. USA 2010, 107, 22196–22201. [Google Scholar]
- Clerc, P.; Avner, P. Role of the region 3' to xist exon 6 in the counting process of x-chromosome inactivation. Nat. Genet. 1998, 19, 249–253. [Google Scholar] [CrossRef]
- Lee, J.T.; Davidow, L.S.; Warshawsky, D. Tsix, a gene antisense to xist at the x-inactivation centre. Nat. Genet. 1999, 21, 400–404. [Google Scholar] [CrossRef]
- Sado, T.; Wang, Z.; Sasaki, H.; Li, E. Regulation of imprinted x-chromosome inactivation in mice by tsix. Development 2001, 128, 1275–1286. [Google Scholar]
- Morey, C.; Arnaud, D.; Avner, P.; Clerc, P. Tsix-mediated repression of xist accumulation is not sufficient for normal random x inactivation. Hum. Mol. Genet. 2001, 10, 1403–1411. [Google Scholar]
- Morey, C.; Navarro, P.; Debrand, E.; Avner, P.; Rougeulle, C.; Clerc, P. The region 3' to xist mediates x chromosome counting and h3 lys-4 dimethylation within the xist gene. EMBO J. 2004, 23, 594–604. [Google Scholar]
- Navarro, P.; Page, D.R.; Avner, P.; Rougeulle, C. Tsix-mediated epigenetic switch of a ctcf-flanked region of the xist promoter determines the xist transcription program. Genes Dev. 2006, 20, 2787–2792. [Google Scholar] [CrossRef]
- Sun, B.K.; Deaton, A.M.; Lee, J.T. A transient heterochromatic state in xist preempts x inactivation choice without rna stabilization. Mol. Cell 2006, 21, 617–628. [Google Scholar] [CrossRef]
- Navarro, P.; Pichard, S.; Ciaudo, C.; Avner, P.; Rougeulle, C. Tsix transcription across the xist gene alters chromatin conformation without affecting xist transcription: Implications for x-chromosome inactivation. Genes Dev. 2005, 19, 1474–1484. [Google Scholar] [CrossRef]
- Ohhata, T.; Hoki, Y.; Sasaki, H.; Sado, T. Crucial role of antisense transcription across the xist promoter in tsix-mediated xist chromatin modification. Development 2008, 135, 227–235. [Google Scholar]
- Ogawa, Y.; Sun, B.K.; Lee, J.T. Intersection of the rna interference and x-inactivation pathways. Science 2008, 320, 1336–1341. [Google Scholar]
- Navarro, P.; Oldfield, A.; Legoupi, J.; Festuccia, N.; Dubois, A.; Attia, M.; Schoorlemmer, J.; Rougeulle, C.; Chambers, I.; Avner, P. Molecular coupling of tsix regulation and pluripotency. Nature 2010, 468, 457–460. [Google Scholar]
- Ogawa, Y.; Lee, J.T. Xite, x-inactivation intergenic transcription elements that regulate the probability of choice. Mol. Cell 2003, 11, 731–743. [Google Scholar] [CrossRef]
- Anguera, M.C.; Ma, W.; Clift, D.; Namekawa, S.; Kelleher, R.J., 3rd; Lee, J.T. Tsx produces a long noncoding rna and has general functions in the germline, stem cells, and brain. PLoS Genet. 2011, 7, e1002248. [Google Scholar] [CrossRef]
- Tian, D.; Sun, S.; Lee, J.T. The long noncoding rna, jpx, is a molecular switch for x chromosome inactivation. Cell 2010, 143, 390–403. [Google Scholar]
- Chureau, C.; Chantalat, S.; Romito, A.; Galvani, A.; Duret, L.; Avner, P.; Rougeulle, C. Ftx is a non-coding rna which affects xist expression and chromatin structure within the x-inactivation center region. Hum. Mol. Genet. 2011, 20, 705–718. [Google Scholar] [CrossRef]
- Navarro, P.; Chambers, I.; Karwacki-Neisius, V.; Chureau, C.; Morey, C.; Rougeulle, C.; Avner, P. Molecular coupling of xist regulation and pluripotency. Science 2008, 321, 1693–1695. [Google Scholar] [CrossRef]
- Donohoe, M.E.; Silva, S.S.; Pinter, S.F.; Xu, N.; Lee, J.T. The pluripotency factor oct4 interacts with ctcf and also controls x-chromosome pairing and counting. Nature 2009, 460, 128–132. [Google Scholar]
- Ma, Z.; Swigut, T.; Valouev, A.; Rada-Iglesias, A.; Wysocka, J. Sequence-specific regulator prdm14 safeguards mouse escs from entering extraembryonic endoderm fates. Nat. Struct. Mol. Biol. 2011, 18, 120–127. [Google Scholar] [CrossRef]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microrna genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef]
- Nesterova, T.B.; Senner, C.E.; Schneider, J.; Alcayna-Stevens, T.; Tattermusch, A.; Hemberger, M.; Brockdorff, N. Pluripotency factor binding and tsix expression act synergistically to repress xist in undifferentiated embryonic stem cells. Epigenet. Chromatin 2011, 4, 17. [Google Scholar] [CrossRef]
- Jonkers, I.; Barakat, T.S.; Achame, E.M.; Monkhorst, K.; Kenter, A.; Rentmeester, E.; Grosveld, F.; Grootegoed, J.A.; Gribnau, J. Rnf12 is an x-encoded dose-dependent activator of x chromosome inactivation. Cell 2009, 139, 999–1011. [Google Scholar]
- Shin, J.; Bossenz, M.; Chung, Y.; Ma, H.; Byron, M.; Taniguchi-Ishigaki, N.; Zhu, X.; Jiao, B.; Hall, L.L.; Green, M.R.; et al. Maternal rnf12/rlim is required for imprinted x-chromosome inactivation in mice. Nature 2010, 467, 977–981. [Google Scholar]
- Surralles, J.; Natarajan, A.T. Position effect of translocations involving the inactive x chromosome: Physical linkage to xic/xist does not lead to long-range de novo inactivation in human differentiated cells. Cytogenet. Cell Genet. 1998, 82, 58–66. [Google Scholar]
- Popova, B.C.; Tada, T.; Takagi, N.; Brockdorff, N.; Nesterova, T.B. Attenuated spread of x-inactivation in an x;autosome translocation. Proc. Natl. Acad. Sci. USA 2006, 103, 7706–7711. [Google Scholar]
- Lyon, M.F. X-chromosome inactivation: A repeat hypothesis. Cytogenet. Cell Genet. 1998, 80, 133–137. [Google Scholar] [CrossRef]
- Chow, J.C.; Ciaudo, C.; Fazzari, M.J.; Mise, N.; Servant, N.; Glass, J.L.; Attreed, M.; Avner, P.; Wutz, A.; Barillot, E.; et al. Line-1 activity in facultative heterochromatin formation during x chromosome inactivation. Cell 2010, 141, 956–969. [Google Scholar]
- Helbig, R.; Fackelmayer, F.O. Scaffold attachment factor a (saf-a) is concentrated in inactive x chromosome territories through its rgg domain. Chromosoma 2003, 112, 173–182. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Brockdorff, N.; Kawano, S.; Tsutui, K.; Nakagawa, S. The matrix protein hnrnp u is required for chromosomal localization of xist rna. Dev. Cell 2010, 19, 469–476. [Google Scholar] [CrossRef]
- Pullirsch, D.; Hartel, R.; Kishimoto, H.; Leeb, M.; Steiner, G.; Wutz, A. The trithorax group protein ash2l and saf-a are recruited to the inactive x chromosome at the onset of stable x inactivation. Development 2010, 137, 935–943. [Google Scholar] [CrossRef]
- Lucchesi, J.C.; Kelly, W.G.; Panning, B. Chromatin remodeling in dosage compensation. Annu. Rev. Genet. 2005, 39, 615–651. [Google Scholar]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.; de la Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of histone h3 lysine 27 methylation in x inactivation. Science 2003, 300, 131–135. [Google Scholar]
- Silva, J.; Mak, W.; Zvetkova, I.; Appanah, R.; Nesterova, T.B.; Webster, Z.; Peters, A.H.; Jenuwein, T.; Otte, A.P.; Brockdorff, N. Establishment of histone h3 methylation on the inactive x chromosome requires transient recruitment of eed-enx1 polycomb group complexes. Dev. Cell 2003, 4, 481–495. [Google Scholar]
- Fang, J.; Chen, T.; Chadwick, B.; Li, E.; Zhang, Y. Ring1b-mediated h2a ubiquitination associates with inactive x chromosomes and is involved in initiation of x inactivation. J. Biol. Chem. 2004, 279, 52812–52815. [Google Scholar] [CrossRef]
- de Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins ring1a/b link ubiquitylation of histone h2a to heritable gene silencing and x inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar]
- Okamoto, I.; Otte, A.P.; Allis, C.D.; Reinberg, D.; Heard, E. Epigenetic dynamics of imprinted x inactivation during early mouse development. Science 2004, 303, 644–649. [Google Scholar] [CrossRef]
- Kalantry, S.; Mills, K.C.; Yee, D.; Otte, A.P.; Panning, B.; Magnuson, T. The polycomb group protein eed protects the inactive x-chromosome from differentiation-induced reactivation. Nat. Cell Biol. 2006, 8, 195–202. [Google Scholar] [CrossRef]
- Kalantry, S.; Magnuson, T. The polycomb group protein eed is dispensable for the initiation of random x-chromosome inactivation. PLoS Genet. 2006, 2, e66. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The polycomb complex prc2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Kohlmaier, A.; Savarese, F.; Lachner, M.; Martens, J.; Jenuwein, T.; Wutz, A. A chromosomal memory triggered by xist regulates histone methylation in x inactivation. PLoS Biol. 2004, 2, E171. [Google Scholar] [CrossRef] [Green Version]
- Schoeftner, S.; Sengupta, A.K.; Kubicek, S.; Mechtler, K.; Spahn, L.; Koseki, H.; Jenuwein, T.; Wutz, A. Recruitment of prc1 function at the initiation of x inactivation independent of prc2 and silencing. EMBO J. 2006, 25, 3110–3122. [Google Scholar] [CrossRef]
- Kanhere, A.; Viiri, K.; Araujo, C.C.; Rasaiyaah, J.; Bouwman, R.D.; Whyte, W.A.; Pereira, C.F.; Brookes, E.; Walker, K.; Bell, G.W.; et al. Short rnas are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell 2010, 38, 675–688. [Google Scholar]
- Kaneko, S.; Li, G.; Son, J.; Xu, C.F.; Margueron, R.; Neubert, T.A.; Reinberg, D. Phosphorylation of the prc2 component ezh2 is cell cycle-regulated and up-regulates its binding to ncrna. Genes Dev. 2010, 24, 2615–2620. [Google Scholar] [CrossRef]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. Rybp-prc1 complexes mediate h2a ubiquitylation at polycomb target sites independently of prc2 and h3k27me3. Cell 2012, 148, 664–678. [Google Scholar] [CrossRef]
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [CrossRef]
- Mohammad, F.; Mondal, T.; Kanduri, C. Epigenetics of imprinted long noncoding rnas. Epigenetics 2009, 4, 277–286. [Google Scholar] [CrossRef]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The air noncoding rna epigenetically silences transcription by targeting g9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef]
- Korostowski, L.; Sedlak, N.; Engel, N. The kcnq1ot1 long non-coding rna affects chromatin conformation and expression of kcnq1, but does not regulate its imprinting in the developing heart. PLoS Genet. 2012, 8, e1002956. [Google Scholar] [CrossRef]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding rna mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar]
- Schuster-Gossler, K.; Simon-Chazottes, D.; Guenet, J.L.; Zachgo, J.; Gossler, A. Gtl2lacz, an insertional mutation on mouse chromosome 12 with parental origin-dependent phenotype. Mammal. Genome : Official J. Int. Mammal. Genome Soc. 1996, 7, 20–24. [Google Scholar] [CrossRef]
- Schuster-Gossler, K.; Bilinski, P.; Sado, T.; Ferguson-Smith, A.; Gossler, A. The mouse gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an rna. Dev. Dyn. 1998, 212, 214–228. [Google Scholar] [CrossRef]
- Cattanach, B.M.; Rasberry, C. Evidence of imprinting involving the distal region of chr. 12. Mouse Genome 1994, 91, 858. [Google Scholar]
- Schmidt, J.V.; Matteson, P.G.; Jones, B.K.; Guan, X.J.; Tilghman, S.M. The dlk1 and gtl2 genes are linked and reciprocally imprinted. GenesDevelop. 2000, 14, 1997–2002. [Google Scholar]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, meg3/gtl2 and its human homologue meg3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000, 5, 211–220. [Google Scholar] [CrossRef]
- Takada, S.; Tevendale, M.; Baker, J.; Georgiades, P.; Campbell, E.; Freeman, T.; Johnson, M.H.; Paulsen, M.; Ferguson-Smith, A.C. Delta-like and gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr. Biol. 2000, 10, 1135–1138. [Google Scholar] [CrossRef]
- Sekita, Y.; Wagatsuma, H.; Irie, M.; Kobayashi, S.; Kohda, T.; Matsuda, J.; Yokoyama, M.; Ogura, A.; Schuster-Gossler, K.; Gossler, A.; et al. Aberrant regulation of imprinted gene expression in gtl2lacz mice. Cytogenet. Genome Res. 2006, 113, 223–229. [Google Scholar] [CrossRef]
- Steshina, E.Y.; Carr, M.S.; Glick, E.A.; Yevtodiyenko, A.; Appelbe, O.K.; Schmidt, J.V. Loss of imprinting at the dlk1-gtl2 locus caused by insertional mutagenesis in the gtl2 5' region. BMC Genet. 2006, 7, 44. [Google Scholar]
- Seitz, H.; Youngson, N.; Lin, S.P.; Dalbert, S.; Paulsen, M.; Bachellerie, J.P.; Ferguson-Smith, A.C.; Cavaille, J. Imprinted microrna genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat. Genet. 2003, 34, 261–262. [Google Scholar] [CrossRef]
- Tsai, C.E.; Lin, S.P.; Ito, M.; Takagi, N.; Takada, S.; Ferguson-Smith, A.C. Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr. Biol. 2002, 12, 1221–1226. [Google Scholar]
- Yevtodiyenko, A.; Carr, M.S.; Patel, N.; Schmidt, J.V. Analysis of candidate imprinted genes linked to dlk1-gtl2 using a congenic mouse line. Mammal. Genome: Official J. Int. Mammal. Genome Soc. 2002, 13, 633–638. [Google Scholar] [CrossRef]
- Cavaille, J.; Seitz, H.; Paulsen, M.; Ferguson-Smith, A.C.; Bachellerie, J.P. Identification of tandemly-repeated c/d snorna genes at the imprinted human 14q32 domain reminiscent of those at the prader-willi/angelman syndrome region. Hum. Mol. Genet. 2002, 11, 1527–1538. [Google Scholar]
- Tierling, S.; Dalbert, S.; Schoppenhorst, S.; Tsai, C.E.; Oliger, S.; Ferguson-Smith, A.C.; Paulsen, M.; Walter, J. High-resolution map and imprinting analysis of the gtl2-dnchc1 domain on mouse chromosome 12. Genomics 2006, 87, 225–235. [Google Scholar]
- Geuns, E.; De Temmerman, N.; Hilven, P.; Van Steirteghem, A.; Liebaers, I.; De Rycke, M. Methylation analysis of the intergenic differentially methylated region of dlk1-gtl2 in human. Eur. J. Hum. Genet. 2007, 15, 352–361. [Google Scholar] [CrossRef]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar]
- Lin, S.P.; Youngson, N.; Takada, S.; Seitz, H.; Reik, W.; Paulsen, M.; Cavaille, J.; Ferguson-Smith, A.C. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the dlk1-gtl2 imprinted cluster on mouse chromosome 12. Nat. Genet. 2003, 35, 97–102. [Google Scholar] [CrossRef]
- Takahashi, N.; Okamoto, A.; Kobayashi, R.; Shirai, M.; Obata, Y.; Ogawa, H.; Sotomaru, Y.; Kono, T. Deletion of gtl2, imprinted non-coding rna, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 2009, 18, 1879–1888. [Google Scholar]
- Stadtfeld, M.; Apostolou, E.; Akutsu, H.; Fukuda, A.; Follett, P.; Natesan, S.; Kono, T.; Shioda, T.; Hochedlinger, K. Aberrant silencing of imprinted genes on chromosome 12qf1 in mouse induced pluripotent stem cells. Nature 2010, 465, 175–181. [Google Scholar]
- Stadtfeld, M.; Apostolou, E.; Ferrari, F.; Choi, J.; Walsh, R.M.; Chen, T.; Ooi, S.S.; Kim, S.Y.; Bestor, T.H.; Shioda, T.; et al. Ascorbic acid prevents loss of dlk1-dio3 imprinting and facilitates generation of all-ips cell mice from terminally differentiated b cells. Nat. Genet. 2012, 44, 398-405, S391-S392. [Google Scholar] [CrossRef]
- Lempiainen, H.; Couttet, P.; Bolognani, F.; Muller, A.; Dubost, V.; Luisier, R.; Del Rio Espinola, A.; Vitry, V.; Unterberger, E.; Thomson, J.P.; et al. Identification of dlk1-dio3 imprinted gene cluster non-coding rnas as novel candidate biomarkers for liver tumor promotion. Toxicol. Sci.: Official J. Soc. Toxicol. 2012, 131, 375–386. [Google Scholar]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived meg3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metabol. 2003, 88, 5119–5126. [Google Scholar]
- Wang, P.; Ren, Z.; Sun, P. Overexpression of the long non-coding rna meg3 impairs in vitro glioma cell proliferation. J. Cell. Biochem. 2012, 113, 1868–1874. [Google Scholar] [CrossRef]
- Zhang, X.; Gejman, R.; Mahta, A.; Zhong, Y.; Rice, K.A.; Zhou, Y.; Cheunsuchon, P.; Louis, D.N.; Klibanski, A. Maternally expressed gene 3, an imprinted noncoding rna gene, is associated with meningioma pathogenesis and progression. CancerRes. 2010, 70, 2350–2358. [Google Scholar]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by meg3 non-coding rna. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar]
- Braconi, C.; Kogure, T.; Valeri, N.; Huang, N.; Nuovo, G.; Costinean, S.; Negrini, M.; Miotto, E.; Croce, C.M.; Patel, T. Microrna-29 can regulate expression of the long non-coding rna gene meg3 in hepatocellular cancer. Oncogene 2011, 30, 4750–4756. [Google Scholar] [CrossRef]
- Gordon, F.E.; Nutt, C.L.; Cheunsuchon, P.; Nakayama, Y.; Provencher, K.A.; Rice, K.A.; Zhou, Y.; Zhang, X.; Klibanski, A. Increased expression of angiogenic genes in the brains of mouse meg3-null embryos. Endocrinology 2010, 151, 2443–2452. [Google Scholar]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding rnas. Mol. Cell 2011, 43, 904–914. [Google Scholar]
- Carr, M.S.; Yevtodiyenko, A.; Schmidt, C.L.; Schmidt, J.V. Allele-specific histone modifications regulate expression of the dlk1-gtl2 imprinted domain. Genomics 2007, 89, 280–290. [Google Scholar] [CrossRef]
- McMurray, E.N.; Schmidt, J.V. Identification of imprinting regulators at the meg3 differentially methylated region. Genomics 2012, 100, 184–194. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated rnas by rip-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef]
- Seitz, H. Redefining microrna targets. Curr. Biol. 2009, 19, 870–873. [Google Scholar] [CrossRef]
- Gu, S.; Jin, L.; Zhang, F.; Sarnow, P.; Kay, M.A. Biological basis for restriction of microrna targets to the 3' untranslated region in mammalian mrnas. Nat. Struct. Mol. Biol. 2009, 16, 144–150. [Google Scholar]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding rna controls muscle differentiation by functioning as a competing endogenous rna. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Hung, K.-H.; Wang, Y.; Zhao, J.C. Regulation of Mammalian Gene Dosage by Long Noncoding RNAs. Biomolecules 2013, 3, 124-142. https://doi.org/10.3390/biom3010124
AMA Style
Hung K-H, Wang Y, Zhao JC. Regulation of Mammalian Gene Dosage by Long Noncoding RNAs. Biomolecules. 2013; 3(1):124-142. https://doi.org/10.3390/biom3010124
Chicago/Turabian StyleHung, Ko-Hsuan, Yang Wang, and Jing Crystal Zhao. 2013. "Regulation of Mammalian Gene Dosage by Long Noncoding RNAs" Biomolecules 3, no. 1: 124-142. https://doi.org/10.3390/biom3010124