
A Novel Anti-Hepatitis C Virus and Antiproliferative Agent Alters Metabolic Networks in HepG2 and Hep3B Cells

 ,
,
Abstract
:1. Introduction
2. Results
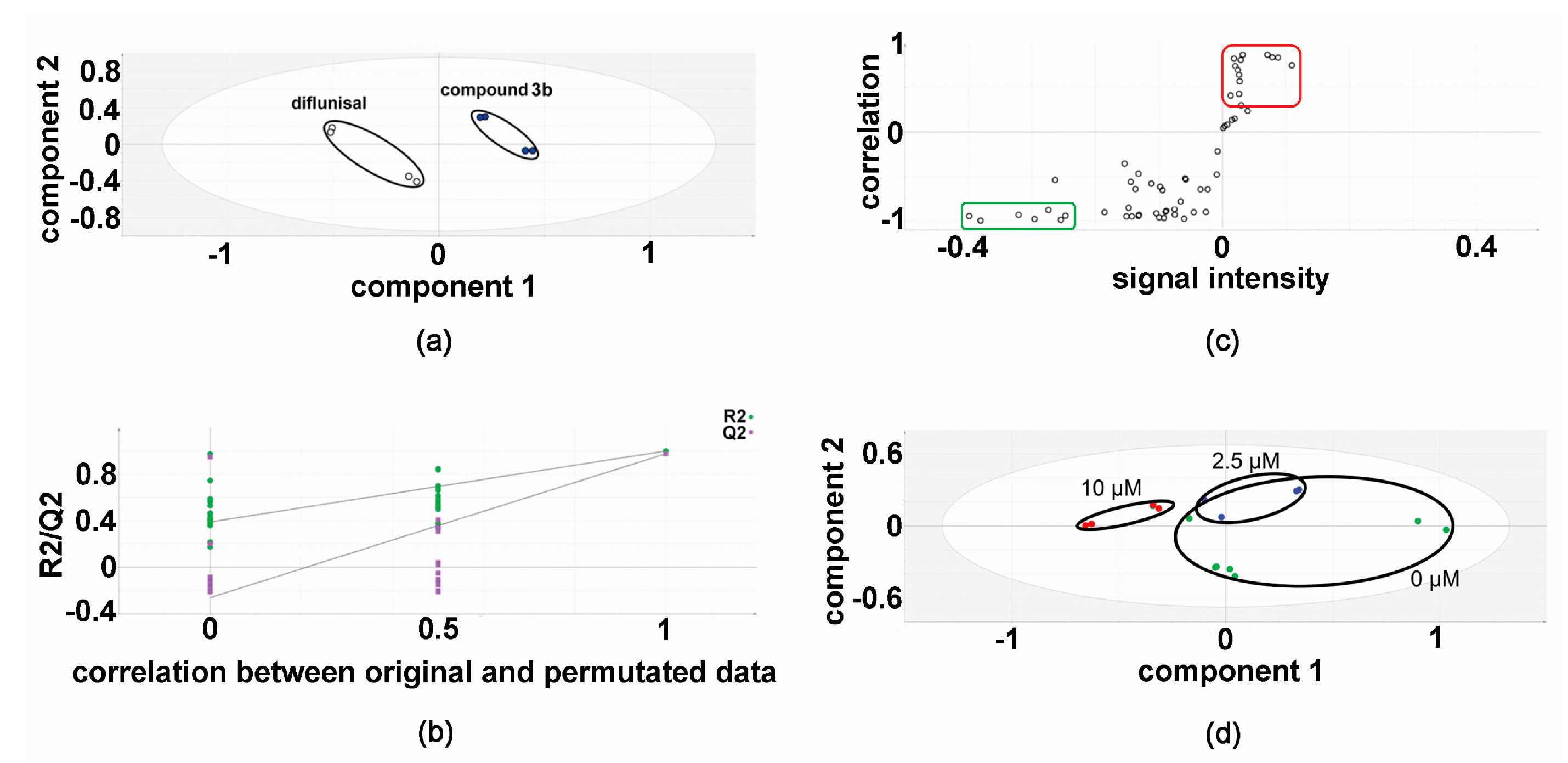
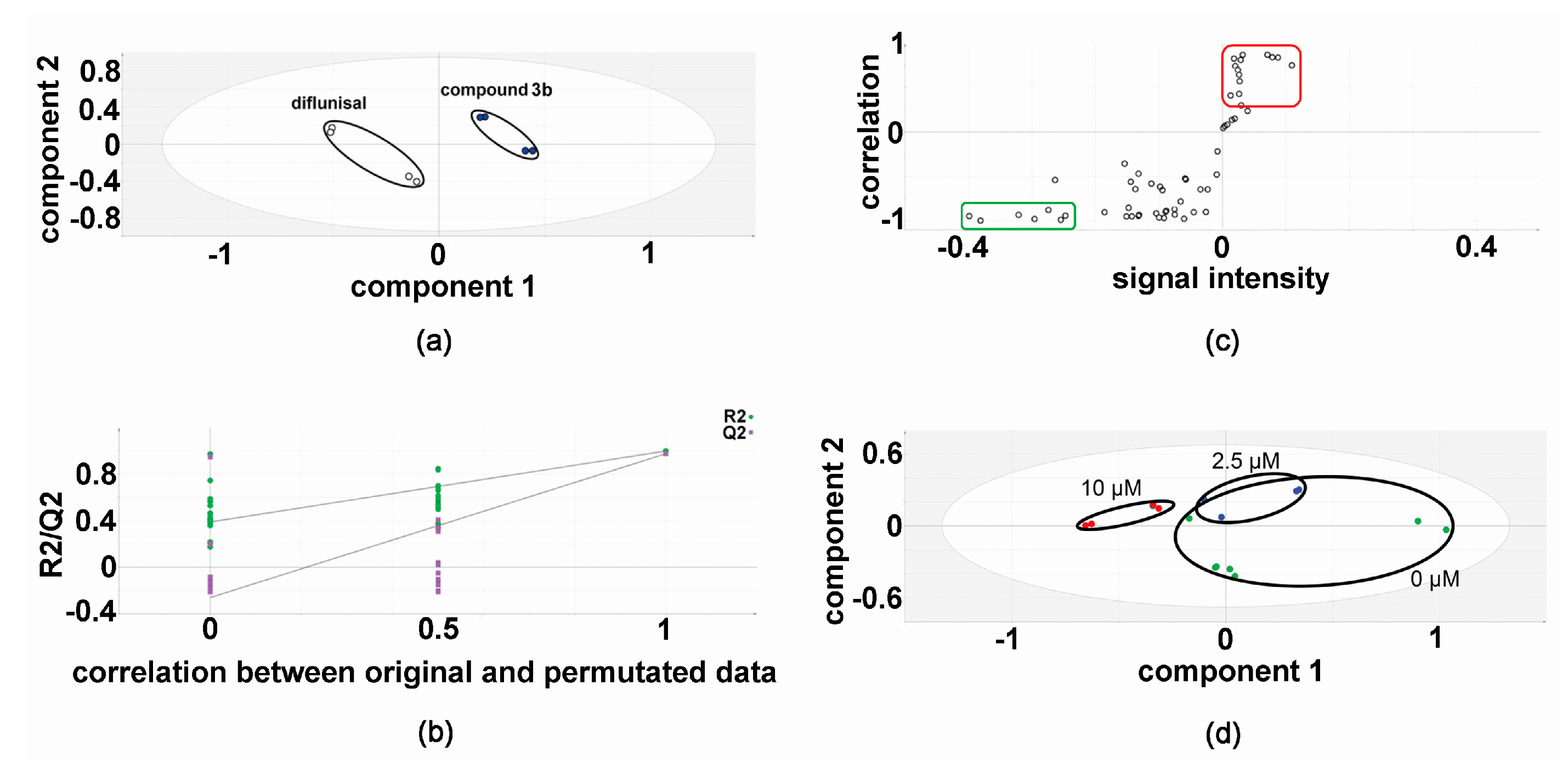
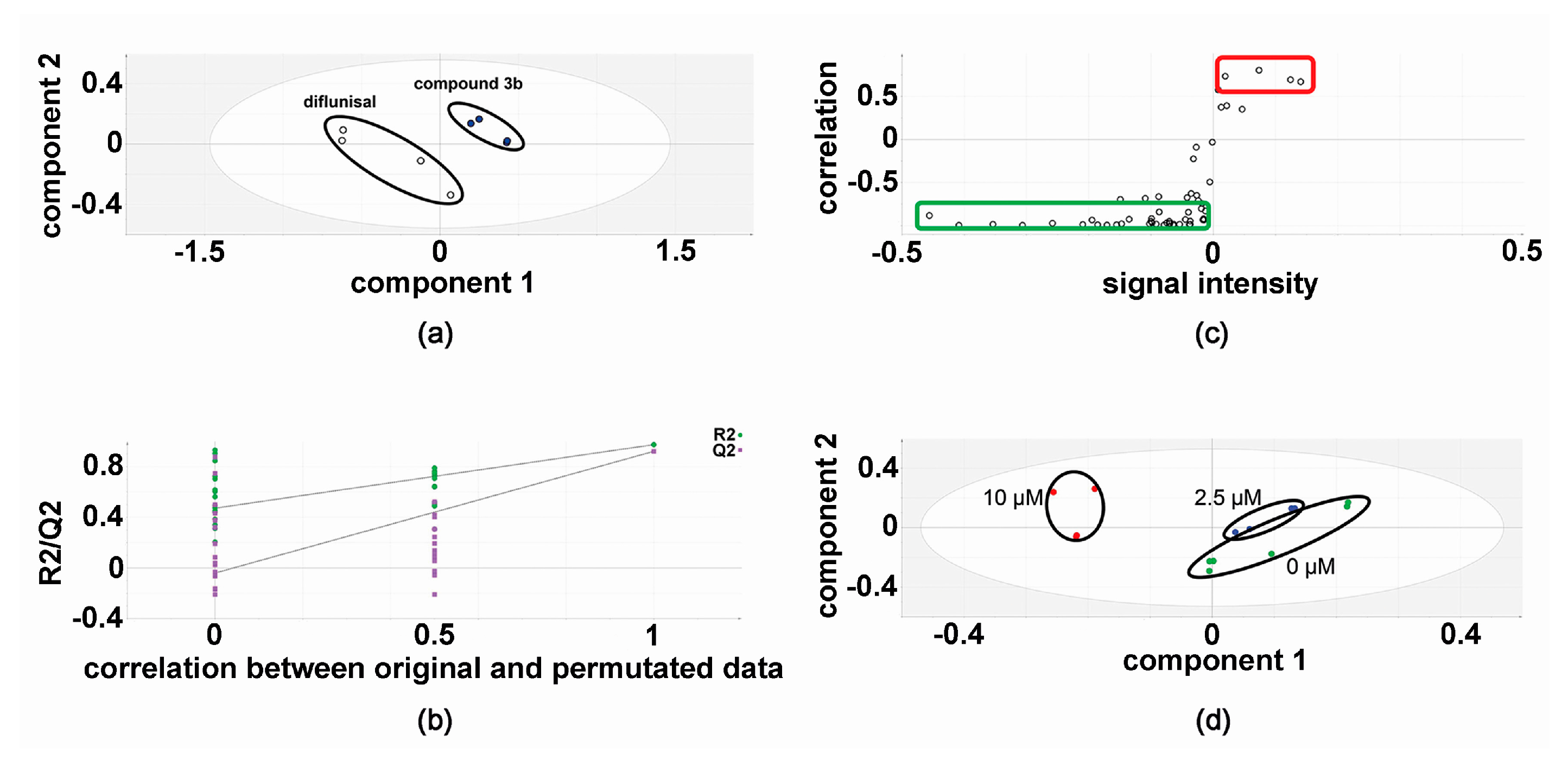
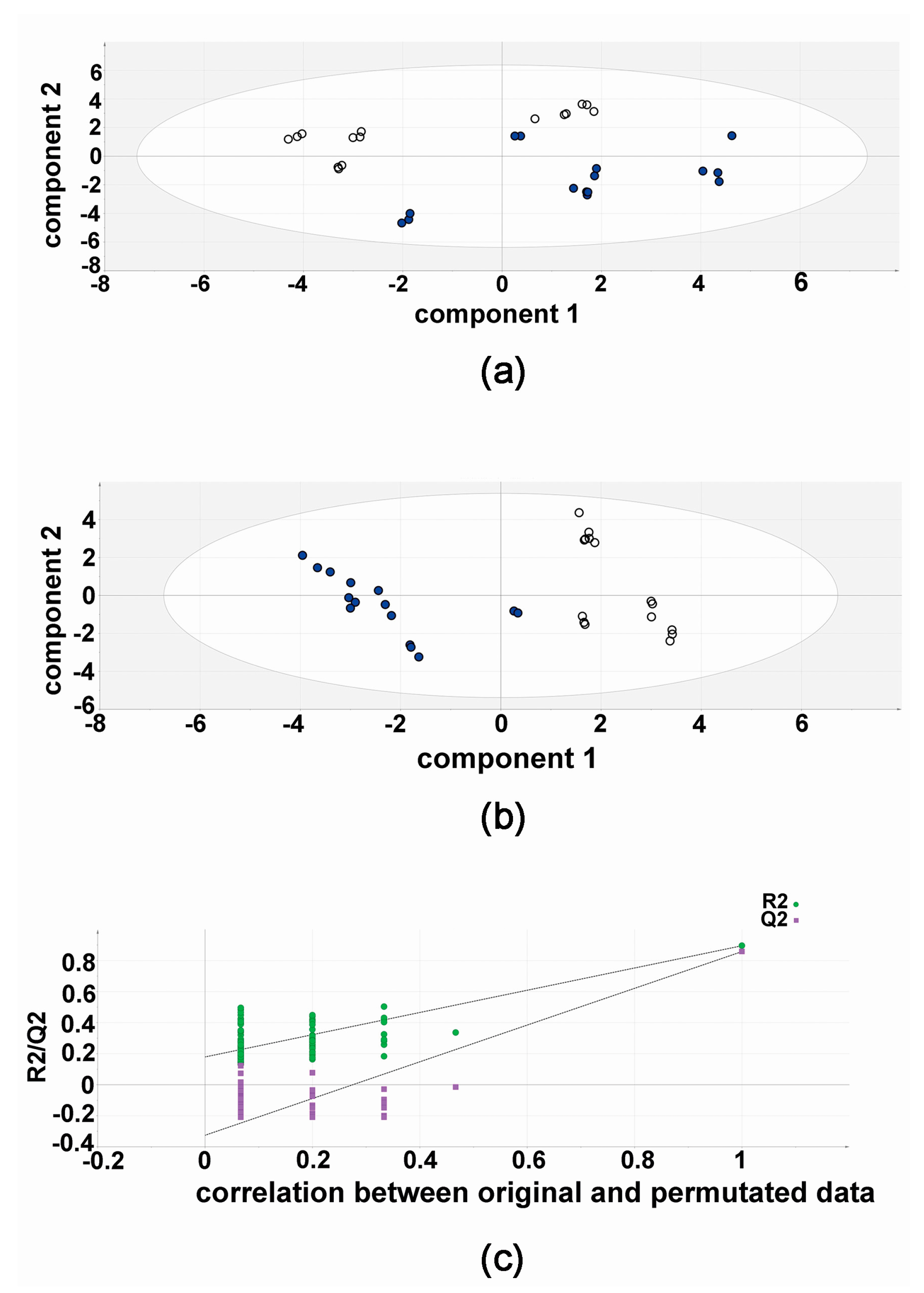
2.1. Metabolomic Analysis—Compound 3b versus Diflunisal
2.2. Metabolic Networks Modified by Compound 3b
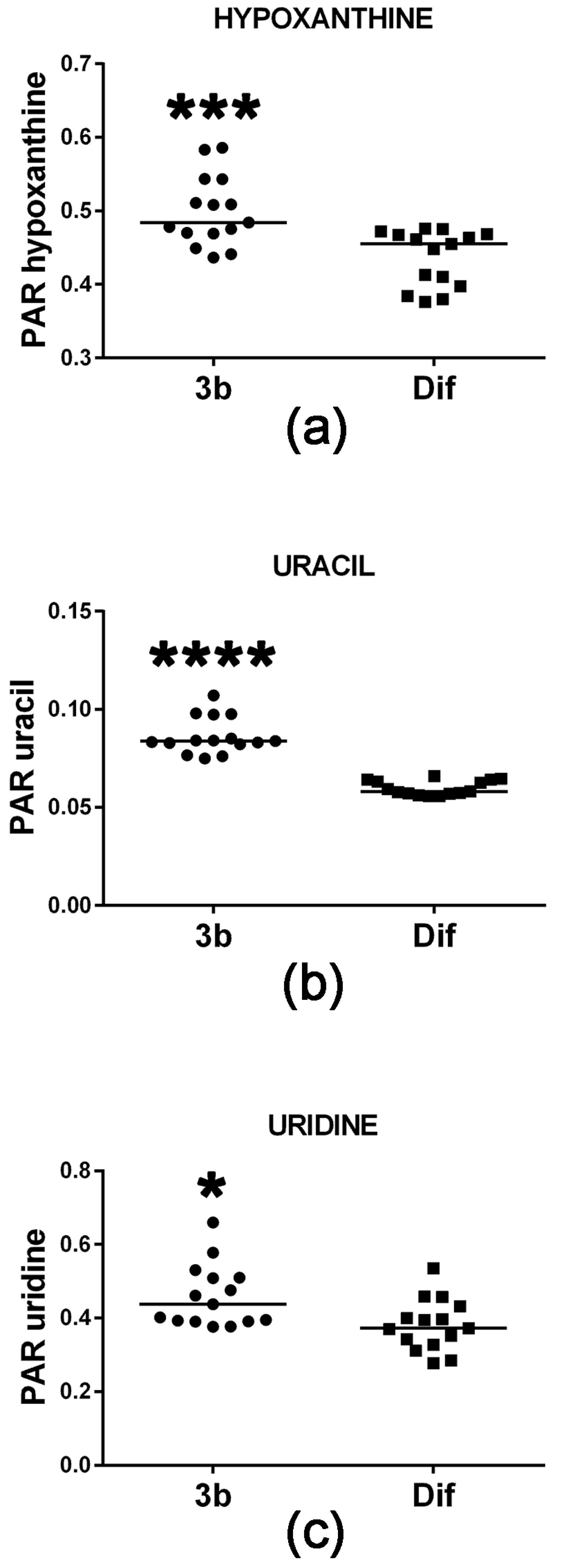
2.2.1. Purine and Pyrimidine Metabolism
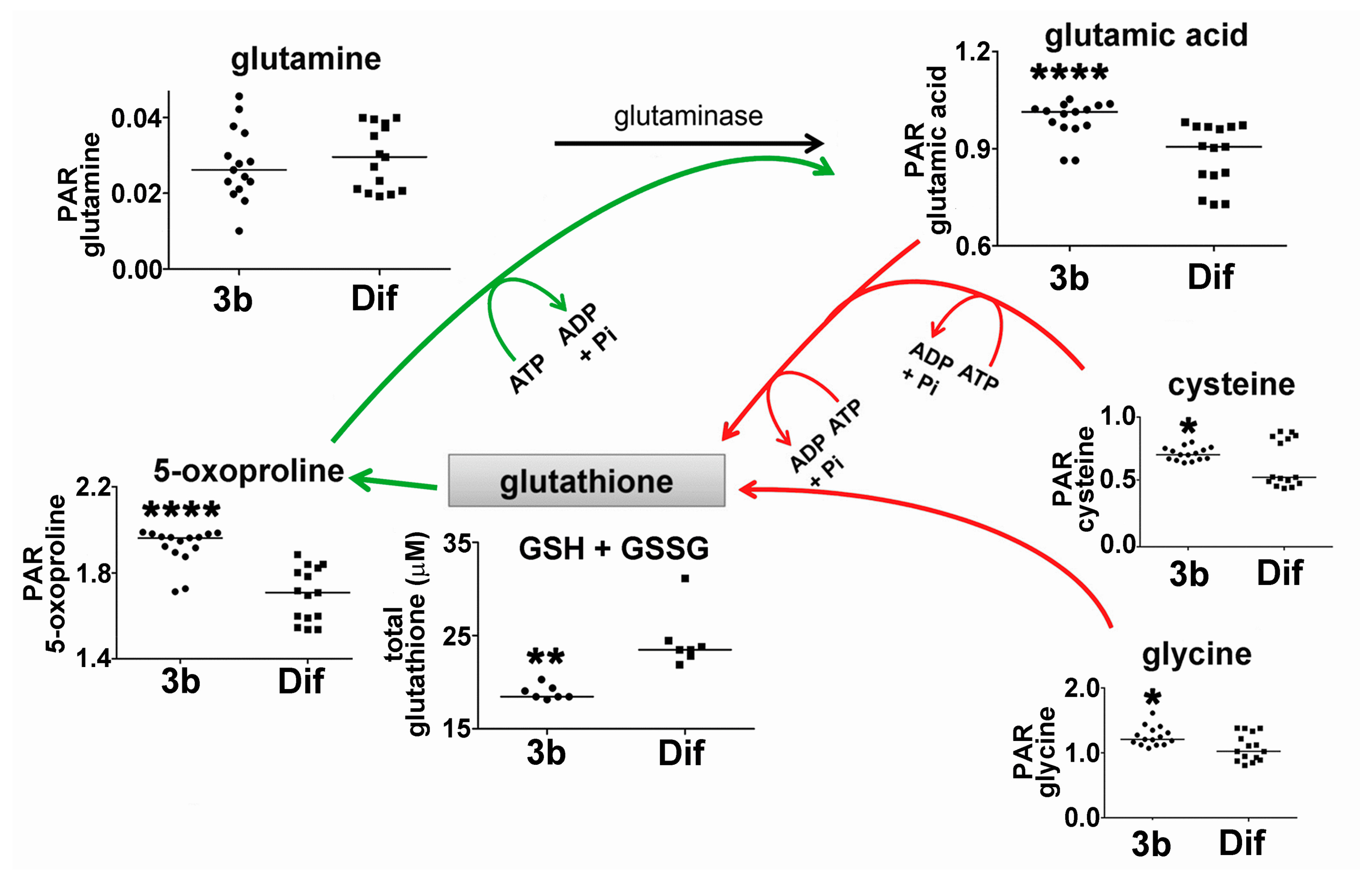
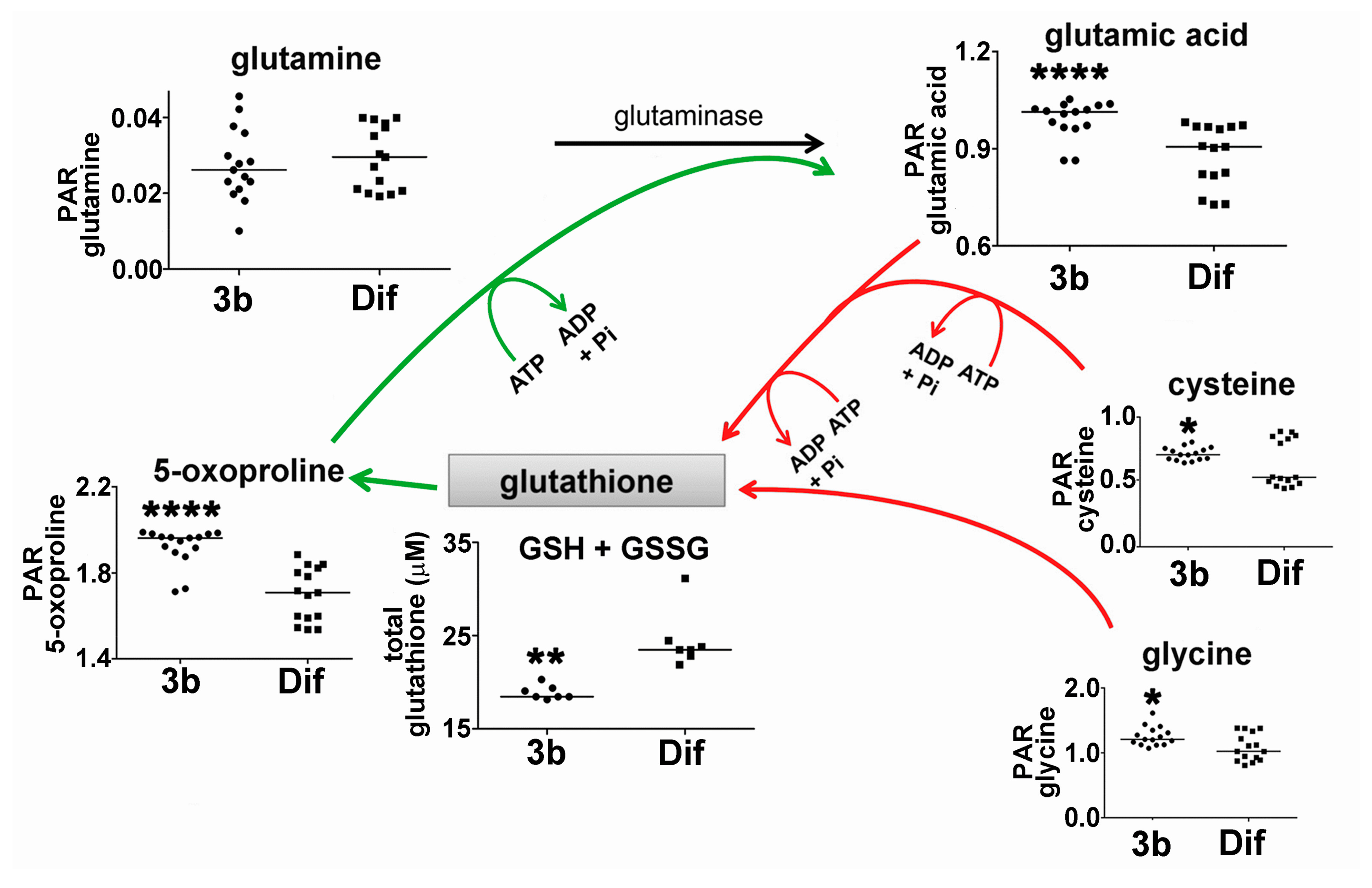
2.2.2. Glutathione Synthesis and Breakdown
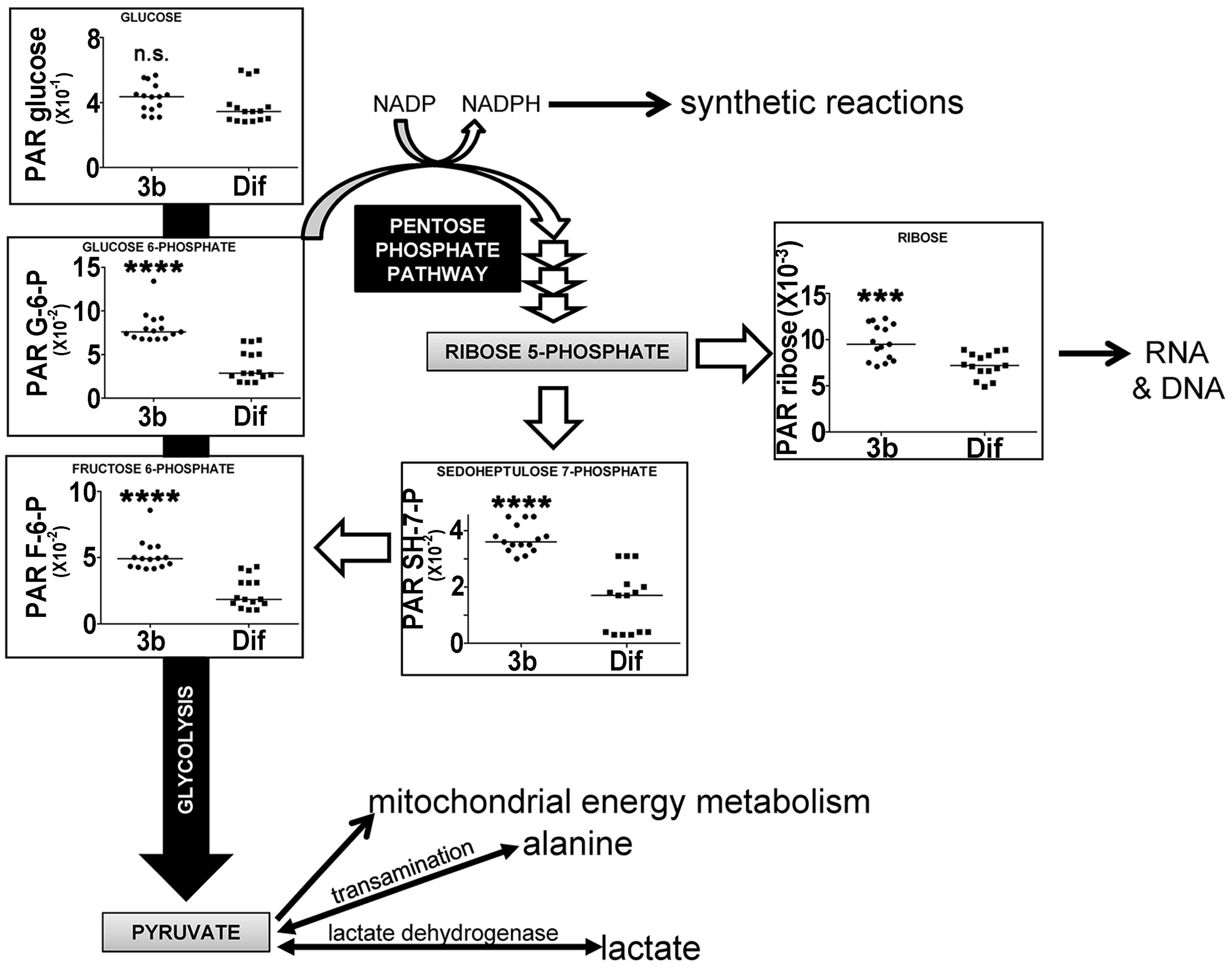
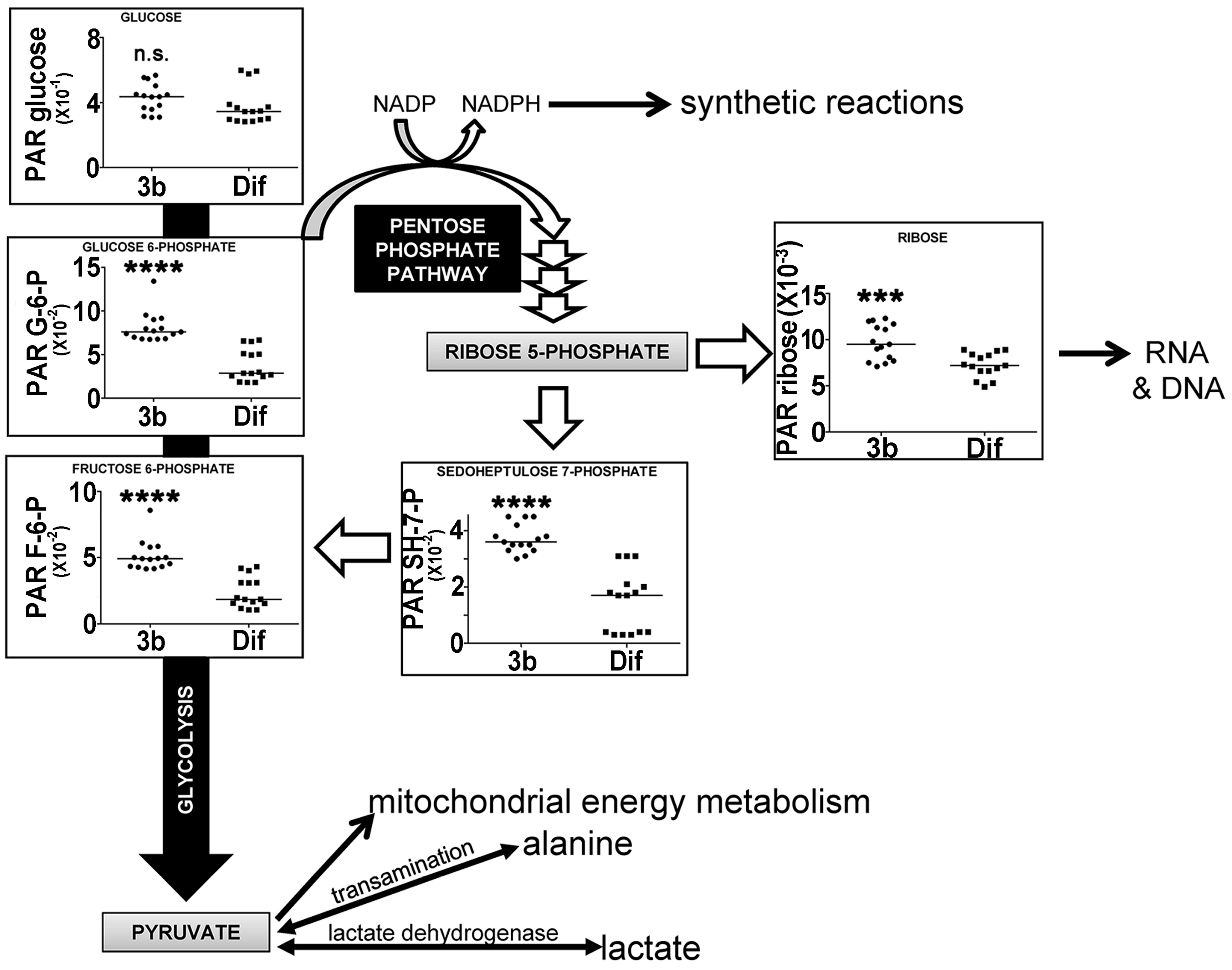
2.2.3. Energy Metabolism by Glycolysis and the Pentose Phosphate Pathway
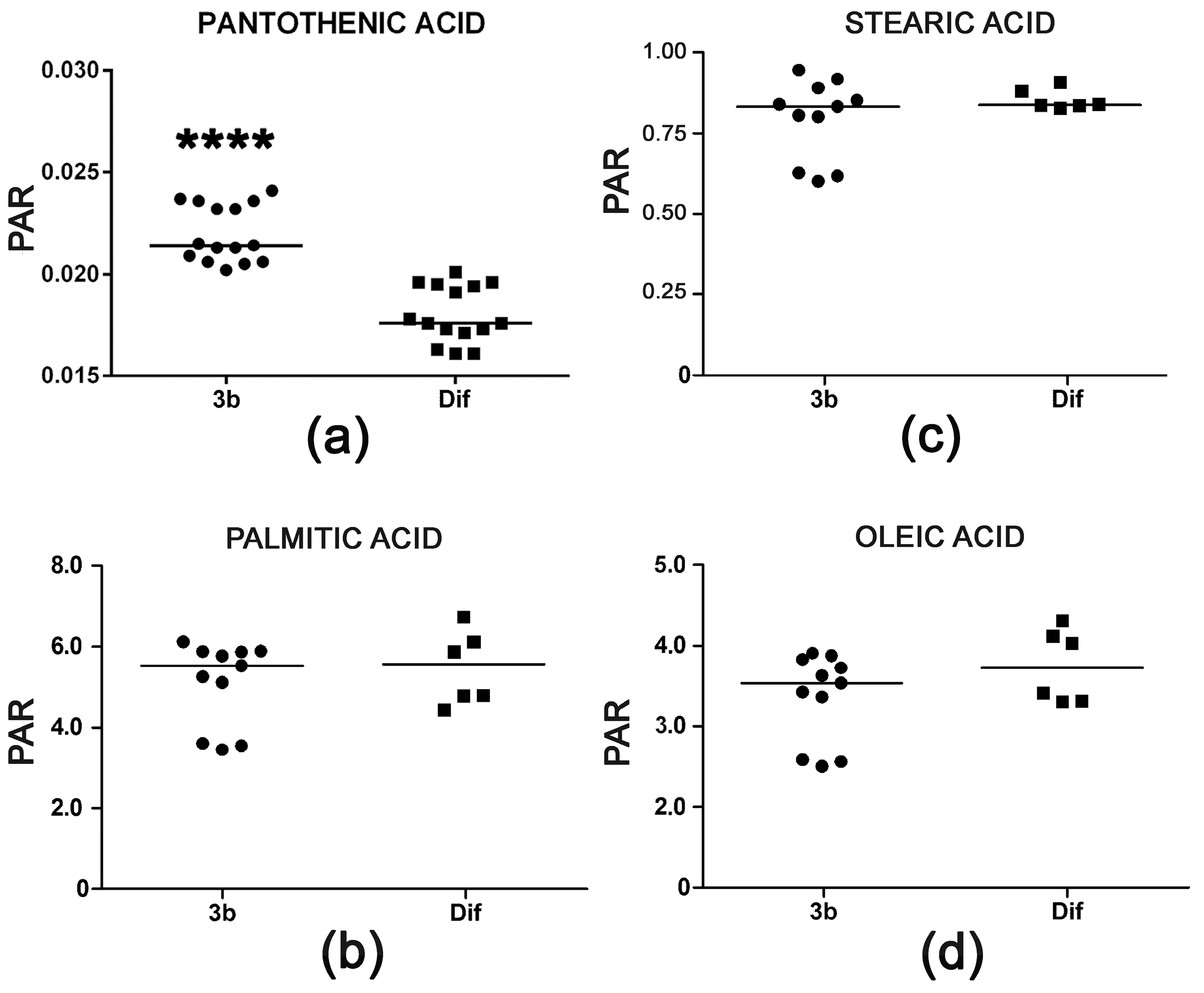
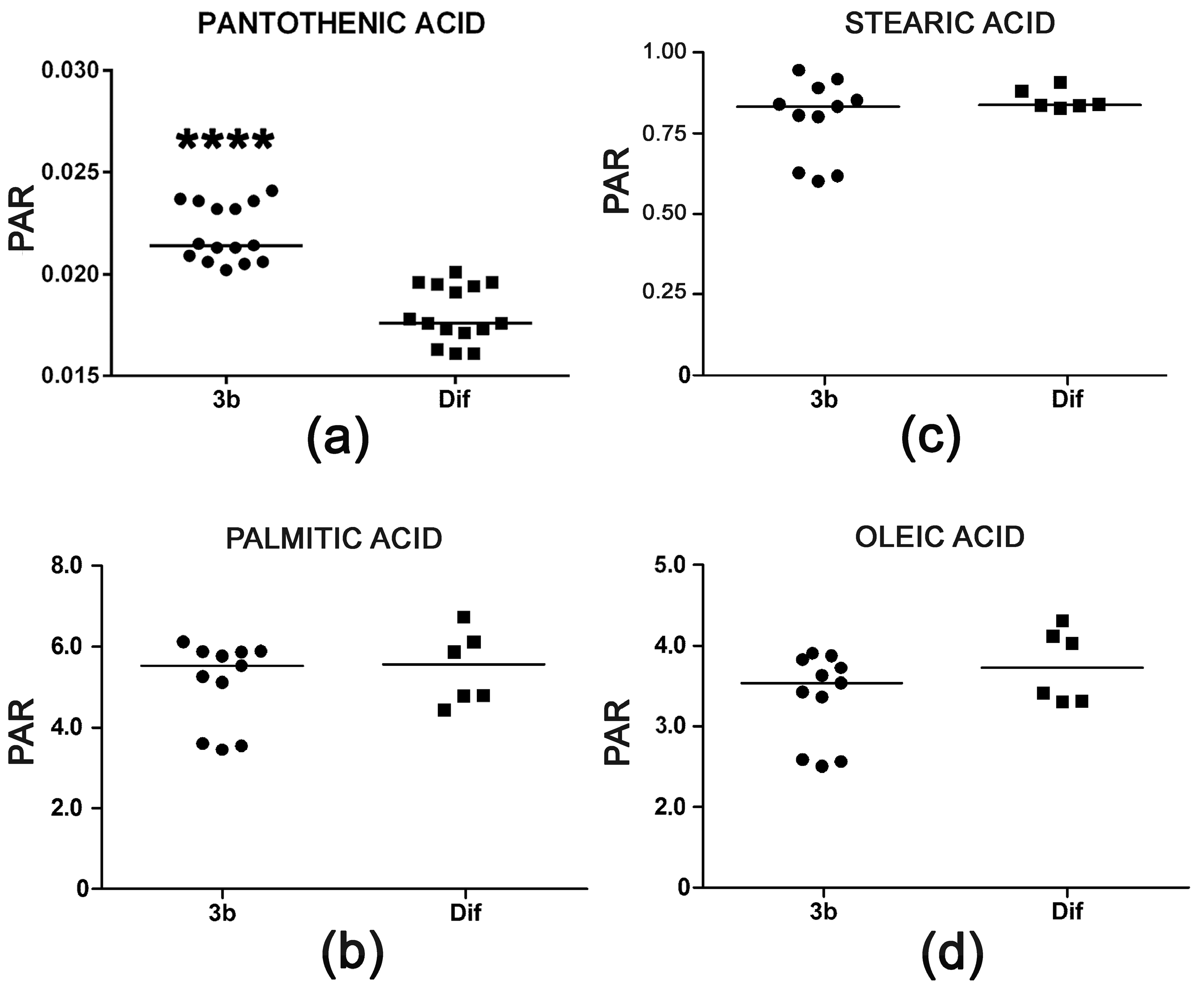
2.2.4. Pantothenic Acid Transport and Fatty Acid β-Oxidation
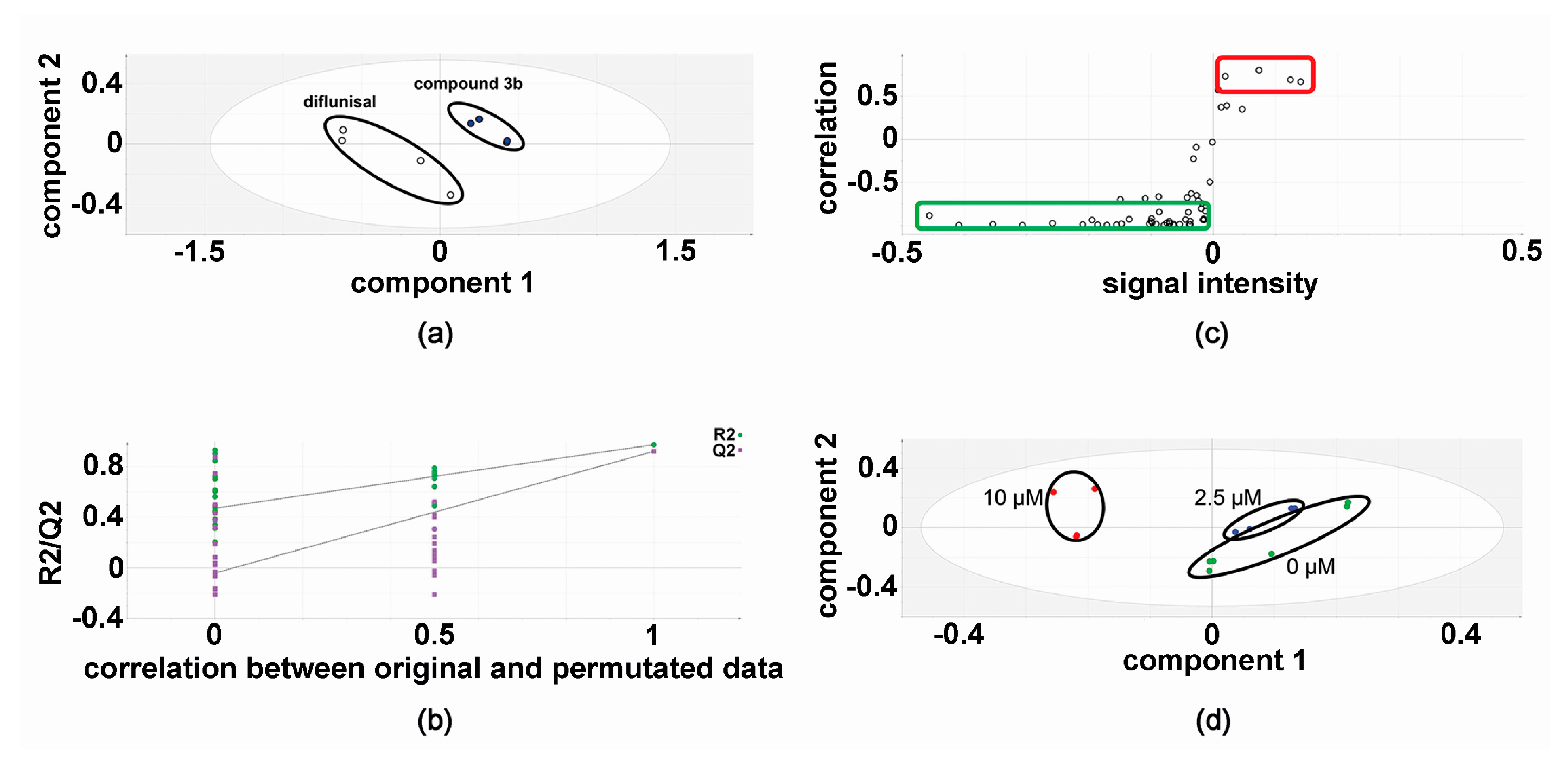
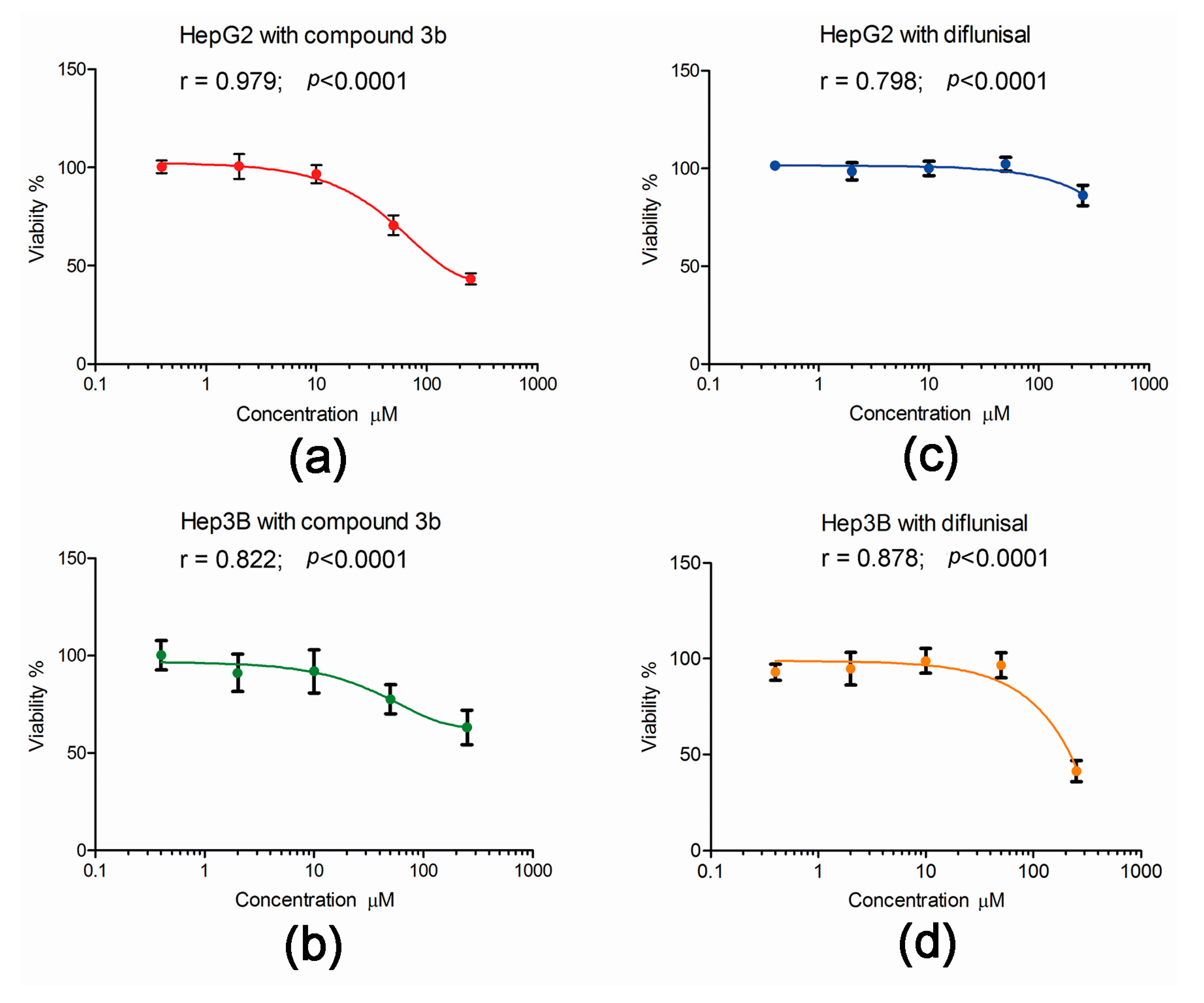
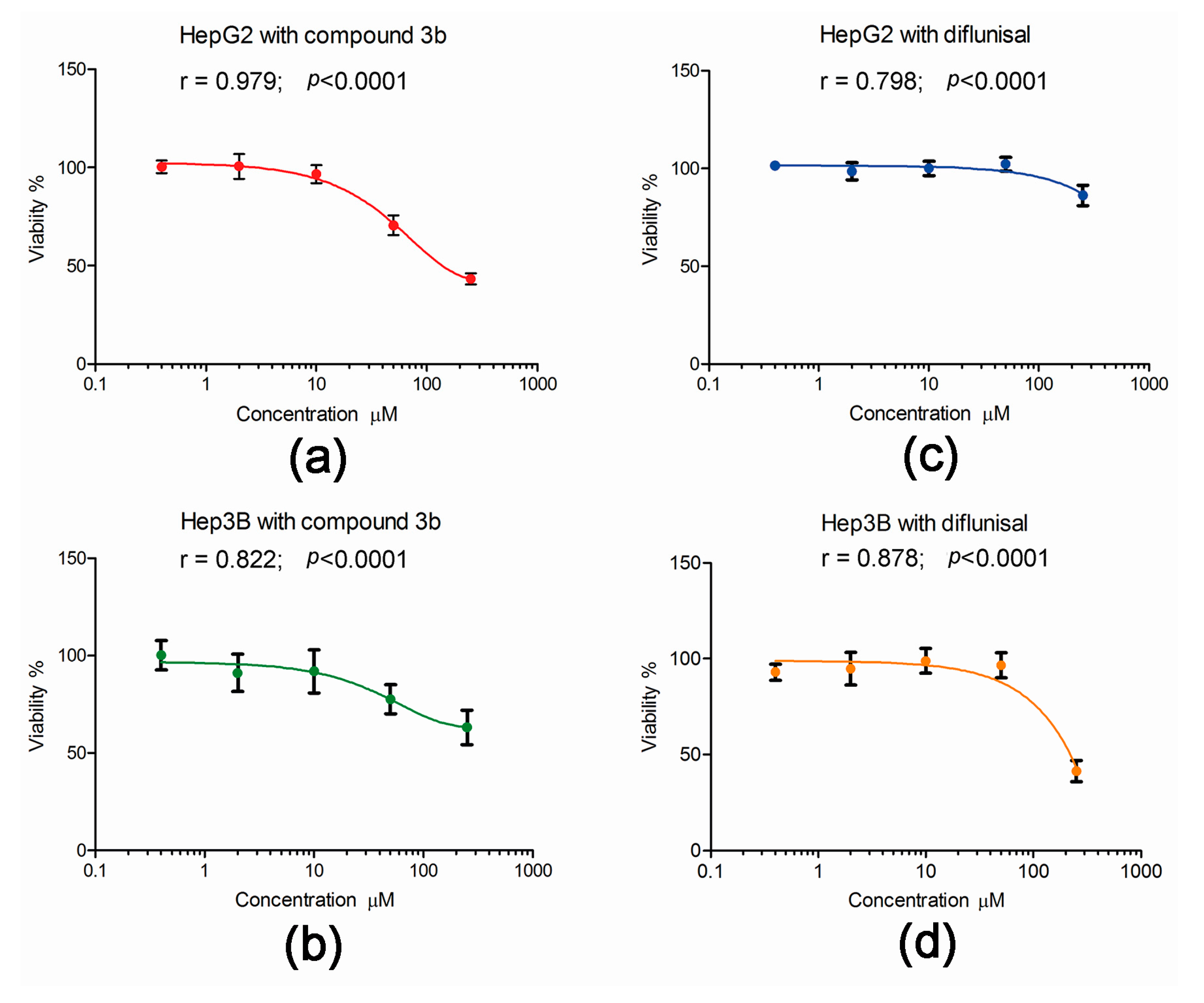
2.3. Effect of Compound 3b and Diflunisal on HepG2 and Hep3B Cell Viability
3. Discussion
4. Materials and Methods
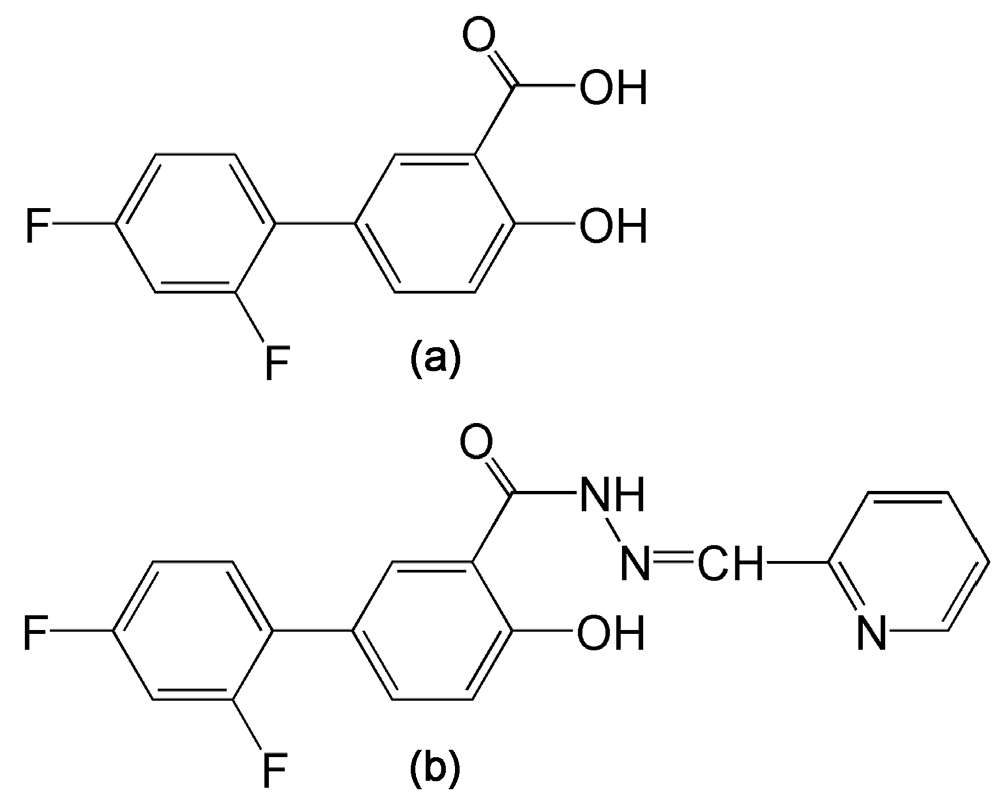
4.1. Drugs
4.2. Cell Culture, Drug Treatment, and Harvesting of Intracellular Contents
4.3. Cell Viability Assay and Cellular Protein Determination
4.4. Gas Chromatography-Mass Spectrometry (GCMS) Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.A.; Hossain, S.; Chowdhury, E.H. Potential therapeutic targets in energy metabolism pathways of breast cancer. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Pabst, T.; Kortz, L.; Fiedler, G.M.; Ceglarek, U.; Idle, J.R.; Beyoglu, D. The plasma lipidome in acute myeloid leukemia at diagnosis in relation to clinical disease features. BBA Clin. 2017, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.L. Metabolic alterations and hepatitis c: From bench to bedside. World J. Gastroenterol. 2016, 22, 1461–1476. [Google Scholar] [CrossRef] [PubMed]
- Semmo, N.; Weber, T.; Idle, J.R.; Beyoglu, D. Metabolomics reveals that aldose reductase activity due to AKR1B10 is upregulated in hepatitis C virus infection. J. Viral Hepat. 2015, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Nishiguchi, S. Factors associated with the response to interferon-based antiviral therapies for chronic hepatitis c. World J. Hepatol. 2015, 7, 2681–2687. [Google Scholar] [CrossRef] [PubMed]
- Stasi, C.; Silvestri, C.; Voller, F.; Cipriani, F. The epidemiological changes of HCV and HBV infections in the era of new antiviral therapies and the anti-HBV vaccine. J. Infect. Public Health 2016, 9, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Wyles, D.L.; Mena, A.; Pedreira, J.D.; Castro-Iglesias, A.; Cachay, E. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antiviral Res. 2014, 108, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, M.; Umemura, A.; Itoh, Y. Current status and future prospects of chemotherapy for advanced hepatocellular carcinoma. Clin. J. Gastroenterol. 2016, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Adnane, J.; Trail, P.A.; Levy, J.; Henderson, A.; Xue, D.; Bortolon, E.; Ichetovkin, M.; Chen, C.; McNabola, A.; et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother. Pharmacol. 2007, 59, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Senkardes, S.; Kaushik-Basu, N.; Durmaz, I.; Manvar, D.; Basu, A.; Atalay, R.; Kucukguzel, S.G. Synthesis of novel diflunisal hydrazide-hydrazones as anti-hepatitis C virus agents and hepatocellular carcinoma inhibitors. Eur. J. Med. Chem. 2016, 108, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Jetten, M.J.; Ruiz-Aracama, A.; Coonen, M.L.; Claessen, S.M.; van Herwijnen, M.H.; Lommen, A.; van Delft, J.H.; Peijnenburg, A.A.; Kleinjans, J.C. Interindividual variation in gene expression responses and metabolite formation in acetaminophen-exposed primary human hepatocytes. Arch. Toxicol. 2016, 90, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Ullah, S.; Schmidt, S.; Nandania, J.; Velagapudi, V.; Beck, O.; Ingelman-Sundberg, M.; Lauschke, V.M. Endogenous and xenobiotic metabolic stability of primary human hepatocytes in long-term 3D spheroid cultures revealed by a combination of targeted and untargeted metabolomics. FASEB J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman-Rossi, J.; Dufour, J.F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Keogh, A.; Treves, S.; Idle, J.R.; Beyoglu, D. The metabolomic profile of gamma-irradiated human hepatoma and muscle cells reveals metabolic changes consistent with the warburg effect. PeerJ 2016, 4, e1624. [Google Scholar] [CrossRef] [PubMed]
- The Human Metabolome Database. Available online: http://www.hmdb.ca/ (accessed on 2 June 2017).
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, G.; Yang, L.; Gajewski, C.D.; Mattiazzi, M. Measurements of ATP in mammalian cells. Methods 2002, 26, 317–326. [Google Scholar] [CrossRef]
- Sweet, S.; Singh, G. Accumulation of human promyelocytic leukemic (HL-60) cells at two energetic cell cycle checkpoints. Cancer Res. 1995, 55, 5164–5167. [Google Scholar] [PubMed]
- Sigoillot, F.D.; Berkowski, J.A.; Sigoillot, S.M.; Kotsis, D.H.; Guy, H.I. Cell cycle-dependent regulation of pyrimidine biosynthesis. J. Biol. Chem. 2003, 278, 3403–3409. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Idle, J.R. The glycine deportation system and its pharmacological consequences. Pharmacol. Ther. 2012, 135, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Smith, R.L.; Idle, J.R. Dog bites man or man bites dog? The enigma of the amino acid conjugations. Biochem. Pharmacol. 2012, 83, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Jung, H.J.; Hyun, D.H.; Park, E.H.; Kim, Y.M.; Lim, C.J. Glutathione reductase plays an anti-apoptotic role against oxidative stress in human hepatoma cells. Biochimie 2010, 92, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Njalsson, R. Glutathione synthetase deficiency. Cell Mol. Life Sci. 2005, 62, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Hammad, N.; Rosas-Lemus, M.; Uribe-Carvajal, S.; Rigoulet, M.; Devin, A. The crabtree and warburg effects: Do metabolite-induced regulations participate in their induction? Biochim. Biophys. Acta 2016, 1857, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Dashty, M. A quick look at biochemistry: Carbohydrate metabolism. Clin. Biochem. 2013, 46, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Steelman, S.L.; Cirillo, V.J.; Tempero, K.F. The chemistry, pharmacology and clinical pharmacology of diflunisal. Curr. Med. Res. Opin. 1978, 5, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Powers, R. The current state of drug discovery and a potential role for nmr metabolomics. J. Med. Chem. 2014, 57, 5860–5870. [Google Scholar] [CrossRef] [PubMed]
- Frederich, M.; Pirotte, B.; Fillet, M.; de Tullio, P. Metabolomics as a challenging approach for medicinal chemistry and personalized medicine. J. Med. Chem. 2016, 59, 8649–8666. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.H.; Xie, X.; Xu, F.; Shi, X.; Wang, Y.; Deng, L. Distinctive pharmacological differences between liver cancer cell lines HepG2 and Hep3B. Cytotechnology 2015, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K.; Ortiz, A.; Said, H.M. Biotin uptake by human intestinal and liver epithelial cells: Role of the SMVT system. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Milner, R.E. The mechanism of pantothenate transport by rat liver parenchymal cells in primary culture. J. Biol. Chem. 1985, 260, 4823–4831. [Google Scholar] [PubMed]
- Vadlapudi, A.D.; Vadlapatla, R.K.; Mitra, A.K. Sodium dependent multivitamin transporter (SMVT): A potential target for drug delivery. Curr. Drug Targets 2012, 13, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ito, K.; Ohtsuki, S.; Kubo, Y.; Suzuki, T.; Terasaki, T. Major involvement of Na+-dependent multivitamin transporter (SLC5A6/SMVT) in uptake of biotin and pantothenic acid by human brain capillary endothelial cells. J. Neurochem. 2015, 134, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Portmann, S.; Fahrner, R.; Lechleiter, A.; Keogh, A.; Overney, S.; Laemmle, A.; Mikami, K.; Montani, M.; Tschan, M.P.; Candinas, D.; et al. Antitumor effect of sirt1 inhibition in human hcc tumor models in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 499–508. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Retention Time (min) | Derivatives Formed | HMDB Number | HepG2 | Hep3B |
|---|---|---|---|---|---|
| Ethanolamine | 9.26 | 2TMS | 00149 | ↓↓↓ | |
| 12.94 | 3TMS | ||||
| Glycolic acid | 9.97 | 2TMS | 00115 | ↓↓ | |
| Glycine | 10.67 | 2TMS | 00123 | ↑ | |
| 13.54 | 3TMS | ||||
| Valine | 12.18 | 2TMS | 00883 | ↓ | |
| Leucine | 12.99 | 2TMS | 00687 | ↓ | |
| Isoleucine | 13.32 | 2TMS | 00172 | ↓ | |
| Proline | 13.41 | 2TMS | 00162 | ↓ | |
| Succinic acid | 13.53 | 2TMS | 00254 | ↓ | |
| Glyceric acid | 13.83 | 3TMS | 00139 | ↑↑ | |
| Uracil | 13.96 | 2TMS | 00300 | ↑↑↑↑ | |
| Serine | 14.24 | 3TMS | 00187 | ↓ | |
| Threonine | 14.62 | 3TMS | 00167 | ↓ | |
| Aminomalonic acid | 15.73 | 3TMS | 01147 | ↓ | |
| Erythritol | 16.20 | 4TMS | 02994 | ↓↓ | |
| Aspartic acid | 16.32 | 3TMS | 00191 | ↓↓↓ | |
| 5-Oxoproline | 16.40 | 2TMS | 00267 | ↑↑↑↑ | ↓ |
| Erythronic acid | 16.67 | 4TMS | 00613 | ↓↓ | |
| Cysteine | 16.79 | 3TMS | 00574 | ↑ | ↓ |
| Creatinine | 16.87 | 3TMS | 00562 | ↓ | ↓ |
| 2-Oxoglutaric acid | 16.96 | 2TMS; 1MOX | 00208 | ↑ | |
| Ornithine | 17.44 | 3TMS | 00214 | ↓ | |
| Glutamic acid | 17.50 | 3TMS | 00148 | ↑↑↑↑ | ↓ |
| Phenylalanine | 17.68 | 2TMS | 00159 | ↓ | |
| N-Acetylaspartic acid | 18.11 | 3TMS | 00812 | ↓ | |
| Lysine | 18.50 | 3TMS | 00182 | ↑↑↑ | ↓ |
| 20.82 | 4TMS | ||||
| Xylitol | 18.78 | 5TMS | 02917 | ↓ | |
| Glycerol 3-phosphate | 19.19 | 4TMS | 00126 | ↓ | |
| Glutamine | 19.25 | 3TMS | 00641 | ↓↓ | |
| Hypoxanthine | 19.66 | 2TMS | 00157 | ↑↑↑ | |
| Galactose | 20.61 | 5TMS; 1MOX | 00143 | ↑ | |
| 21.08 | 5TMS; 1MOX | ||||
| Tyrosine | 21.02 | 3TMS | 00158 | ↑↑ | ↓ |
| Pantothenic acid | 21.55 | 3TMS | 00210 | ↑↑↑↑ | |
| Myo-inositol | 22.64 | 6TMS | 00211 | ↓ | |
| Phosphoglycolic acid | 22.72 | 3TMS | 00816 | ↓ | |
| Ribose | 22.76 | 5TMS; 1MOX | 00283 | ↑↑↑ | |
| Oleic acid | 23.46 | 1TMS | 00207 | ↓ | |
| Fructose 6-phosphate | 24.59 | 6TMS; 1MOX | 00124 | ↑↑↑↑ | ↑ |
| Glucose 6-phosphate | 24.71 | 6TMS; 1MOX | 01401 | ↑↑↑↑ | ↑ |
| Arachidonic acid | 24.80 | 1TMS | 01043 | ↓ | |
| Sedoheptulose 7-phosphate | 24.88 | 7TMS; 1MOX | 01068 | ↑↑↑↑ | |
| Uridine | 25.63 | 3TMS | 00296 | ↑ | |
| Guanosine | 28.38 | 5TMS | 00133 | ↑↑ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keogh, A.; Şenkardeş, S.; Idle, J.R.; Küçükgüzel, Ş.G.; Beyoğlu, D. A Novel Anti-Hepatitis C Virus and Antiproliferative Agent Alters Metabolic Networks in HepG2 and Hep3B Cells. Metabolites 2017, 7, 23. https://doi.org/10.3390/metabo7020023
Keogh A, Şenkardeş S, Idle JR, Küçükgüzel ŞG, Beyoğlu D. A Novel Anti-Hepatitis C Virus and Antiproliferative Agent Alters Metabolic Networks in HepG2 and Hep3B Cells. Metabolites. 2017; 7(2):23. https://doi.org/10.3390/metabo7020023
Chicago/Turabian StyleKeogh, Adrian, Sevil Şenkardeş, Jeffrey R. Idle, Ş. Güniz Küçükgüzel, and Diren Beyoğlu. 2017. "A Novel Anti-Hepatitis C Virus and Antiproliferative Agent Alters Metabolic Networks in HepG2 and Hep3B Cells" Metabolites 7, no. 2: 23. https://doi.org/10.3390/metabo7020023