Acylcarnitine Profiles in Acetaminophen Toxicity in the Mouse: Comparison to Toxicity, Metabolism and Hepatocyte Regeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
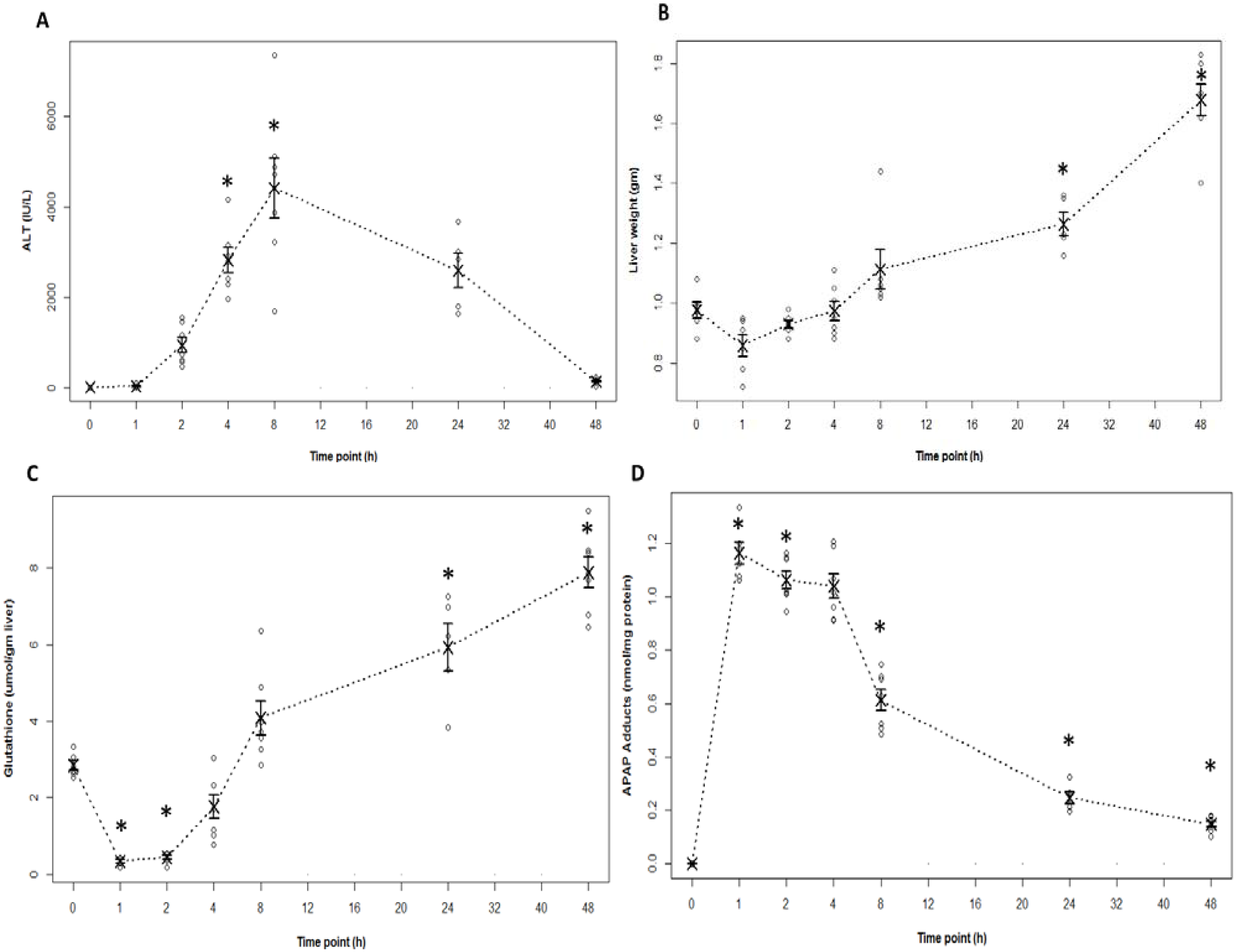
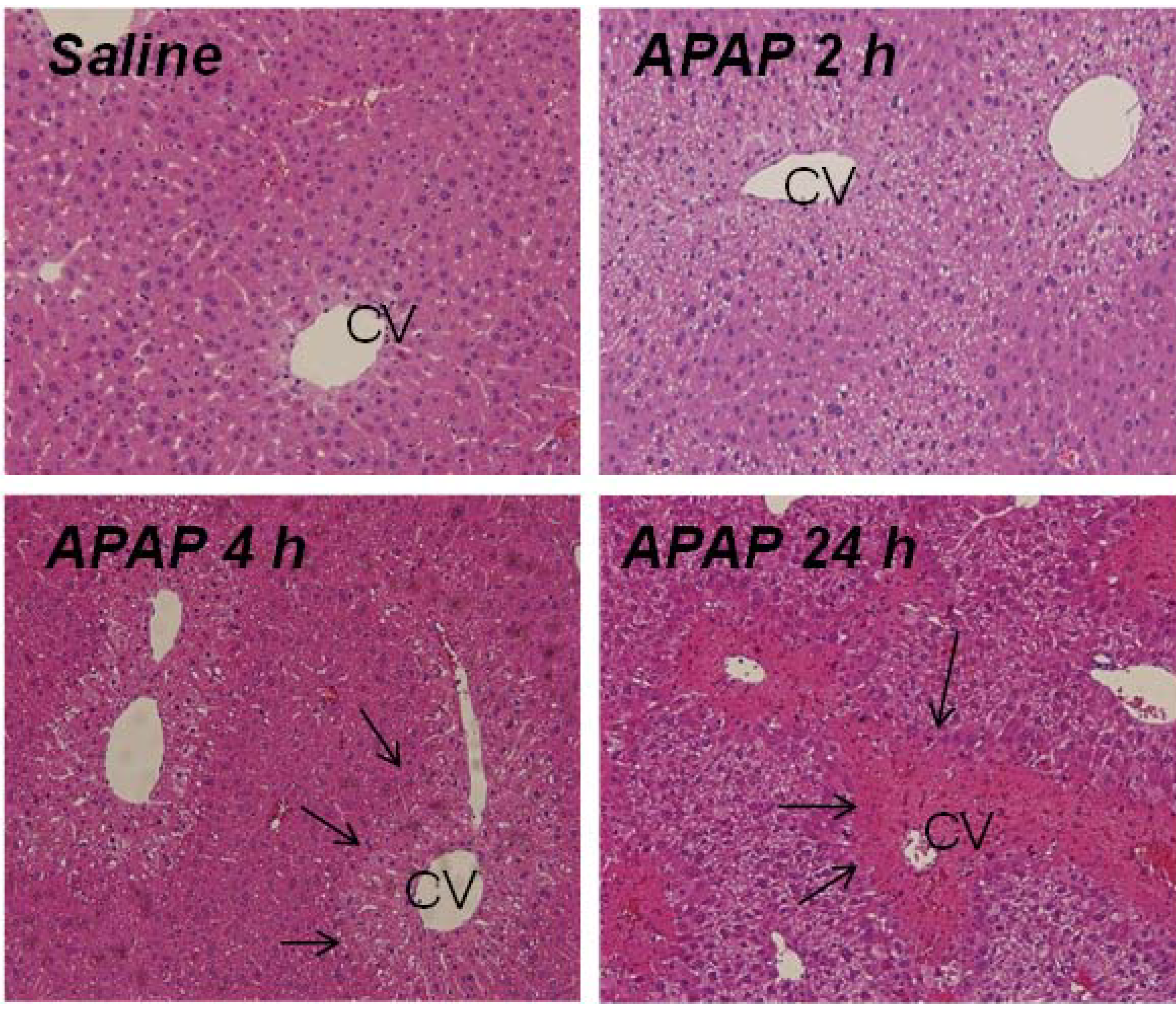
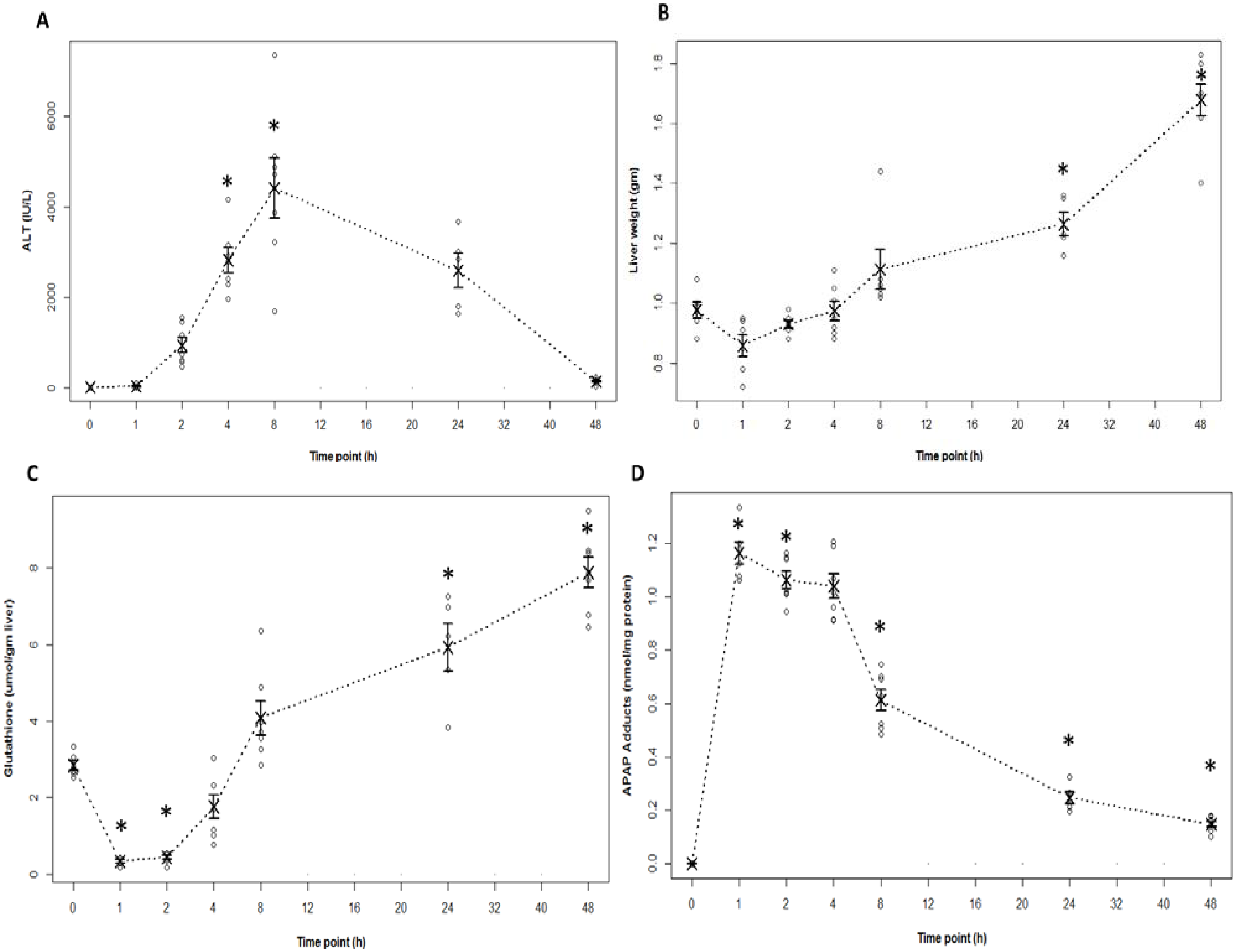
2.1. Indicators of Hepatotoxicity and Oxidative Metabolism in APAP Treated Mice
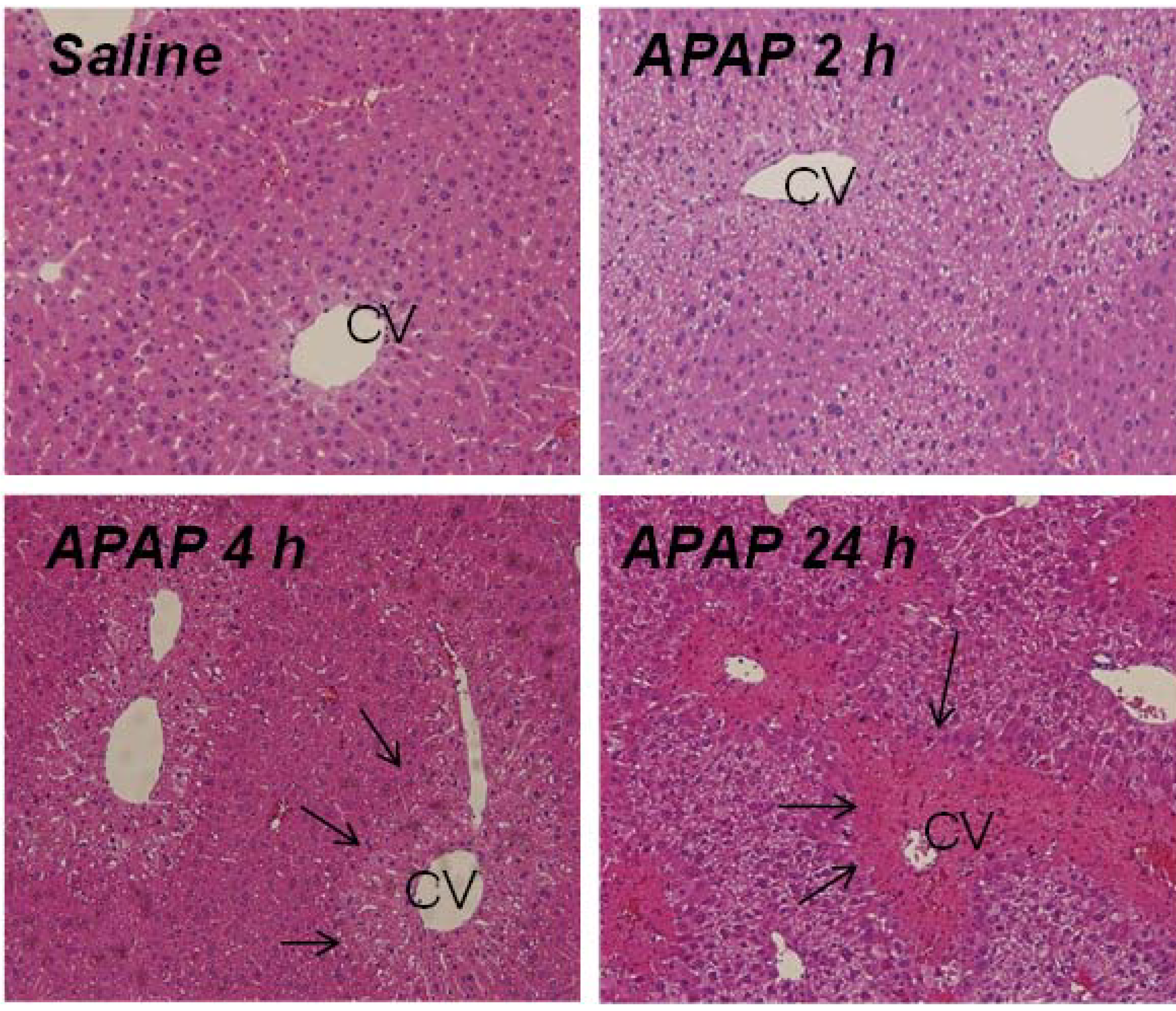
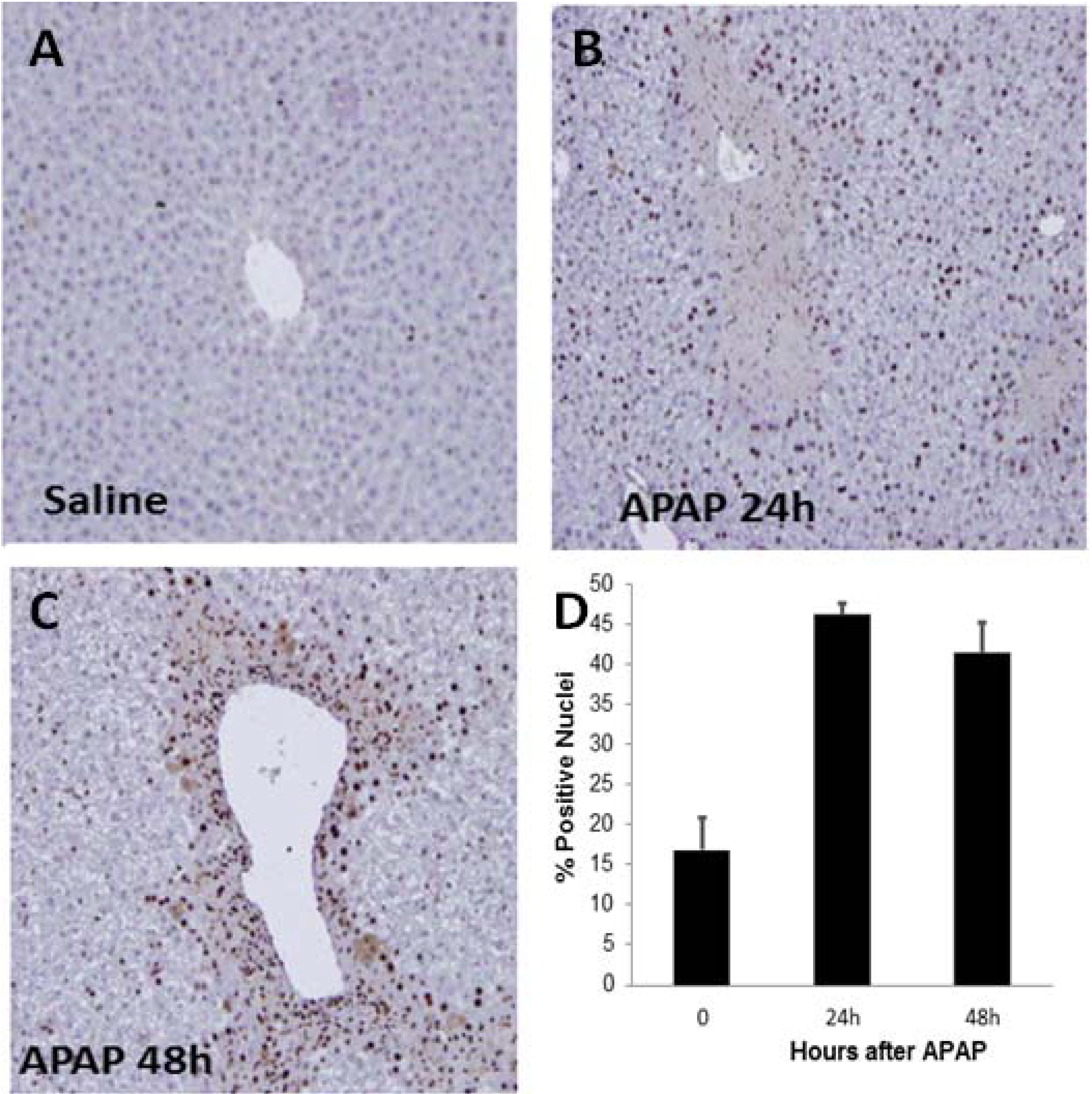
2.2. Indicators of Hepatocyte Regeneration in APAP Toxicity

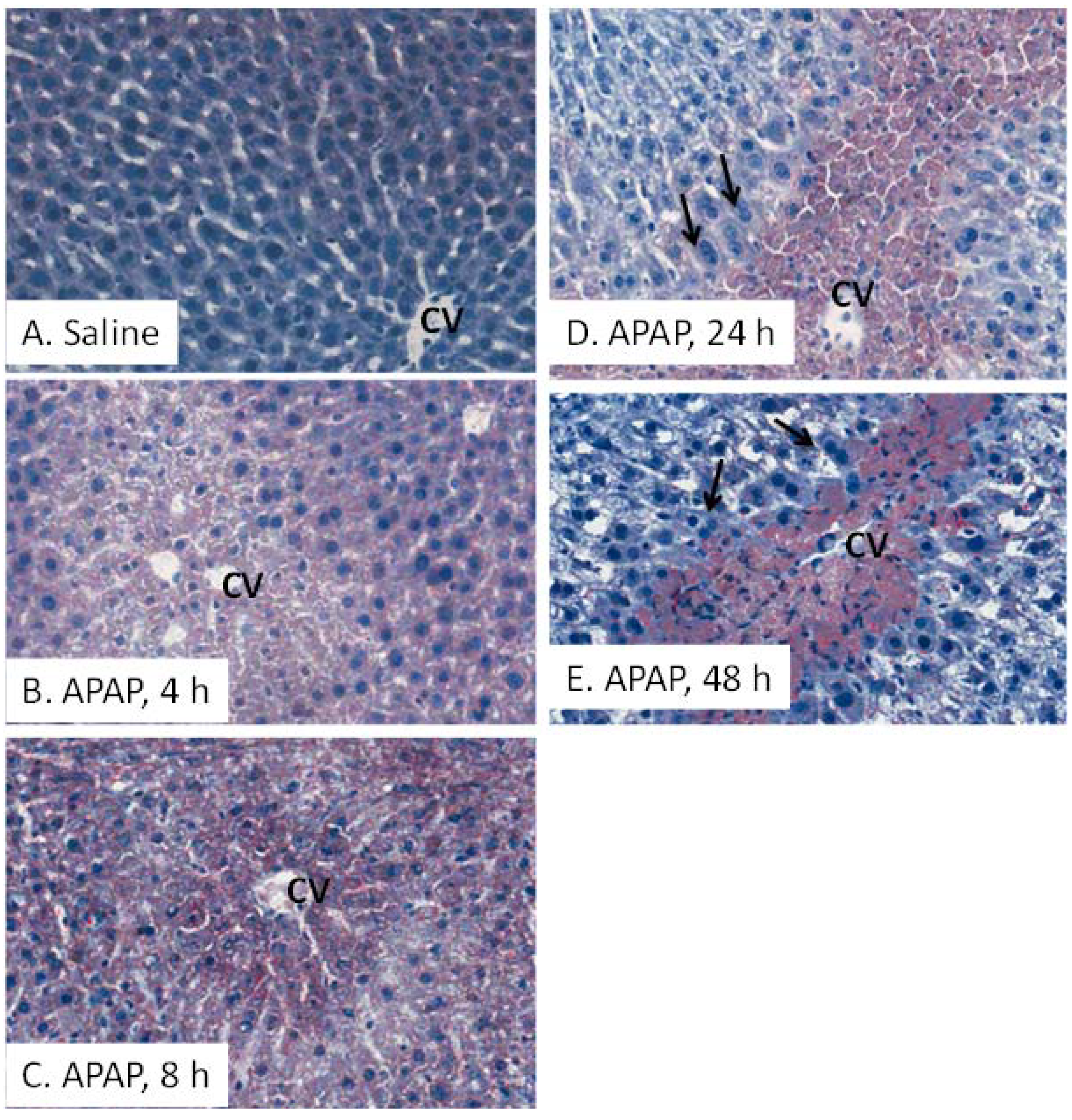
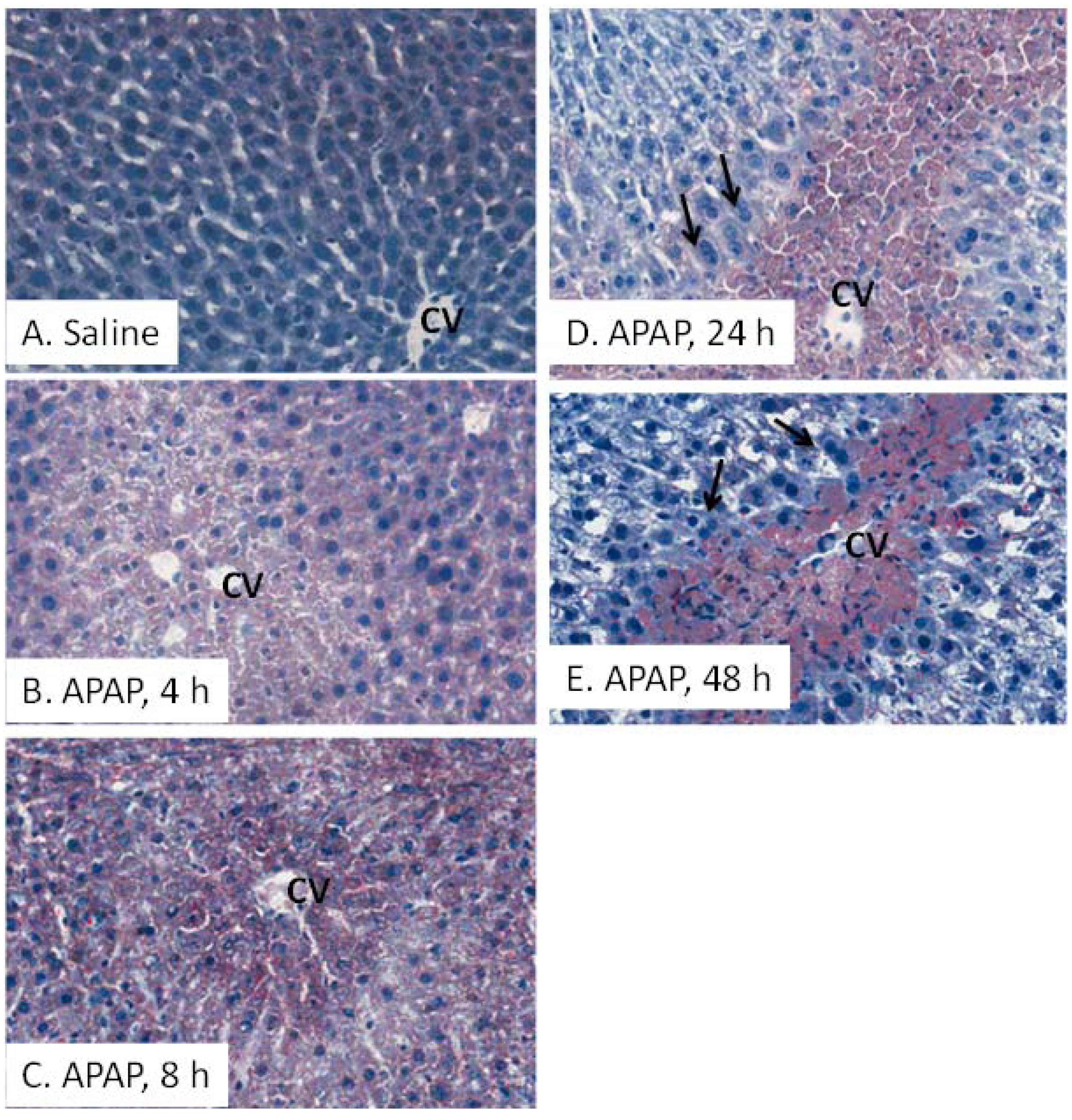
2.3. Oil Red O (ORO) Analysis and Liver Triglycerides
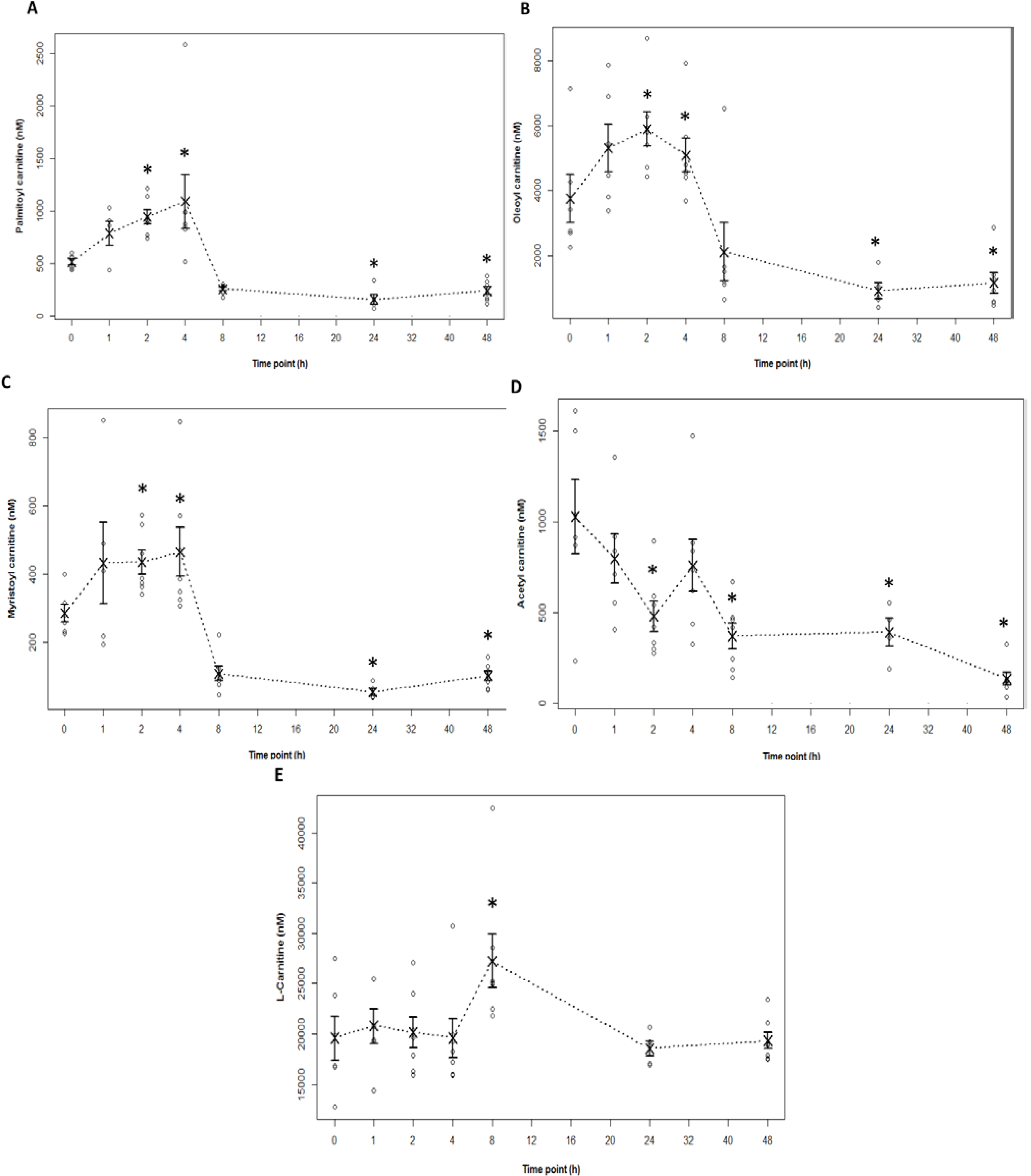
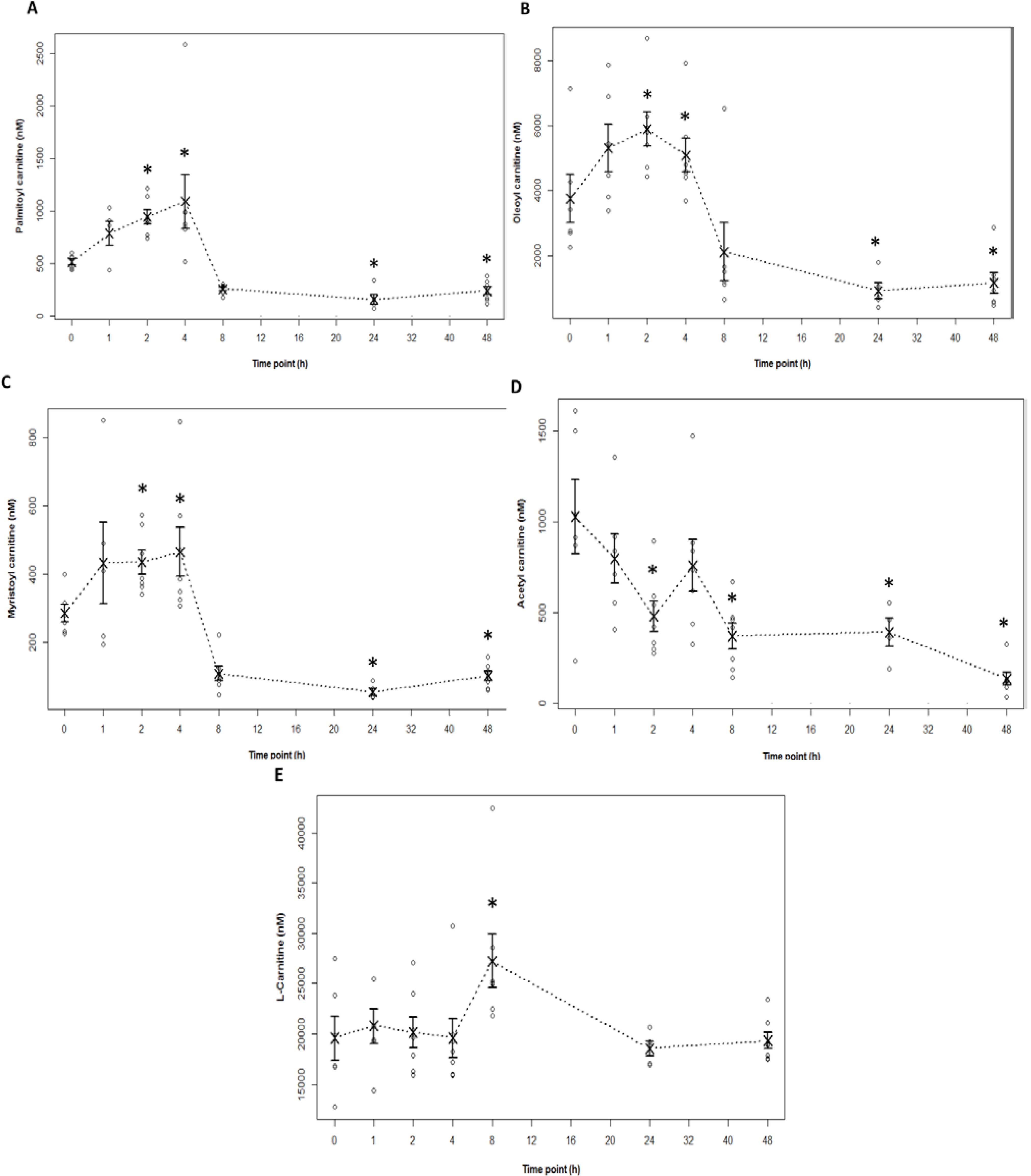
2.4. Acylcarnitine Profiles in APAP Toxicity in the Mouse
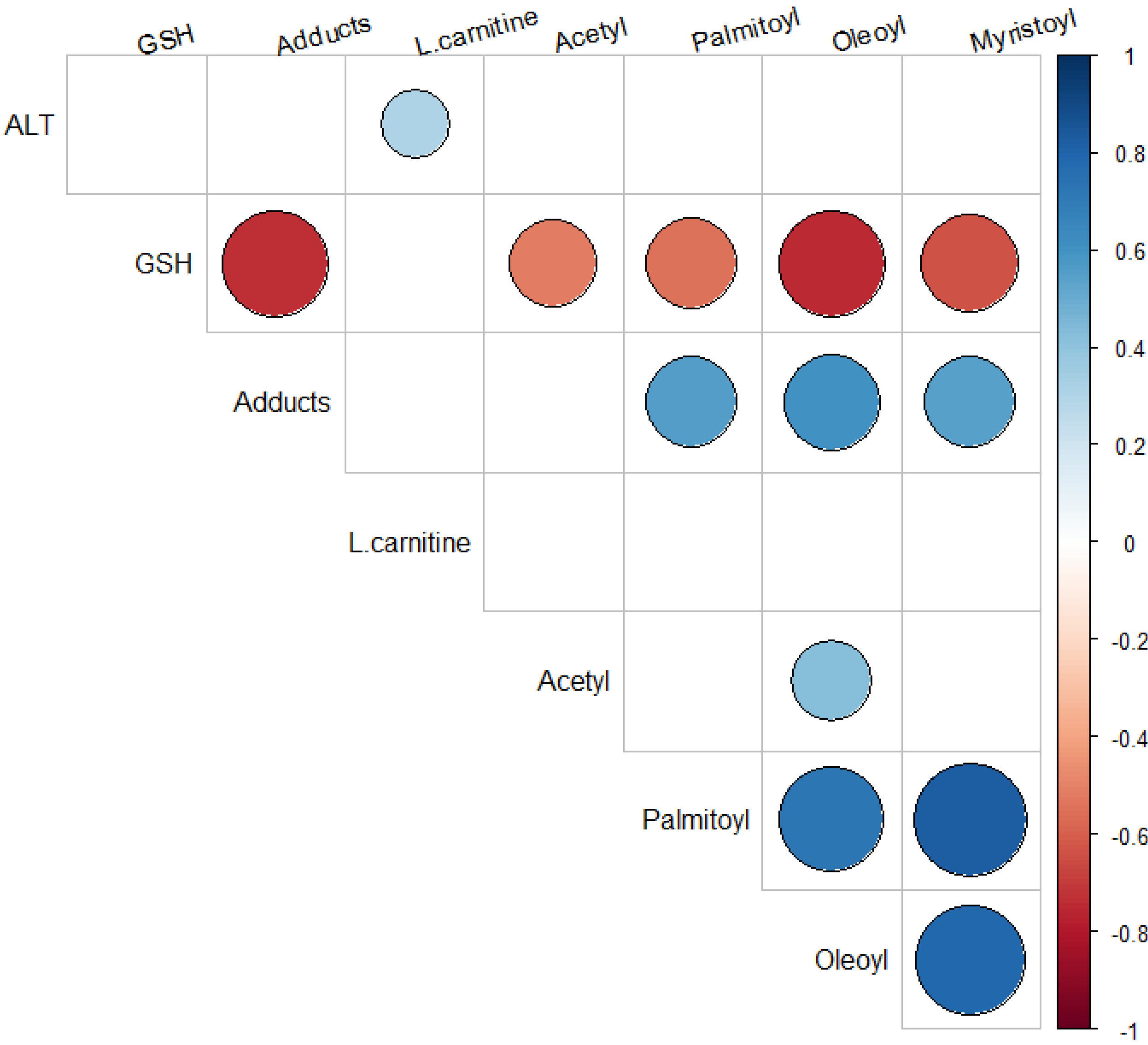
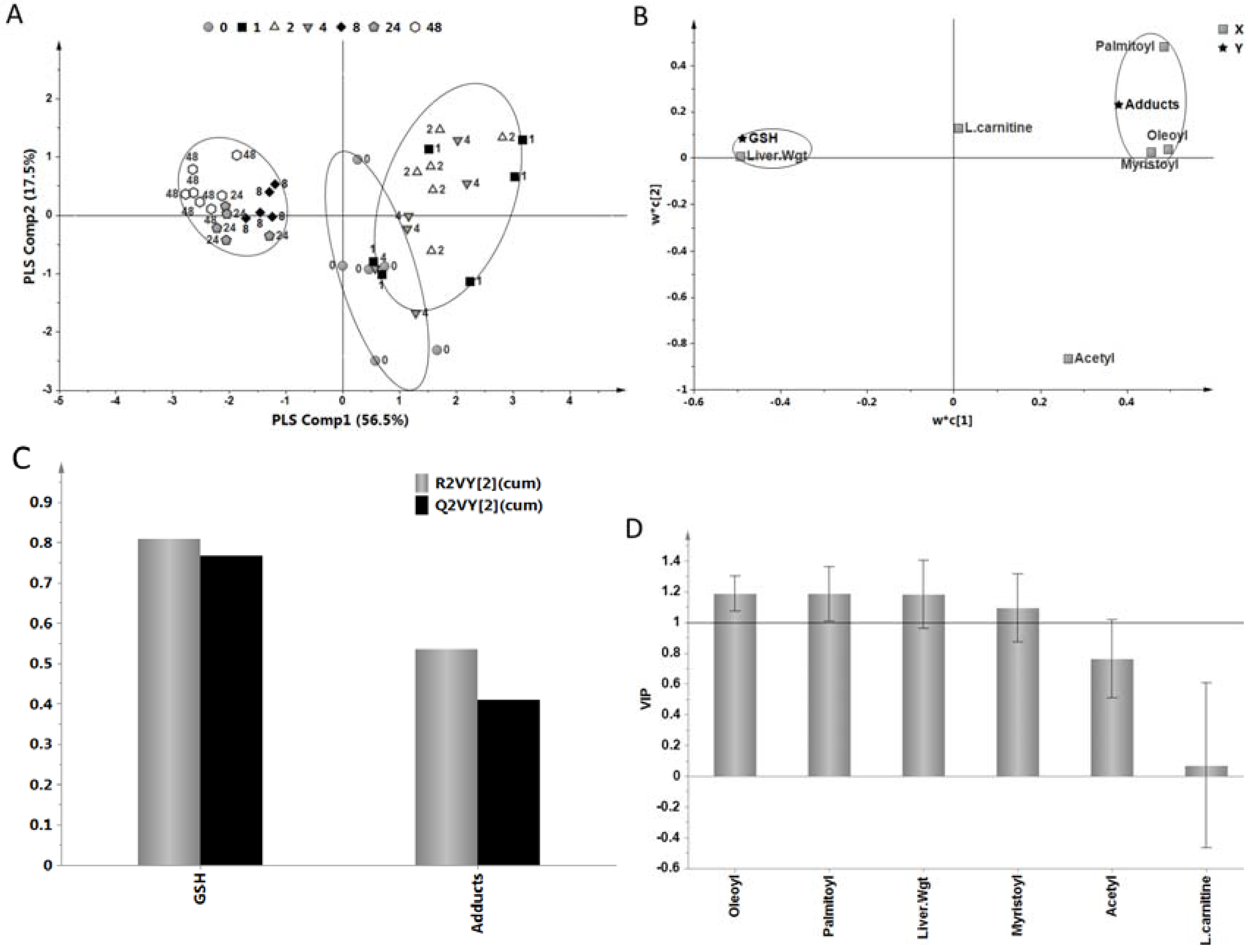
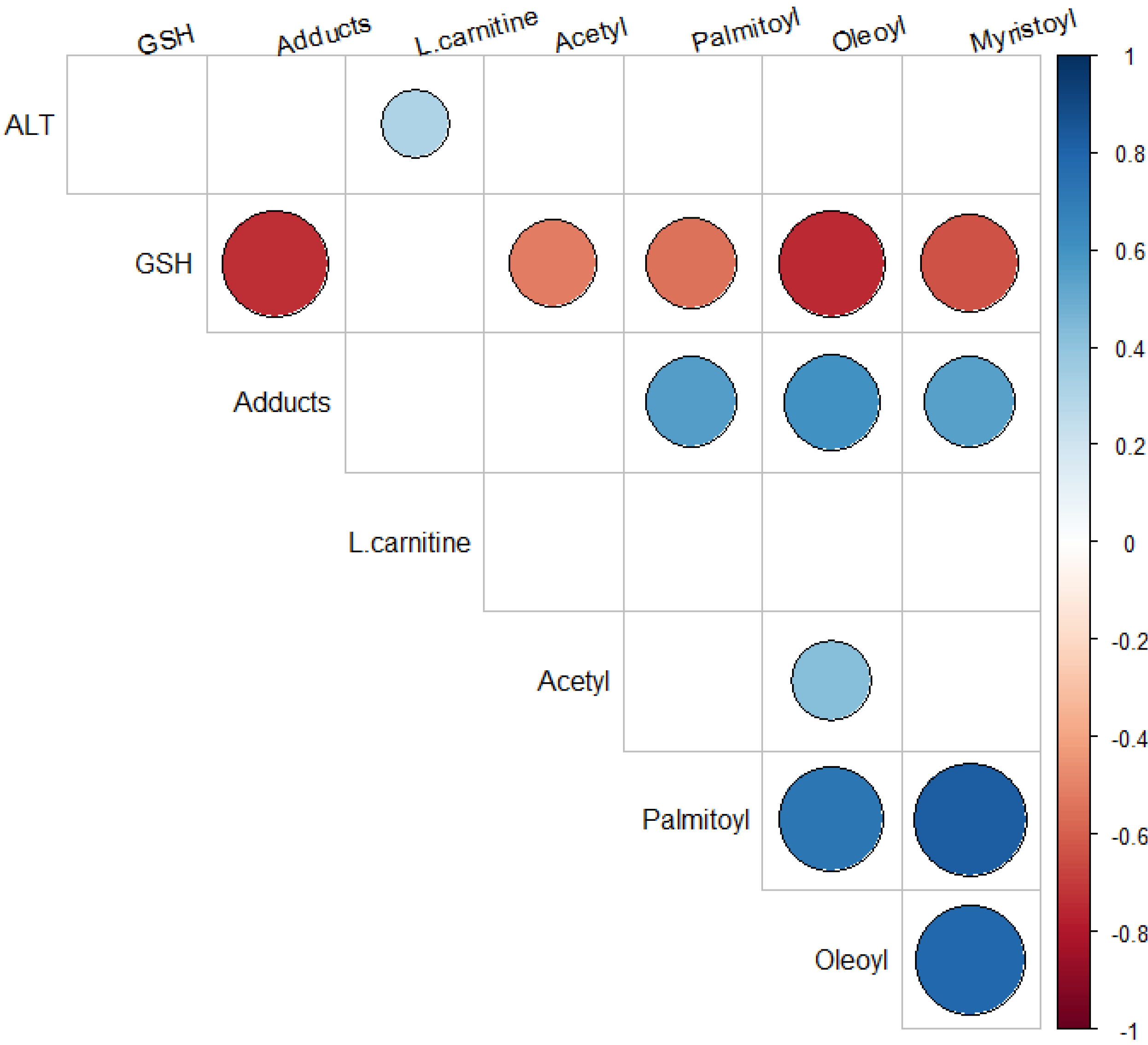
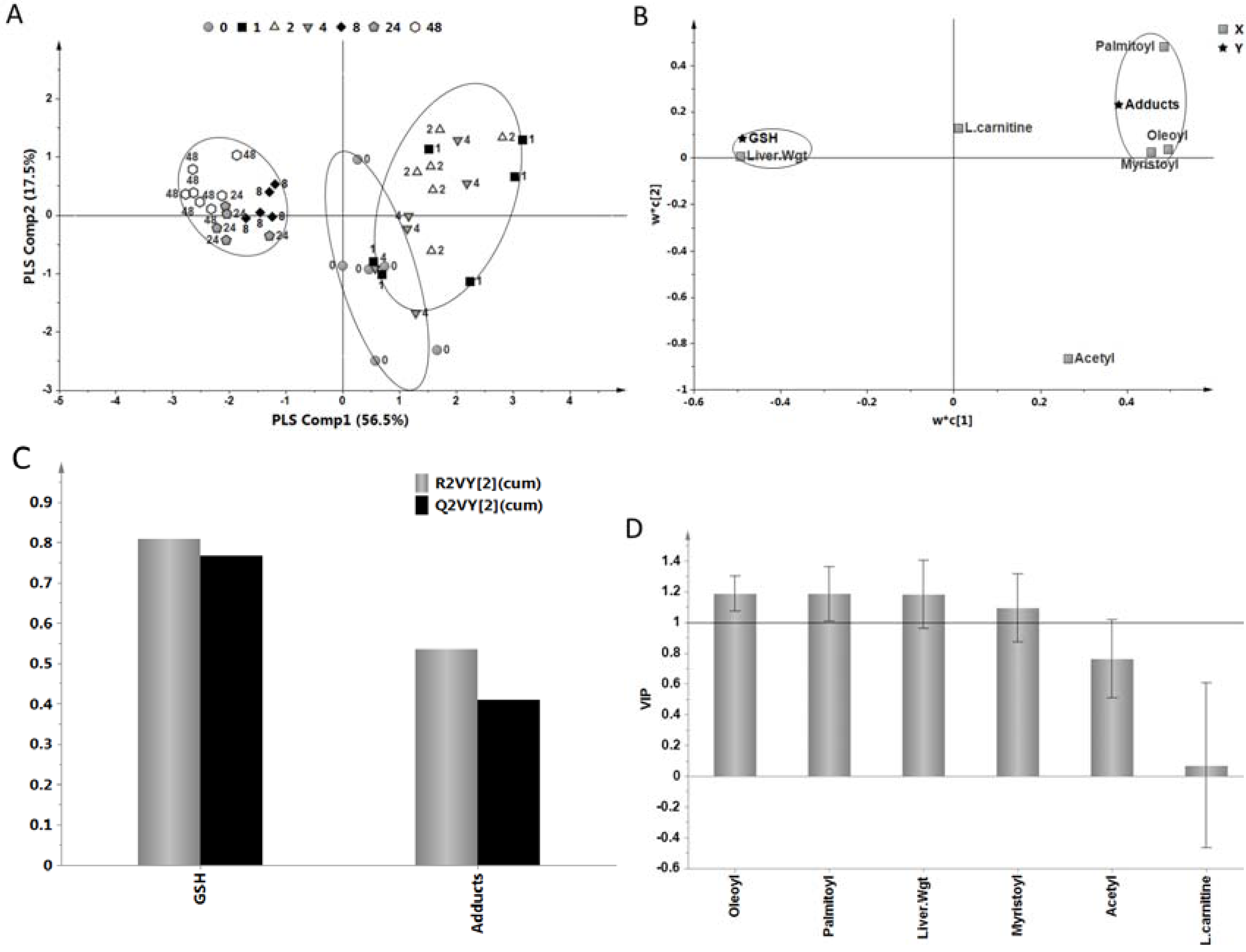
2.5. Relationship of Acylcarnitines to APAP Protein Adducts and Hepatic Glutathione
3. Materials and Methods
3.1. Drugs and Reagents
3.2. Experimental Animals
3.3. Assessment of Hepatotoxicity
3.4. Determination of Hepatic Glutathione and APAP Protein Adducts
3.5. Oil Red O Analysis of Hepatic Lipid Deposition
3.6. Proliferating Cell Nuclear Antigen (PCNA) Immunohistochemistry
3.7. Targeted Analysis of Serum Acylcarnitines Using a UPLC-TQ
3.8. Data Analysis
4. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiodt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef]
- Squires, R.H., Jr.; Shneider, B.L.; Bucuvalas, J.; Alonso, E.; Sokol, R.J.; Narkewicz, M.R.; Dhawan, A.; Rosenthal, P.; Rodriguez-Baez, N.; Murray, K.F.; et al. Acute liver failure in children: the first 348 patients in the pediatric acute liver failure study group. J. Pediatr. 2006, 148, 652–658. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185–194. [Google Scholar]
- Cohen, S.D.; Khairallah, E.A. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab. Rev. 1997, 29, 59–77. [Google Scholar] [CrossRef]
- Roberts, D.W.; Bucci, T.J.; Benson, R.W.; Warbritton, A.R.; McRae, T.A.; Pumford, N.R.; Hinson, J.A. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am. J. Pathol. 1991, 138, 359–371. [Google Scholar]
- Hinson, J.A.; Pike, S.L.; Pumford, N.R.; Mayeux, P.R. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chem. Res. Toxicol. 1998, 11, 604–607. [Google Scholar] [CrossRef]
- Jaeschke, H.; Gores, G.J.; Cederbaum, A.I.; Hinson, J.A.; Pessayre, D.; Lemasters, J.J. Mechanisms of hepatotoxicity. Toxicol. Sci. 2002, 65, 166–176. [Google Scholar] [CrossRef]
- Chen, C.; Hennig, G.E.; Whiteley, H.E.; Corton, J.C.; Manautou, J.E. Peroxisome proliferator-activated receptor alpha-null mice lack resistance to acetaminophen hepatotoxicity following clofibrate exposure. Toxicol. Sci. 2000, 57, 338–344. [Google Scholar] [CrossRef]
- Manautou, J.E.; Hoivik, D.J.; Tveit, A.; Hart, S.G.; Khairallah, E.A.; Cohen, S.D. Clofibrate pretreatment diminishes acetaminophen's selective covalent binding and hepatotoxicity. Toxicol. Appl. Pharmacol. 1994, 129, 252–263. [Google Scholar] [CrossRef]
- Shankar, K.; Vaidya, V.S.; Corton, J.C.; Bucci, T.J.; Liu, J.; Waalkes, M.P.; Mehendale, H.M. Activation of PPAR-alpha in streptozotocin-induced diabetes is essential for resistance against acetaminophen toxicity. Faseb. J. 2003, 17, 1748–1750. [Google Scholar]
- Manautou, J.E.; Tveit, A.; Hoivik, D.J.; Khairallah, E.A.; Cohen, S.D. Protection by clofibrate against acetaminophen hepatotoxicity in male CD-1 mice is associated with an early increase in biliary concentration of acetaminophen-glutathione adducts. Toxicol. Appl. Pharmacol. 1996, 140, 30–38. [Google Scholar] [CrossRef]
- Manautou, J.E.; Emeigh Hart, S.G.; Khairallah, E.A.; Cohen, S.D. Protection against acetaminophen hepatotoxicity by a single dose of clofibrate: Effects on selective protein arylation and glutathione depletion. Fundam. Appl. Toxicol. 1996, 29, 229–237. [Google Scholar] [CrossRef]
- Donthamsetty, S.; Bhave, V.S.; Mitra, M.S.; Latendresse, J.R.; Mehendale, H.M. Nonalcoholic steatohepatitic (NASH) mice are protected from higher hepatotoxicity of acetaminophen upon induction of PPARalpha with clofibrate. Toxicol. Appl. Pharmacol. 2008, 230, 327–337. [Google Scholar] [CrossRef]
- Kleemann, R.; Verschuren, L.; de Rooij, B.J.; Lindeman, J.; de Maat, M.M.; Szalai, A.J.; Princen, H.M.; Kooistra, T. Evidence for anti-inflammatory activity of statins and PPARalpha activators in human C-reactive protein transgenic mice in vivo and in cultured human hepatocytes in vitro. Blood 2004, 103, 4188–4194. [Google Scholar] [CrossRef]
- Chen, C.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J. Biol. Chem. 2008, 283, 4543–4559. [Google Scholar]
- Chen, C.; Krausz, K.W.; Shah, Y.M.; Idle, J.R.; Gonzalez, F.J. Serum metabolomics reveals irreversible inhibition of fatty acid beta-oxidation through the suppression of PPARalpha activation as a contributing mechanism of acetaminophen-induced hepatotoxicity. Chem. Res. Toxicol. 2009, 22, 699–707. [Google Scholar] [CrossRef]
- Coen, M.; Lenz, E.M.; Nicholson, J.K.; Wilson, I.D.; Pognan, F.; Lindon, J.C. An integrated metabonomic investigation of acetaminophen toxicity in the mouse using NMR spectroscopy. Chem. Res. Toxicol. 2003, 16, 295–303. [Google Scholar] [CrossRef]
- Arafa, H.M. Carnitine deficiency: A possible risk factor in paracetamol hepatotoxicity. Arch. Toxicol. 2009, 83, 139–150. [Google Scholar] [CrossRef]
- Yapar, K.; Kart, A.; Karapehlivan, M.; Atakisi, O.; Tunca, R.; Erginsoy, S.; Citil, M. Hepatoprotective effect of L-carnitine against acute acetaminophen toxicity in mice. Exp. Toxicol. Pathol. 2007, 59, 121–128. [Google Scholar] [CrossRef]
- Davern, T.J., II; James, L.P.; Hinson, J.A.; Polson, J.; Larson, A.M.; Fontana, R.J.; Lalani, E.; Munoz, S.; Shakil, A.O.; Lee, W.M. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006, 130, 687–694. [Google Scholar] [CrossRef]
- James, L.P.; Letzig, L.; Simpson, P.M.; Capparelli, E.; Roberts, D.W.; Hinson, J.A.; Davern, T.J.; Lee, W.M. Pharmacokinetics of acetaminophen-protein adducts in adults with acetaminophen overdose and acute liver failure. Drug Metab. Dispos. 2009, 37, 1779–1784. [Google Scholar] [CrossRef]
- Muldrew, K.L.; James, L.P.; Coop, L.; McCullough, S.S.; Hendrickson, H.P.; Hinson, J.A.; Mayeux, P.R. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high- performance liquid chromatography with electrochemical detection. Drug Metab. Dispos. 2002, 30, 446–451. [Google Scholar] [CrossRef]
- Donahower, B.; McCullough, S.S.; Kurten, R.C.; Lamps, L.W.; Simpson, P.M.; Hinson, J.A.; James, L.P. Vascular Endothelial Growth Factor and Hepatocyte Regeneration in Acetaminophen Toxicity. Am. J. Physiol. Gastrointest Liver Physiol. 2006, 291, 102–109. [Google Scholar] [CrossRef]
- James, L.P.; McCullough, S.S.; Lamps, L.W.; Hinson, J.A. Effect of N-acetylcysteine on acetaminophen toxicity in mice: Relationship to reactive nitrogen and cytokine formation. Toxicol. Sci. 2003, 75, 458–467. [Google Scholar] [CrossRef]
- Donahower, B.C.; McCullough, S.S.; Hennings, L.; Simpson, P.M.; Stowe, C.D.; Saad, A.G.; Kurten, R.C.; Hinson, J.A.; James, L.P. Human recombinant vascular endothelial growth factor reduces necrosis and enhances hepatocyte regeneration in a mouse model of acetaminophen toxicity. J. Pharmacol. Exp. Ther. 2010, 334, 33–43. [Google Scholar] [CrossRef]
- Buttar, H.S.; Nera, E.A.; Downie, R.H. Serum enzyme activities and hepatic triglyceride levels in acute and subacute acetaminophen-treated rats. Toxicology 1976, 6, 9–20. [Google Scholar] [CrossRef]
- Nakatani, T.; Ozawa, K.; Asano, M.; Ukikusa, M.; Kamiyama, Y.; Tobe, T. Differences in predominant energy substrate in relation to the resected hepatic mass in the phase immediately after hepatectomy. J. Lab. Clin. Med. 1981, 97, 887–898. [Google Scholar]
- Severino, V.; Locker, J.; Ledda-Columbano, G.M.; Columbano, A.; Parente, A.; Chambery, A. Proteomic characterization of early changes induced by triiodothyronine in rat liver. J. Proteome Res. 2011, 10, 3212–3224. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Thorgeirsson, S.S.; Potter, W.Z.; Jollow, D.J.; Keiser, H. Acetaminophen-induced hepatic injury: protective role of glutathione in man and rationale for therapy. Clin. Pharmacol. Ther. 1974, 16, 676–684. [Google Scholar]
- Chaudhuri, S.; McCullough, S.S.; Hennings, L.; Letzig, L.; Simpson, P.M.; Hinson, J.A.; James, L.P. Acetaminophen hepatotoxicity and HIF-1alpha induction in mice occur without hypoxia. Toxicol. Appl. Pharmacol. 2010, 252, 211–220. [Google Scholar]
- Baumgardner, J.N.; Shankar, K.; Hennings, L.; Albano, E.; Badger, T.M.; Ronis, M.J. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J. Nutr. 2008, 138, 1872–1879. [Google Scholar]
- Varmuza, K.; Filzmoser, P. Introduction to Multivariate Statistical Analysis in Chemometrics; Taylor & Francis-CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bhattacharyya, S.; Pence, L.; Beger, R.; Chaudhuri, S.; McCullough, S.; Yan, K.; Simpson, P.; Hennings, L.; Hinson, J.; James, L. Acylcarnitine Profiles in Acetaminophen Toxicity in the Mouse: Comparison to Toxicity, Metabolism and Hepatocyte Regeneration. Metabolites 2013, 3, 606-622. https://doi.org/10.3390/metabo3030606
Bhattacharyya S, Pence L, Beger R, Chaudhuri S, McCullough S, Yan K, Simpson P, Hennings L, Hinson J, James L. Acylcarnitine Profiles in Acetaminophen Toxicity in the Mouse: Comparison to Toxicity, Metabolism and Hepatocyte Regeneration. Metabolites. 2013; 3(3):606-622. https://doi.org/10.3390/metabo3030606
Chicago/Turabian StyleBhattacharyya, Sudeepa, Lisa Pence, Richard Beger, Shubhra Chaudhuri, Sandra McCullough, Ke Yan, Pippa Simpson, Leah Hennings, Jack Hinson, and Laura James. 2013. "Acylcarnitine Profiles in Acetaminophen Toxicity in the Mouse: Comparison to Toxicity, Metabolism and Hepatocyte Regeneration" Metabolites 3, no. 3: 606-622. https://doi.org/10.3390/metabo3030606