
Accurate Measurement of the in vivo Ammonium Concentration in Saccharomyces cerevisiae

Abstract
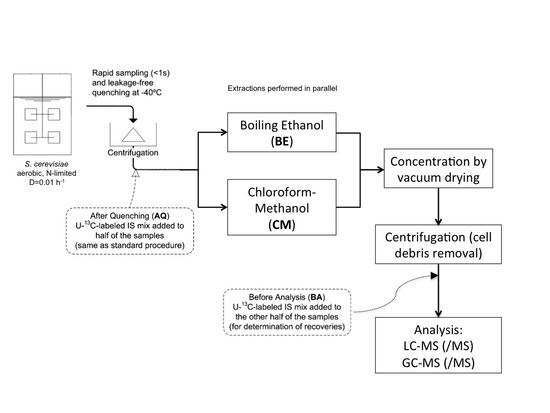
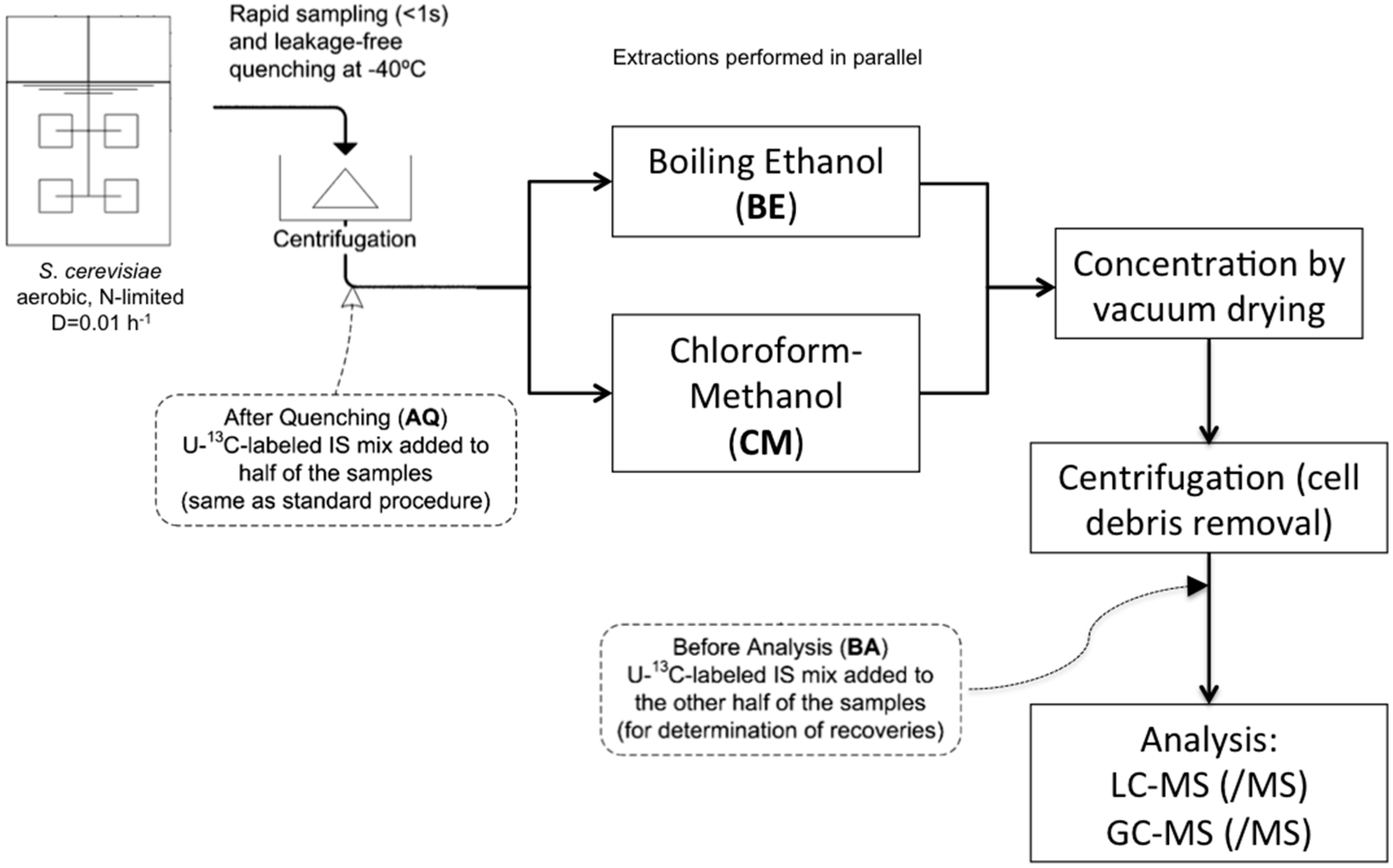
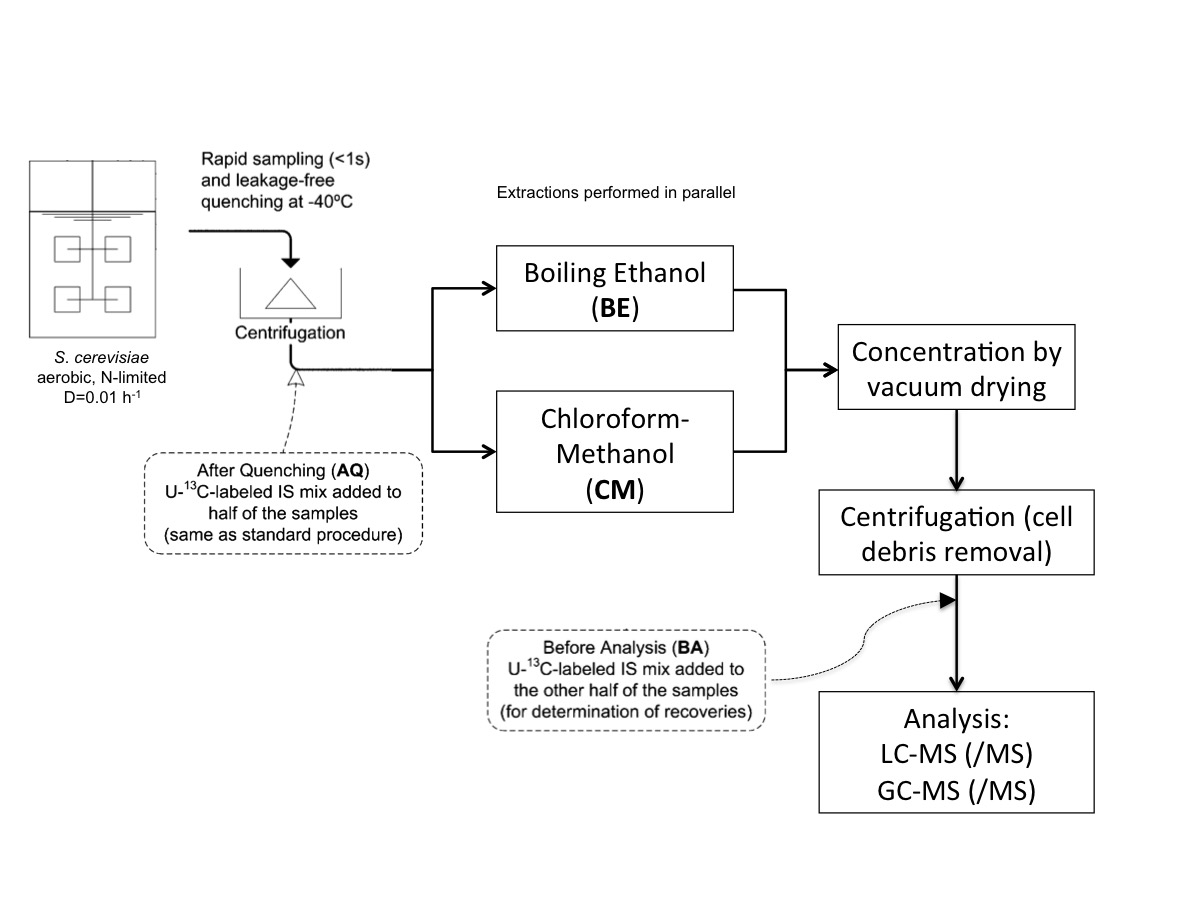
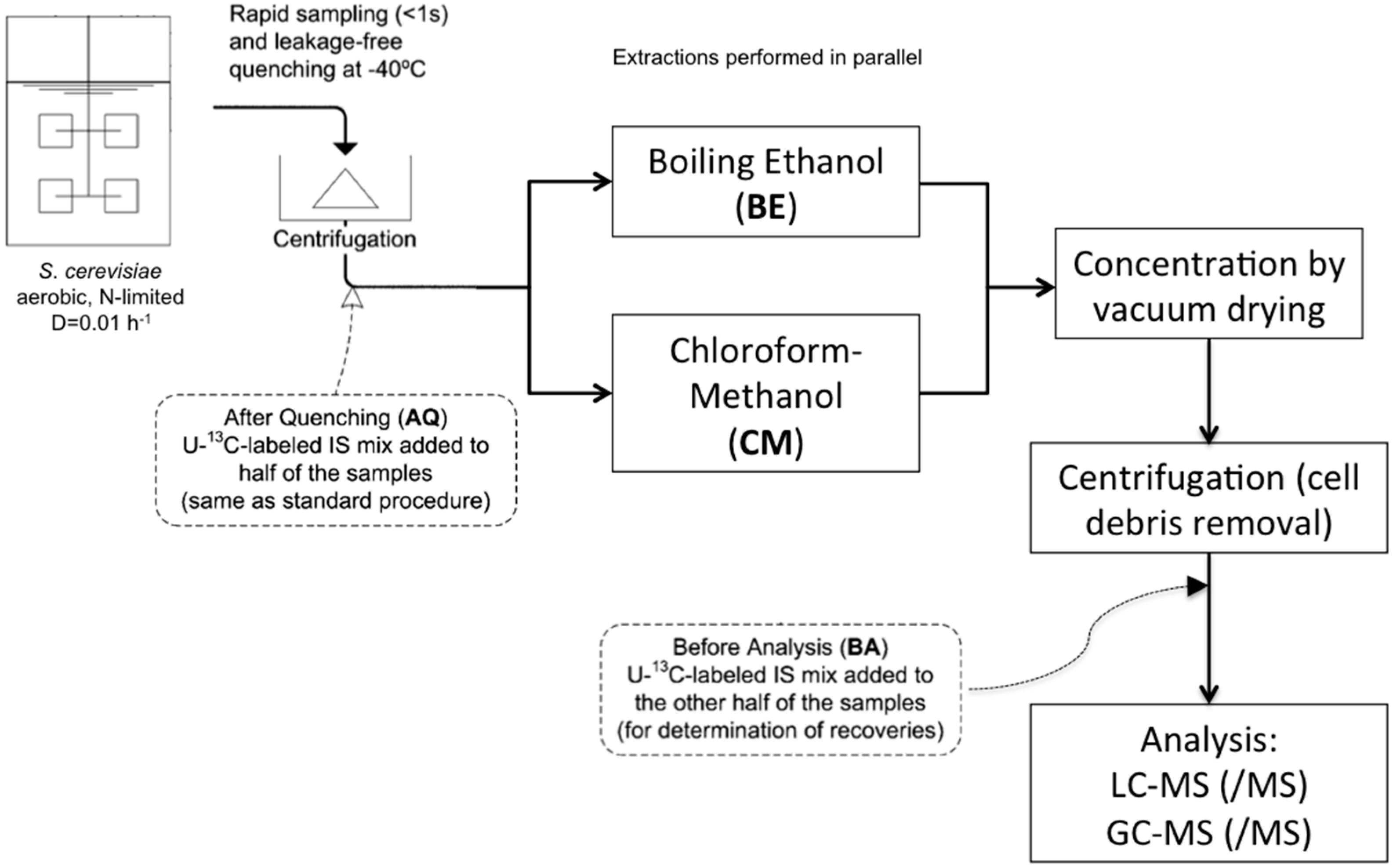
:
1. Introduction
- (1)
- Formation or consumption of NH4+ during sample processing must be avoided.
- (2)
- Extensive leakage of NH4+ must be absent.
- (3)
- Extraction of intracellular NH4+ from biomass must be fast and complete.
- (4)
- Proper internal standards must be applied.
2. Results
2.1. Reproducibility and Linearity of the Method Using Standards
2.2. Comparison between Centrifugation and Filtration for Biomass Separation
2.3. NH3-Evaporation during Rapid Vap Drying
2.4. Absence of Matrix Effects
2.5. Intracellular Ammonium Quantification Requires Extraction with Cold-Buffered Methanol-Chloroform
2.6. Amino Acid Degradation
3. Discussion
4. Materials and Methods
4.1. Strain and Culture Conditions
4.2. Standards
4.3. Sampling and Sample Preparation
4.3.1. Samples for Extracellular Metabolites and Ammonium Analysis
4.3.2. Rapid Sampling and Biomass Extraction for Intracellular Metabolites and Intracellular Ammonium
4.3.3. Boiling Ethanol Extraction of the Biomass Pellet
4.3.4. CM5: Cold Chloroform-Methanol Buffered at pH = 5 Extraction of the Biomass Pellet
4.3.5. CM: Cold Non-Buffered Chloroform-Methanol Extraction of the Biomass Pellet
4.3.6. TBE5: Boiling Buffered Ethanol pH = 5 Extraction of Total Broth
4.3.7. TBE: Boiling Non-Buffered Ethanol Extraction of Total Broth
4.3.8. TCM5: Cold Chloroform-Methanol Buffered at pH = 5 Extraction of Total Broth
4.4. Analytical Methods
4.4.1. Ammonium Quantification
4.4.2. Metabolite Quantification
4.5. Data Reconciliation
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Ethics Approval
Abbreviations
| αKG | 2-Oxoglutarate |
| AQ | After quenching |
| BA | Before analysis |
| BE | Boiling ethanol intracellular metabolite extraction (non-buffered) |
| BE5 | Buffered (pH = 5) boiling ethanol intracellular metabolite extraction |
| CM | Cold chloroform-methanol-water intracellular metabolite extraction (non-buffered) |
| CM5 | Cold-buffered (pH = 5) chloroform-methanol-water intracellular metabolite extraction |
| DEEMM | Diethyl ethoxymethylenemalonate |
| DOT | Dissolved oxygen tension |
| IC | Intracellular |
| IDMS | Isotope dilution mass spectrometry |
| PEP | Phosphoenolpyruvate |
| SIM | Selected ion monitoring |
| TB | Total broth |
| TBE | Boiling non-buffered ethanol extraction of total broth |
| TBE5 | Boiling buffered (pH = 5) ethanol extraction of total broth |
| TCM5 | Cold chloroform-methanol buffered (pH = 5) extraction of total broth |
| UHPLC-IDMS | Ultra high performance liquid chromatography-isotope dilution mass spectrometry |
References
- Kim, M.; Zhang, Z.; Okano, H.; Yan, D.; Groisman, A.; Hwa, T. Need-based activation of ammonium uptake in escherichia coli. Mol. Syst. Biol. 2012, 8, 616. [Google Scholar] [CrossRef] [PubMed]
- Magasanik, B. Ammonia assimilation by saccharomyces cerevisiae. Eukaryot. Cell 2003, 2, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Ljungdahl, P.O.; Daignan-Fornier, B. Regulation of amino acid, nucleotide, and phosphate metabolism in saccharomyces cerevisiae. Genetics 2012, 190, 885–929. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Schothorst, J.; Kankipati, H.N.; Van Zeebroeck, G.; Rubio-Texeira, M.; Thevelein, J.M. Nutrient sensing and signaling in the yeast saccharomyces cerevisiae. FEMS Microbiol. Rev. 2014, 38, 254–299. [Google Scholar] [CrossRef] [PubMed]
- Magasanik, B.; Kaiser, C.A. Nitrogen regulation in saccharomyces cerevisiae. Gene 2002, 290, 1–18. [Google Scholar] [CrossRef]
- Dikicioglu, D.; Karabekmez, E.; Rash, B.; Pir, P.; Kirdar, B.; Oliver, S.G. How yeast re-programmes its transcriptional profile in response to different nutrient impulses. BMC Syst. Biol. 2011, 5, 148. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D. The transport of nh3 and hn4+ across biological membranes. Biochim .Biophys. Acta Rev. Bioenerg. 1981, 639, 41–52. [Google Scholar] [CrossRef]
- Roon, R.J.; Even, H.L.; Dunlop, P.; Larimore, F.L. Methylamine and ammonia transport in saccharomyces cerevisiae. J. Bacteriol. 1975, 122, 502–509. [Google Scholar] [PubMed]
- Van Nuland, A.; Vandormael, P.; Donaton, M.; Alenquer, M.; Lourenco, A.; Quintino, E.; Versele, M.; Thevelein, J.M. Ammonium permease-based sensing mechanism for rapid ammonium activation of the protein kinase a pathway in yeast. Mol. Microbiol. 2006, 59, 1485–1505. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lai, L.; Ouyang, Q.; Tang, C. Flux balance analysis of ammonia assimilation network in e. Coli predicts preferred regulation point. PloS One 2011, 6, e16362. [Google Scholar] [CrossRef] [PubMed]
- Barreto, L.; Canadell, D.; Valverde-Saubi, D.; Casamayor, A.; Arino, J. The short-term response of yeast to potassium starvation. Environ. Microbiol. 2012, 14, 3026–3042. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.J.; Cooper, T.G. Tor1/2 regulation of retrograde gene expression in saccharomyces cerevisiae derives indirectly as a consequence of alterations in ammonia metabolism. J. Biol. Chem. 2003, 278, 36924–36933. [Google Scholar] [CrossRef] [PubMed]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Maleki Seifar, R.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 2009, 81, 7379–7389. [Google Scholar] [CrossRef] [PubMed]
- Niedenfuhr, S.; Pierick, A.T.; van Dam, P.T.; Suarez-Mendez, C.A.; Noh, K.; Wahl, S.A. Natural isotope correction of ms/ms measurements for metabolomics and c fluxomics. Biotechnol. Bioeng. 2015. [Google Scholar]
- Suarez-Mendez, C.A.; Sousa, A.; Heijnen, J.J.; Wahl, A. Fast “feast/famine” cycles for studying microbial physiology under dynamic conditions: A case study with saccharomyces cerevisiae. Metabolites 2014, 4, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Jules, M.; Beltran, G.; Francois, J.; Parrou, J.L. New insights into trehalose metabolism by saccharomyces cerevisiae: Nth2 encodes a functional cytosolic trehalase, and deletion of tps1 reveals ath1p-dependent trehalose mobilization. Appl. Environ. Microbiol. 2008, 74, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Boer, V.M.; de Winde, J.H.; Pronk, J.T.; Piper, M.D. The genome-wide transcriptional responses of saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J. Biol. Chem. 2003, 278, 3265–3274. [Google Scholar] [CrossRef] [PubMed]
- Mashego, M.R.; van Gulik, W.M.; Vinke, J.L.; Visser, D.; Heijnen, J.J. In vivo kinetics with rapid perturbation experiments in saccharomyces cerevisiae using a second-generation bioscope. Metab. Eng. 2006, 8, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.C.; Eman, M.; van Zuijlen, G.; Visser, D.; van Dam, J.C.; Frank, J.; de Mattos, M.J.; Heijnen, J.J. Improved rapid sampling for in vivo kinetics of intracellular metabolites in saccharomyces cerevisiae. Biotechnol. Bioeng. 2001, 75, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Redruello, B.; Ladero, V.; Cuesta, I.; Alvarez-Buylla, J.R.; Martin, M.C.; Fernandez, M.; Alvarez, M.A. A fast, reliable, ultra high performance liquid chromatography method for the simultaneous determination of amino acids, biogenic amines and ammonium ions in cheese, using diethyl ethoxymethylenemalonate as a derivatising agent. Food Chem. 2013, 139, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Maleki Seifar, R.; Ras, C.; Deshmukh, A.T.; Bekers, K.M.; Suarez-Mendez, C.A.; da Cruz, A.L.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of intracellular coenzymes in saccharomyces cerevisiae using ion pair reversed phase ultra high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2013, 1311, 115–120. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, L.P.; Buijs, N.A.; ten Pierick, A.; Deshmukh, A.; Zhao, Z.; Kiel, J.A.; Heijnen, J.J.; van Gulik, W.M. Scale-down of penicillin production in penicillium chrysogenum. Biotechnol. J. 2011, 6, 944–958. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, P.J.T. Data reconciliation and error detection. In The Metabolic Pathway Engineering Handbook; Smolke, C.D., Ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 8.1–8.13. [Google Scholar]

{kind=link}
{kind=link}
| Sample | Measured Concentration µMsample (measurement-spike) | Average ± St. Dev. µMsample |
|---|---|---|
| Extracellular 1 (Oct 14) | 6.1 | 4.2 ± 2.8 |
| Extracellular 2 (Oct 14) | 2.2 | |
| Extracellular 3 (Oct 14) | N.D. | |
| Spiked extracellular 1 (Oct 14) | 61.1 (11.1) | 9.2 ± 2.1 1 |
| Spiked extracellular 2 (Oct 14) | 57.0 (7.0) | |
| Spiked extracellular 3 (Oct 14) | 59.5 (9.5) | |
| Intracellular 4 (Oct 14) | 35.9 | 31.2 ± 4.1 |
| Intracellular 5 (Oct 14) | 28.7 | |
| Intracellular 6 (Oct 14) | 28.9 | |
| Spiked intracellular 4 (Oct 14) | 94.4 (44.4) | 34.3 ± 9.0 1 |
| Spiked intracellular 5 (Oct 14) | 81.3 (31.3) | |
| Spiked intracellular 6 (Oct 14) | 77.2 (27.2) | |
| Total broth 4 (Oct 14) | 21.8 | 22.7 ± 1.3 |
| Total broth 5 (Oct 14) | 22.1 | |
| Total broth 6 (Oct 14) | 24.2 | |
| Spiked total broth 4 (Oct 14) | 77.8 (27.8) | 29.1 ± 6.5 1 |
| Spiked total broth 5 (Oct 14) | 73.3 (23.3) | |
| Spiked total broth 6 (Oct 14) | 86.2 (36.2) |
| Type of Sample | Concentration Measured 1 (µMbroth) | Recovery (%) | Reconciled Concentration * (µMbroth) |
|---|---|---|---|
| Intracellular BE (Aug 14) | 24.51 ± 2.15 | 124.50% | N.A. |
| Extracellular (Aug 14) | 6.23 ± 1.15 | ||
| Total broth BE (Aug 14) | 24.69 ± 5.25 | ||
| Intracellular BE5 (Oct 14) | 10.04 ± 1.93 | 77.93% | N.A. |
| Extracellular (Oct 14) | 1.05 ± 0.49 | ||
| Total broth BE5 (Oct 14) | 14.23 ± 0.23 | ||
| Intracellular CM5 (Oct 14) | 15.16 ± 1.24 | 96.66% | 15.57 ± 0.64 |
| Extracellular (Oct 14) | 1.05 ± 0.49 | 1.12 ± 0.46 | |
| Total broth CM5 (Oct 14) | 16.77 ± 0.57 | 16.68 ± 0.52 |
| 13C Addition | Extraction Solution | Intracellular Metabolite Concentration (µmol/gCDW) (% Recovery with Respect to 13C Addition after Quenching) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ala | Glu | Asp | Gln | PyrGlu | Pyruvate | αKG | NH4+ | ||
| AQ | BE5 | 6.97 ± 0.21 | 52.79 ± 0.36 | 11.51 ± 0.14 | 10.77 ± 0.25 | 0.78 ± 0.02 | 1.96 ± 0.17 | 3.01 ± 0.05 | 3.46 ± 0.28 |
| BA | BE5 | 5.73 ± 0.25 (82.3%) 1 | 51.05 ± 0.75 (96.7%) | 10.80 ± 0.33 (93.9%) | 9.81 ± 0.19 (91.2%) | 1.11 ± 0.02 (141.9%) 1 | N.M. | N.M. | N.M. |
| AQ | CM5 | 6.50 ± 0.07 | 51.95 ± 0.65 | 9.03 ± 0.09 2 | 10.85 ± 0.16 | 0.51 ± 0.02 2 | 0.21 ± 0.01 | 0.50 ± 0.01 | 2.29 ± 0.44 |
| BA | CM5 | 6.00 ± 0.05 (92.4%) 1 | 49.95 ± 0.30 (96.2%) | 8.90 ± 0.07 (98.5%) | 10.68 ± 0.07 (98.4%) | 0.57 ± 0.06 (112.2%) | N.M. | N.M. | N.M. |
| AQ | BE | 7.86 ± 0.13 | 56.00 ± 0.95 | 11.54 ± 0.28 | 11.28 ± 0.14 | N.M. | 2.17 ± 0.24 | 2.81 ± 0.06 | 5.78 ± 0.20 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cueto-Rojas, H.F.; Maleki Seifar, R.; Ten Pierick, A.; Heijnen, S.J.; Wahl, A. Accurate Measurement of the in vivo Ammonium Concentration in Saccharomyces cerevisiae. Metabolites 2016, 6, 12. https://doi.org/10.3390/metabo6020012
Cueto-Rojas HF, Maleki Seifar R, Ten Pierick A, Heijnen SJ, Wahl A. Accurate Measurement of the in vivo Ammonium Concentration in Saccharomyces cerevisiae. Metabolites. 2016; 6(2):12. https://doi.org/10.3390/metabo6020012
Chicago/Turabian StyleCueto-Rojas, Hugo F., Reza Maleki Seifar, Angela Ten Pierick, Sef J. Heijnen, and Aljoscha Wahl. 2016. "Accurate Measurement of the in vivo Ammonium Concentration in Saccharomyces cerevisiae" Metabolites 6, no. 2: 12. https://doi.org/10.3390/metabo6020012