Optimal Regimen of N-Acetylcysteine on Chromium-Induced Renal Cell Damage

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
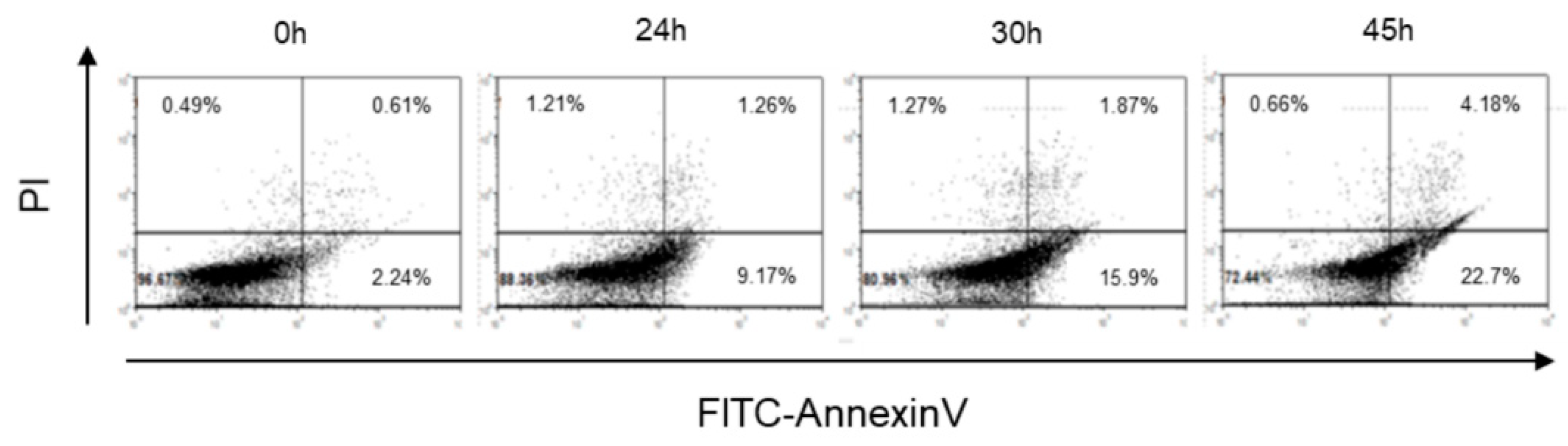
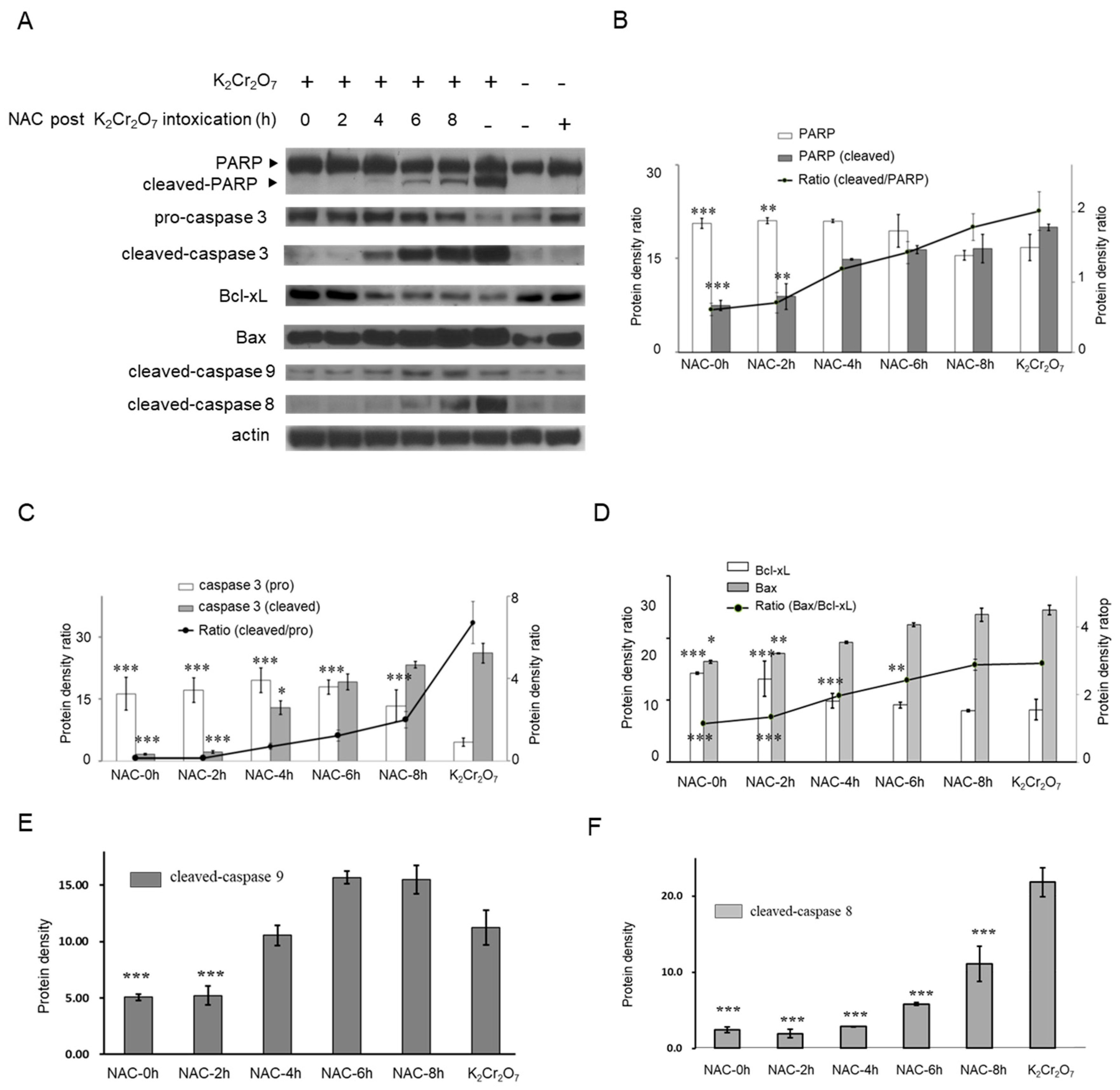
2.1. Cr(VI)-Induced Toxicity Results in HK-2 Apoptosis
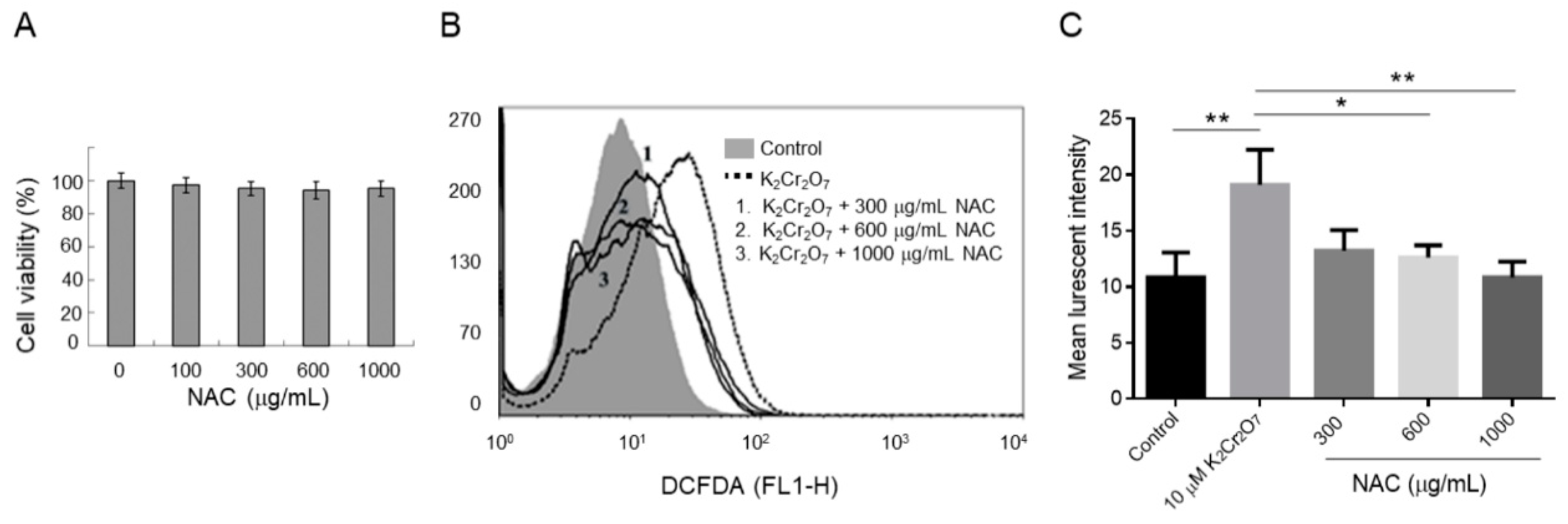
2.2. NAC Treatment Protects HK-2 from Cr(VI)-Induced ROS
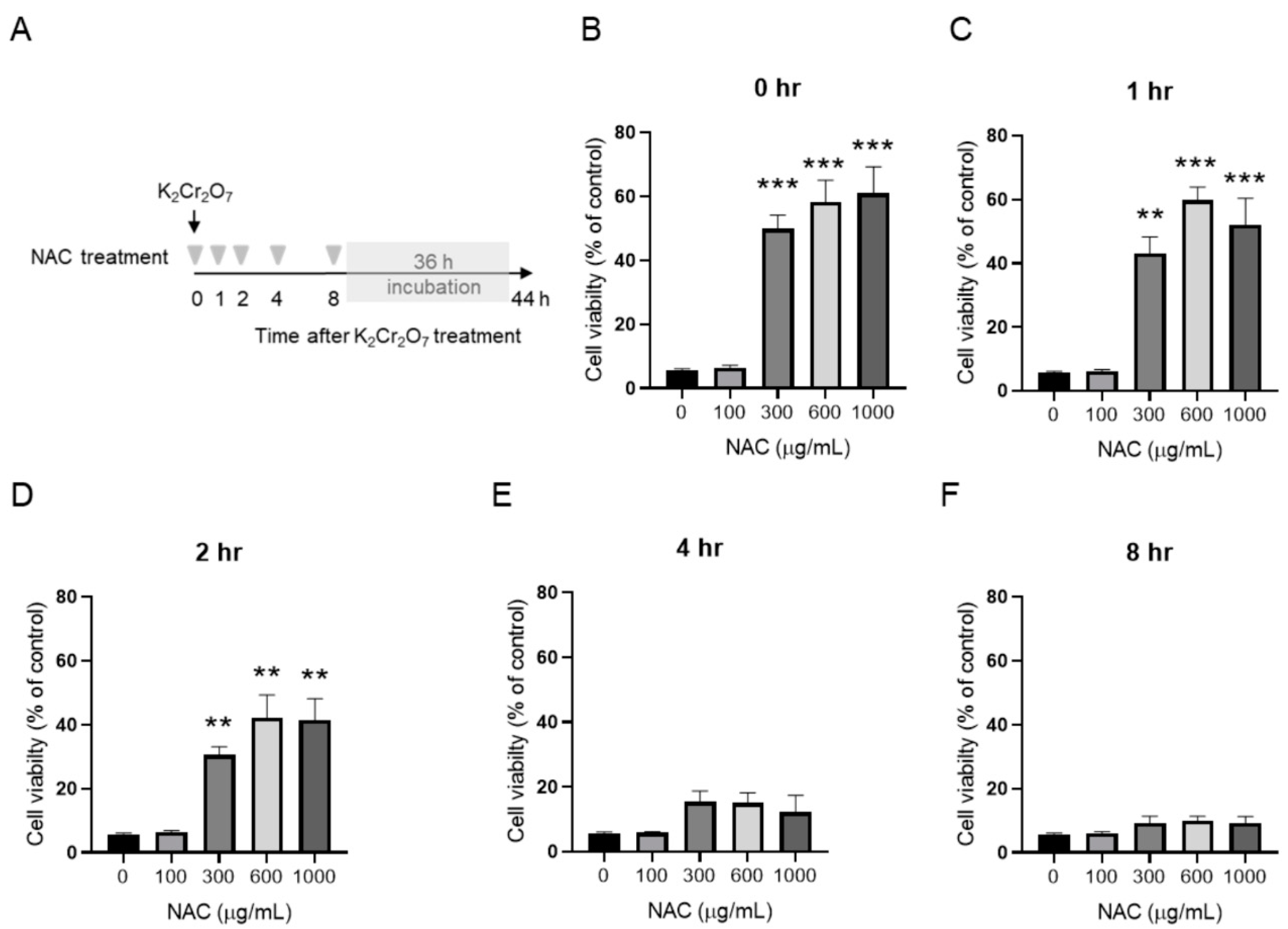
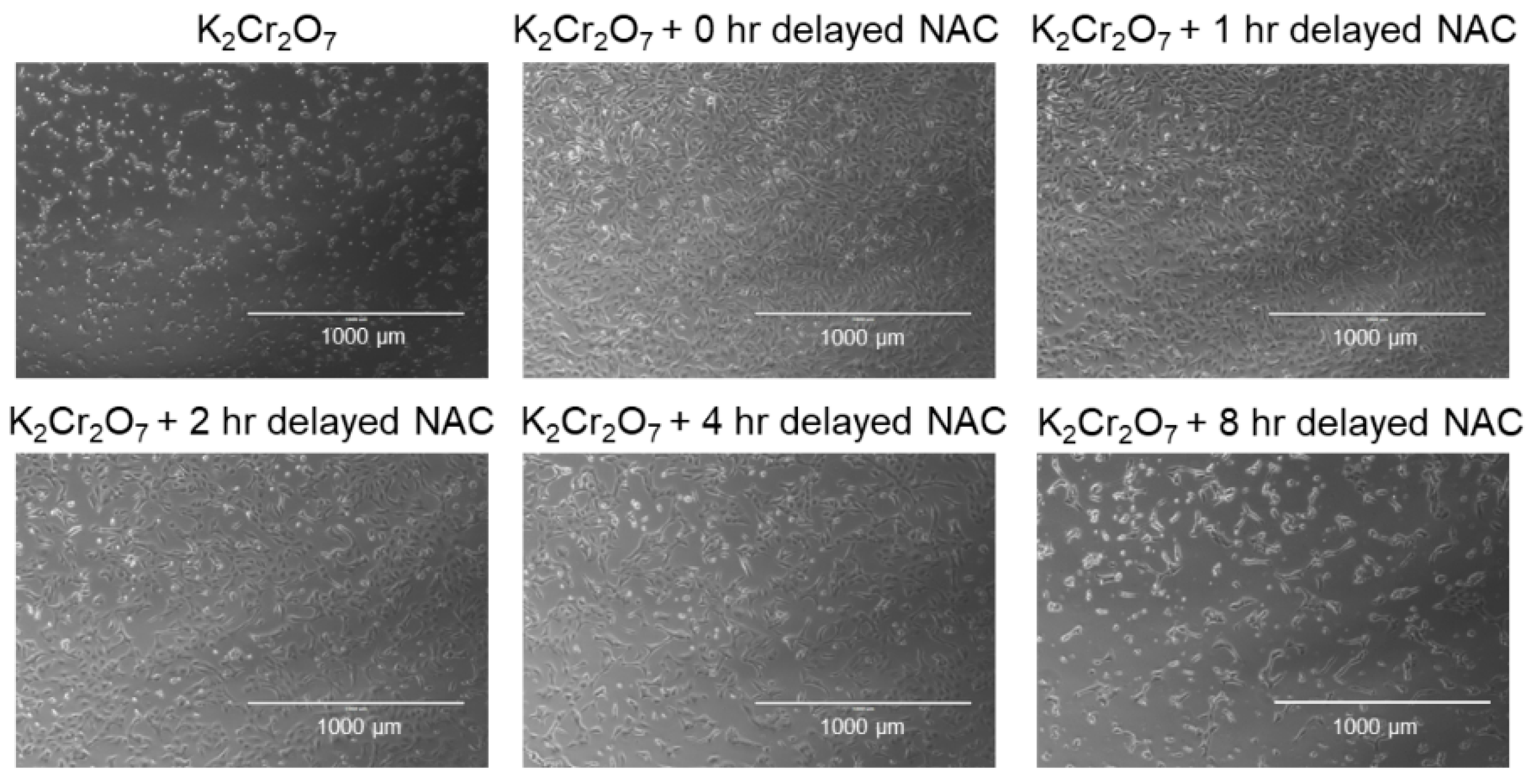
2.3. NAC Treatment Protects HK-2 from Cr(VI)-Induced Cell Death
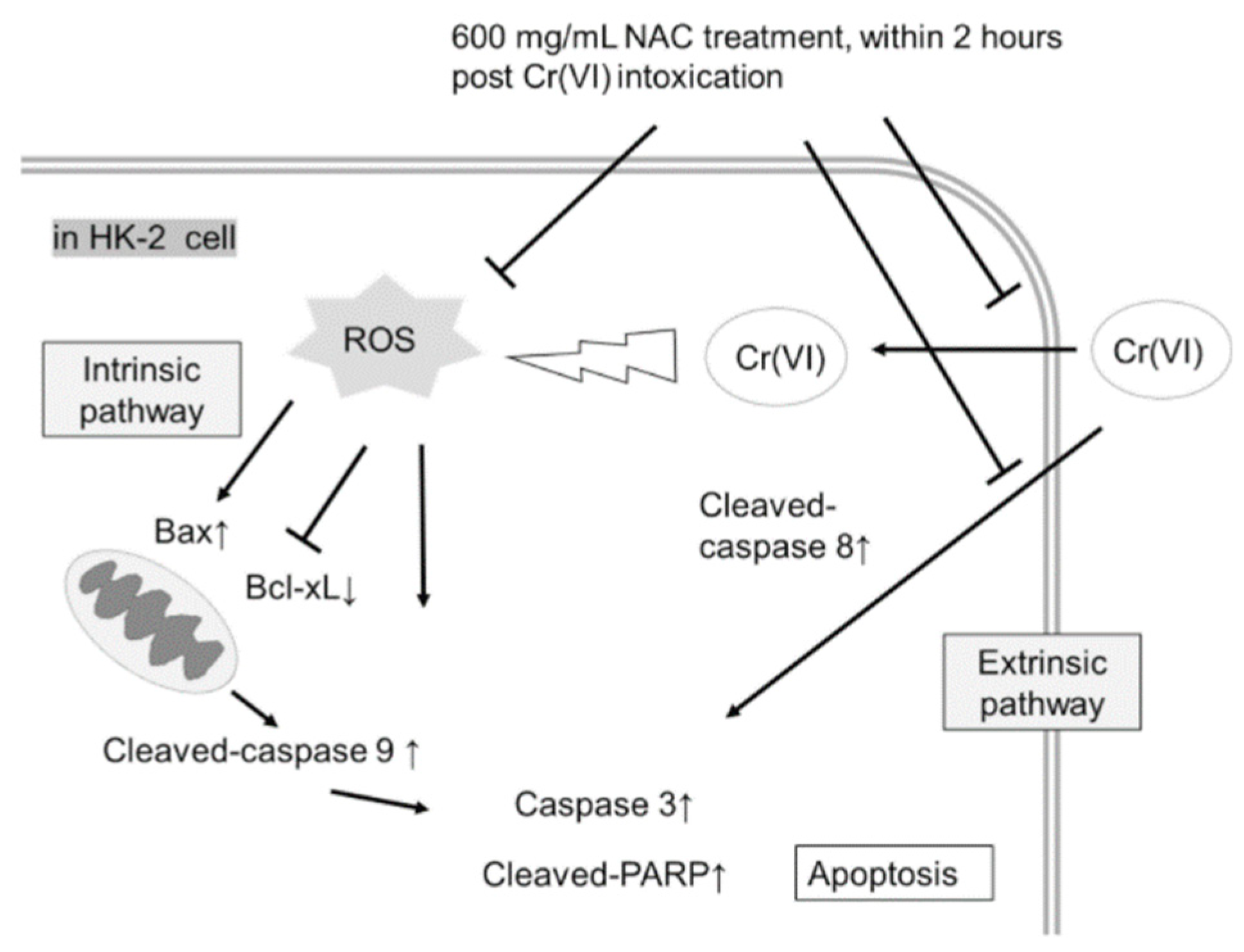
2.4. NAC Treatment Altered Cr(VI)-Induced Apoptotic Pathways
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. HK-2 Cell Culture
5.2. MTT Assay for Cell Viability
5.3. Annexin V/Propidium Iodide (PI) Staining
5.4. Oxidative Stress Assays
5.5. Apoptotic Assay by Western Blot Analysis
5.6. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Stohs, S.J.; Bagchi, D. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Xing, M.L.; Shen, Y.; Zhu, X.; Xu, L.H. Oral administration of Cr(VI) induced oxidative stress, DNA damage and apoptotic cell death in mice. Toxicology 2006, 228, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Brocato, J.; Costa, M. Oral Chromium Exposure and Toxicity. Curr. Environ. Health Rep. 2015, 2, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Bihari, V.; Agarwal, S.K.; Verma, V.; Kesavachandran, C.N.; Pangtey, B.S.; Mathur, N.; Singh, K.P.; Srivastava, M.; Goel, S.K. Groundwater contaminated with hexavalent chromium [Cr (VI)]: A health survey and clinical examination of community inhabitants (Kanpur, India). PloS ONE 2012, 7, e47877. [Google Scholar] [CrossRef] [PubMed]
- Zhitkovich, A. Chromium in drinking water: Sources, metabolism, and cancer risks. Chem. Res. Toxicol. 2011, 24, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.D. Role of active oxygen species in metal-induced DNA strand breakage in human diploid fibroblasts. Mutat. Res. 1988, 193, 237–246. [Google Scholar] [CrossRef]
- Wakeman, T.P.; Kim, W.J.; Callens, S.; Chiu, A.; Brown, K.D.; Xu, B. The ATM-SMC1 pathway is essential for activation of the chromium [VI]-induced S-phase checkpoint. Mutat. Res. 2004, 554, 241–251. [Google Scholar] [CrossRef]
- Ha, L.; Ceryak, S.; Patierno, S.R. Generation of S phase-dependent DNA double-strand breaks by Cr (VI) exposure: Involvement of ATM in Cr (VI) induction of gamma-H2AX. Carcinogenesis 2004, 25, 2265–2274. [Google Scholar] [CrossRef]
- Shil, K.; Pal, S. Metabolic adaptability in hexavalent chromium-treated renal tissue: An in vivo study. Clin. Kidney J. 2018, 11, 222–229. [Google Scholar] [CrossRef]
- Sanz, P.; Nogue, S.; Munne, P.; Torra, R.; Marques, F. Acute potassium dichromate poisoning. Hum. Exp. Toxicol. 1991, 10, 228–229. [Google Scholar] [CrossRef]
- Ellis, E.N.; Brouhard, B.H.; Lynch, R.E.; Dawson, E.B.; Tisdell, R.; Nichols, M.M.; Ramirez, F. Effects of hemodialysis and dimercaprol in acute dichromate poisoning. J. Toxicol. Clin. Toxicol. 1982, 19, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Meert, K.L.; Ellis, J.; Aronow, R.; Perrin, E. Acute ammonium dichromate poisoning. Ann. Emerg. Med. 1994, 24, 748–750. [Google Scholar] [CrossRef]
- Sunilkumar, M.N.; Ajith, T.A.; Parvathy, V.K. Acute ammonium dichromate poisoning in a 2 year-old child. Indian J. Crit. Care Med. 2014, 18, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.B.; DiNicola, W.; McIntosh, R. Acute potassium dichromate poisoning. Treated by peritoneal dialysis. Am. J. Dis. Child. 1970, 119, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.; Shi, X.L.; Lee, W.K.; Hill, R.; Wakeman, T.P.; Katz, A.; Xu, B.; Dalal, N.S.; Robertson, J.D.; Chen, C.; et al. Review of chromium (VI) apoptosis, cell-cycle-arrest, and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2010, 28, 188–230. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Zheng, P.; Feng, H.; Jia, G. Imbalance of oxidative and reductive species involved in chromium(VI)-induced toxic effects. React. Oxyg. Species 2017, 3, 1–11. [Google Scholar] [CrossRef]
- Lin, T.J.; Huang, Y.L.; Chang, J.S.; Liu, K.T.; Yen, M.C.; Chen, F.W.; Shih, Y.L.; Jeo, J.C.; Huang, P.C.; Yeh, I.J. Optimal dosage and early intervention of L-ascorbic acid inhibiting K2Cr2O7-induced renal tubular cell damage. J. Trace Elem. Med. Biol. 2018, 48, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cotgreave, I.A. N-acetylcysteine: Pharmacological considerations and experimental and clinical applications. Adv. Pharmacol. 1997, 38, 205–227. [Google Scholar] [PubMed]
- Scholz, R.W.; Graham, K.S.; Gumpricht, E.; Reddy, C.C. Mechanism of interaction of vitamin-E and glutathione in the protection against membrane lipid-peroxidation. Ann. N. Y. Acad. Sci. 1989, 570, 514–517. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Quinteros, F.A.; Machiavelli, L.I.; Miler, E.A.; Cabilla, J.P.; Duvilanski, B.H. Mechanisms of chromium (VI)-induced apoptosis in anterior pituitary cells. Toxicology 2008, 249, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Saito, C.; Zwingmann, C.; Jaeschke, H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 2010, 51, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Louie, B.; Rajamahanty, S.; Pyo, P.; Choudhury, M.; Konno, S. Mode of cytotoxic action of nephrotoxic agents: Oxidative stress and glutathione-dependent enzyme. BJU Int. 2010, 105, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Blanusa, M.; Varnai, V.M.; Piasek, M.; Kostial, K. Chelators as antidotes of metal toxicity: Therapeutic and experimental aspects. Curr. Med. Chem. 2005, 12, 2771–2794. [Google Scholar] [CrossRef] [PubMed]
- Boşgelmez, İ.İ.; Güvendik, G. N-Acetyl-L-Cysteine Protects Liver and Kidney Against Chromium(VI)-Induced Oxidative Stress in Mice. Biol. Trace Elem. Res. 2017, 178, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.J.; Shi, X. In vivo reduction of chromium (VI) and its related free radical generation. In Molecular Mechanisms of Metal Toxicity and Carcinogenesis; Springer: Boston, MA, USA, 2001; pp. 41–47. [Google Scholar]
- Leonard, S.S.; Roberts, J.R.; Antonini, J.M.; Castranova, V.; Shi, X. PbCrO4 mediates cellular responses via reactive oxygen species. Mol. Cell. Biochem. 2004, 255, 171–179. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Pedersen, R.S.; Morch, P.T. Chromic acid poisoning treated with acute hemodialysis. Nephron 1978, 22, 592–595. [Google Scholar] [CrossRef]
- KhanSaif, R.; Aparna, Q.; Shahzad, S.; Haque, F. Chromium induced AKI: Case with protean implications. Ann. Trop. Med. Public Health 2014, 7, 136–138. [Google Scholar]
- Karaytug, S.; Sevgiler, Y.; Karayakar, F. Comparison of the protective effects of antioxidant compounds in the liver and kidney of Cd- and Cr-exposed common carp. Environ. Toxicol. 2014, 29, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Banner, W., Jr.; Koch, M.; Capin, D.M.; Hopf, S.B.; Chang, S.; Tong, T.G. Experimental chelation therapy in chromium, lead, and boron intoxication with N-acetylcysteine and other compounds. Toxicol. Appl. Pharmacol. 1986, 83, 142–147. [Google Scholar] [CrossRef]
- Nitescu, N.; Ricksten, S.E.; Marcussen, N.; Haraldsson, B.; Nilsson, U.; Basu, S.; Guron, G. N-acetylcysteine attenuates kidney injury in rats subjected to renal ischaemia-reperfusion. Nephrol. Dial. Transplant. 2006, 21, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobashi, K.; Singh, I.; Orak, J.K.; Asayama, K.; Singh, A.K. Combination therapy of N-acetylcysteine, sodium nitroprusside and phosphoramidon attenuates ischemia-reperfusion injury in rat kidney. Mol. Cell. Biochem. 2002, 240, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tsuji, T.; Yasuda, H.; Sun, Y.; Fujigaki, Y.; Hishida, A. The molecular mechanisms of the attenuation of cisplatin-induced acute renal failure by N-acetylcysteine in rats. Nephrol. Dial. Transplant. 2008, 23, 2198–2205. [Google Scholar] [PubMed] [Green Version]
- Kim, J.H.; Lee, S.S.; Jung, M.H.; Yeo, H.D.; Kim, H.J.; Yang, J.I.; Roh, G.S.; Chang, S.H.; Park, D.J. N-acetylcysteine attenuates glycerol-induced acute kidney injury by regulating MAPKs and Bcl-2 family proteins. Nephrol. Dial. Transplant. 2010, 25, 1435–1443. [Google Scholar] [PubMed]
- Fishman, A.I.; Alexander, B.; Eshghi, M.; Choudhury, M.; Konno, S. Nephrotoxin-induced renal cell injury involving biochemical alterations and its prevention with antioxidant. J. Clin. Med. Res. 2012, 4, 95–101. [Google Scholar] [CrossRef]
- Shimizu, M.H.; Coimbra, T.M.; de Araujo, M.; Menezes, L.F.; Seguro, A.C. N-acetylcysteine attenuates the progression of chronic renal failure. Kidney Int. 2005, 68, 2208–2217. [Google Scholar] [Green Version]
- Prescott, L.F.; Donovan, J.W.; Jarvie, D.R.; Proudfoot, A.T. The disposition and kinetics of intravenous N-acetylcysteine in patients with paracetamol overdosage. Eur. J. Clin. Pharmacol. 1989, 37, 501–506. [Google Scholar] [CrossRef]
- Lin, C.C.; Wu, M.L.; Yang, C.C.; Ger, J.; Tsai, W.J.; Deng, J.F. Acute severe chromium poisoning after dermal exposure to hexavalent chromium. J. Chin. Med. Assoc. 2009, 72, 219–221. [Google Scholar] [CrossRef]
- Quinteros, F.A.; Poliandri, A.H.; Machiavelli, L.I.; Cabilla, J.P.; Duvilanski, B.H. In vivo and in vitro effects of chromium VI on anterior pituitary hormone release and cell viability. Toxicol. Appl. Pharmacol. 2007, 218, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Li, Y.; Dai, L.; Deng, Y.; Zou, Y.; Li, P.; Yang, Y.; Zhong, C. Hexavalent chromium targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent caspase-3 activation in L-02 hepatocytes. Int. J. Mol. Med. 2012, 30, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, S.; Bagchi, J.; Mandal, L.; Paul, K.; Ganguly, D.; Bhattacharjee, S.; Bandyopadhyay, S.; Biswas, N.; Chaudhuri, U.; Ghosh, M.; et al. N-acetyl cysteine enhances imatinib-induced apoptosis of Bcr-Abl+ cells by endothelial nitric oxide synthase-mediated production of nitric oxide. Apoptosis 2009, 14, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, K.; Wang, N.; Zhang, H. Nacetylcysteine induces apoptosis via the mitochondriadependent pathway but not via endoplasmic reticulum stress in H9c2 cells. Mol. Med. Rep. 2017, 16, 6626–6633. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Johnson, G.; Kirk, J.; Fuerstenberg, S.M.; Zager, R.A.; Torok-Storb, B. HK-2: An immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1994, 45, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, I.-J.; Wang, T.-Y.; Lin, J.-C.; Lin, T.-J.; Chang, J.-S.; Yen, M.-C.; Liu, Y.-H.; Wu, P.-L.; Chen, F.-W.; Shih, Y.-L.; et al. Optimal Regimen of N-Acetylcysteine on Chromium-Induced Renal Cell Damage. Metabolites 2019, 9, 172. https://doi.org/10.3390/metabo9090172
Yeh I-J, Wang T-Y, Lin J-C, Lin T-J, Chang J-S, Yen M-C, Liu Y-H, Wu P-L, Chen F-W, Shih Y-L, et al. Optimal Regimen of N-Acetylcysteine on Chromium-Induced Renal Cell Damage. Metabolites. 2019; 9(9):172. https://doi.org/10.3390/metabo9090172
Chicago/Turabian StyleYeh, I-Jeng, Tzu-Yi Wang, Jhong-Ching Lin, Tzeng-Jih Lin, Jung-San Chang, Meng-Chi Yen, Yao-Hua Liu, Pei-Lin Wu, Fen-Wei Chen, Yueh-Lun Shih, and et al. 2019. "Optimal Regimen of N-Acetylcysteine on Chromium-Induced Renal Cell Damage" Metabolites 9, no. 9: 172. https://doi.org/10.3390/metabo9090172