Validated and Predictive Processing of Gas Chromatography-Mass Spectrometry Based Metabolomics Data for Large Scale Screening Studies, Diagnostics and Metabolite Pattern Verification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| CV | Cross validation or Cross validated |
| GC/TOFMS | Gas chromatography time of flight mass spectrometry |
| H-MCR | Hierarchical multivariate curve resolution |
| MCR | Multivariate curve resolution |
| OPLS | Orthogonal projections to latent structures |
| OPLS-DA | Orthogonal projections to latent structures discriminant analysis |
| OSC | Orthogonal signal correction |
| PCA | Principal component analysis |
| PLS | Partial least squares projections to latent structures |
| UPLC/MS | Ultra performance liquid chromatography mass spectrometry |
1. Introduction
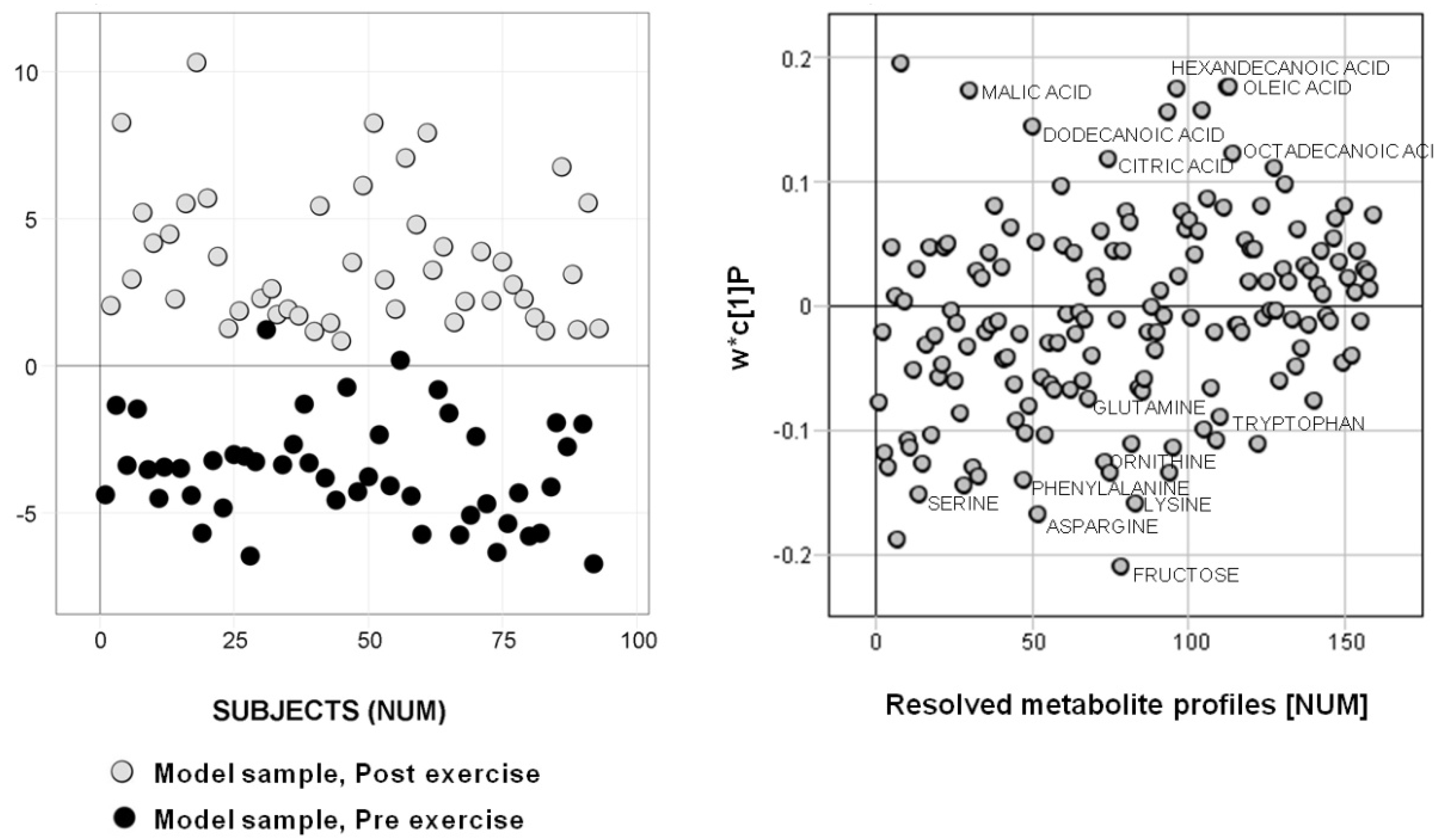
2. Results
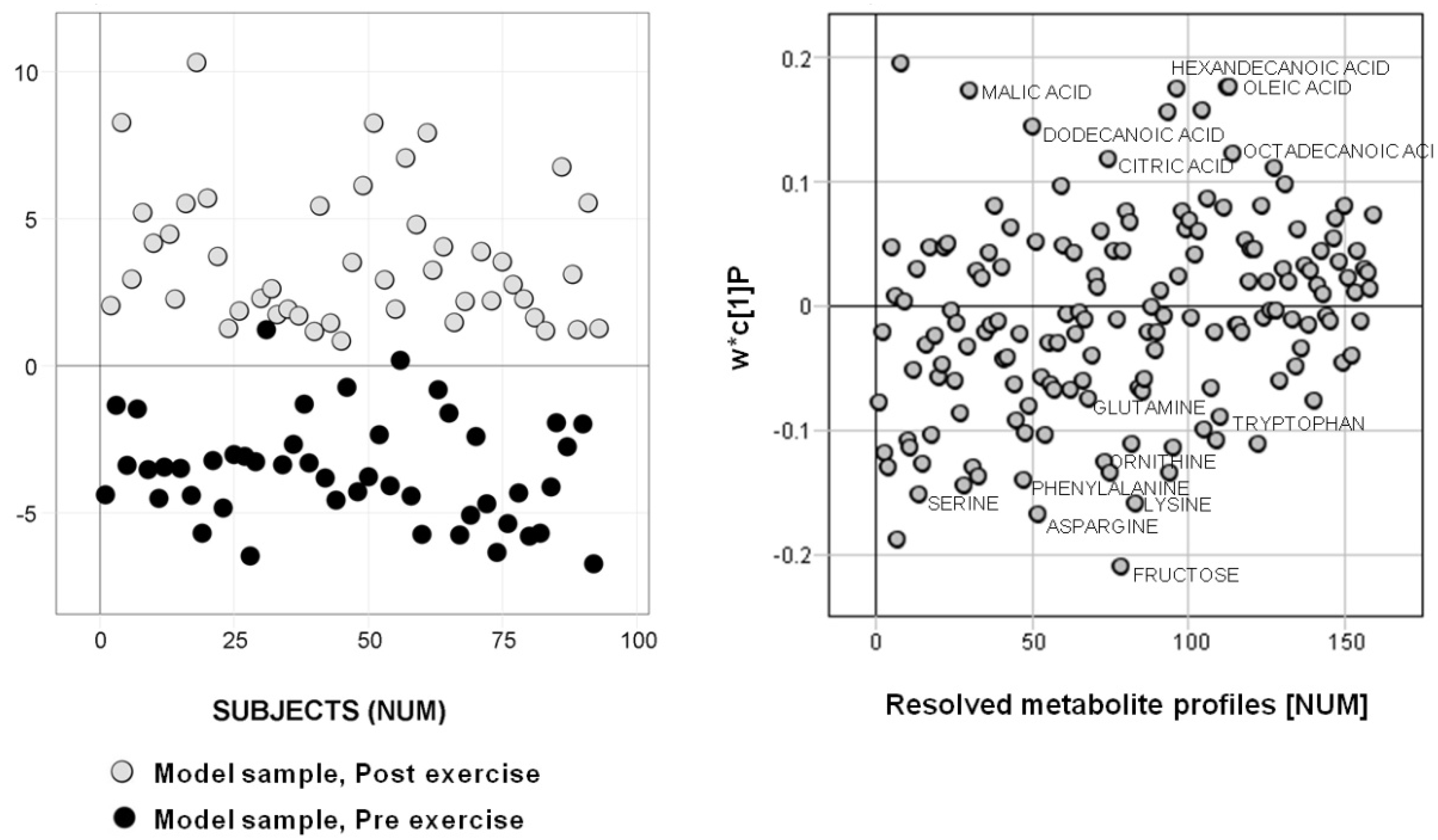
2.1. Subset Selection 1 — Metadata
2.2. Subset Selection 2 — Analytical Data


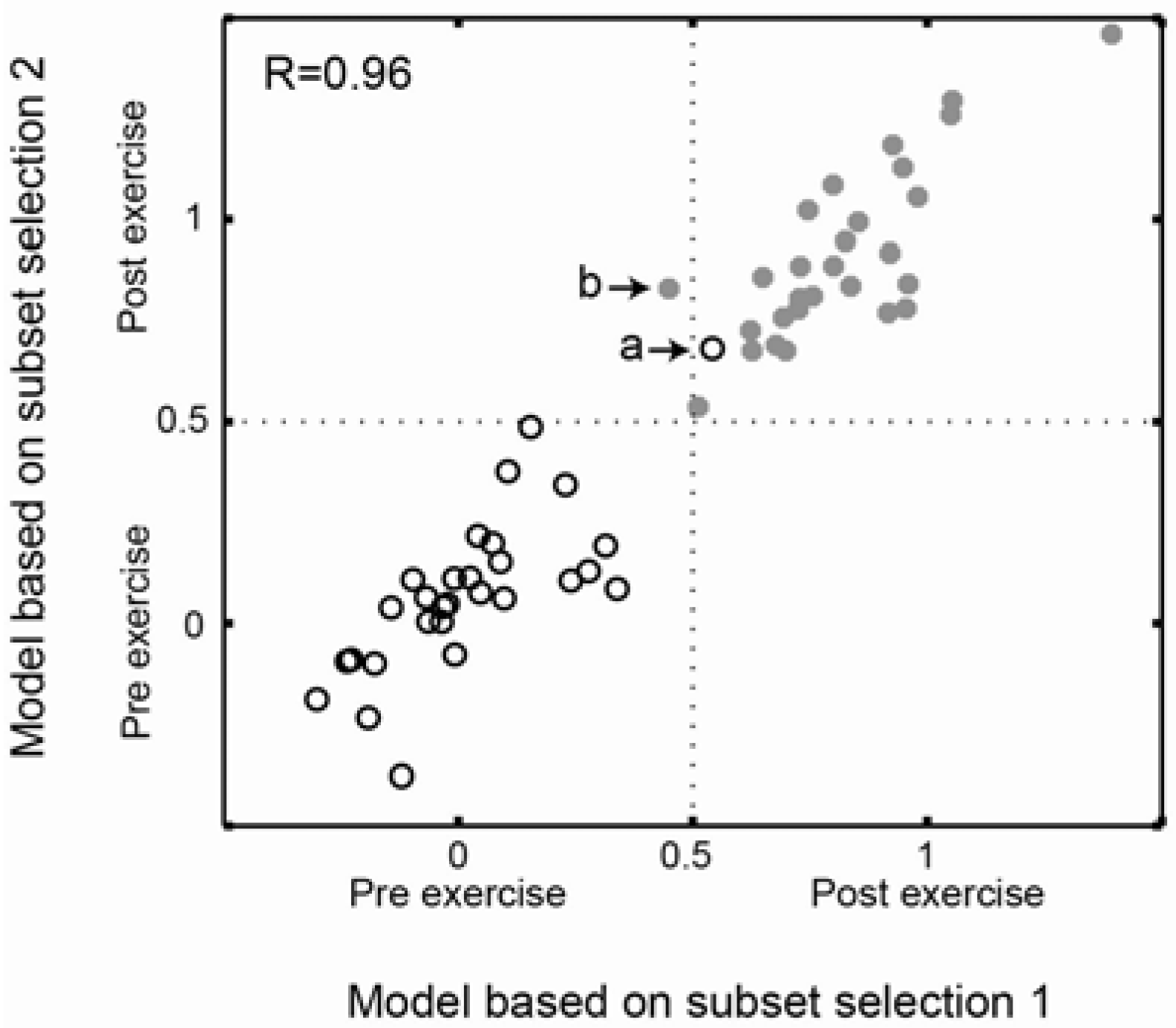
2.3. Comparison of Prediction Similarity of Models Based on Subset Selections
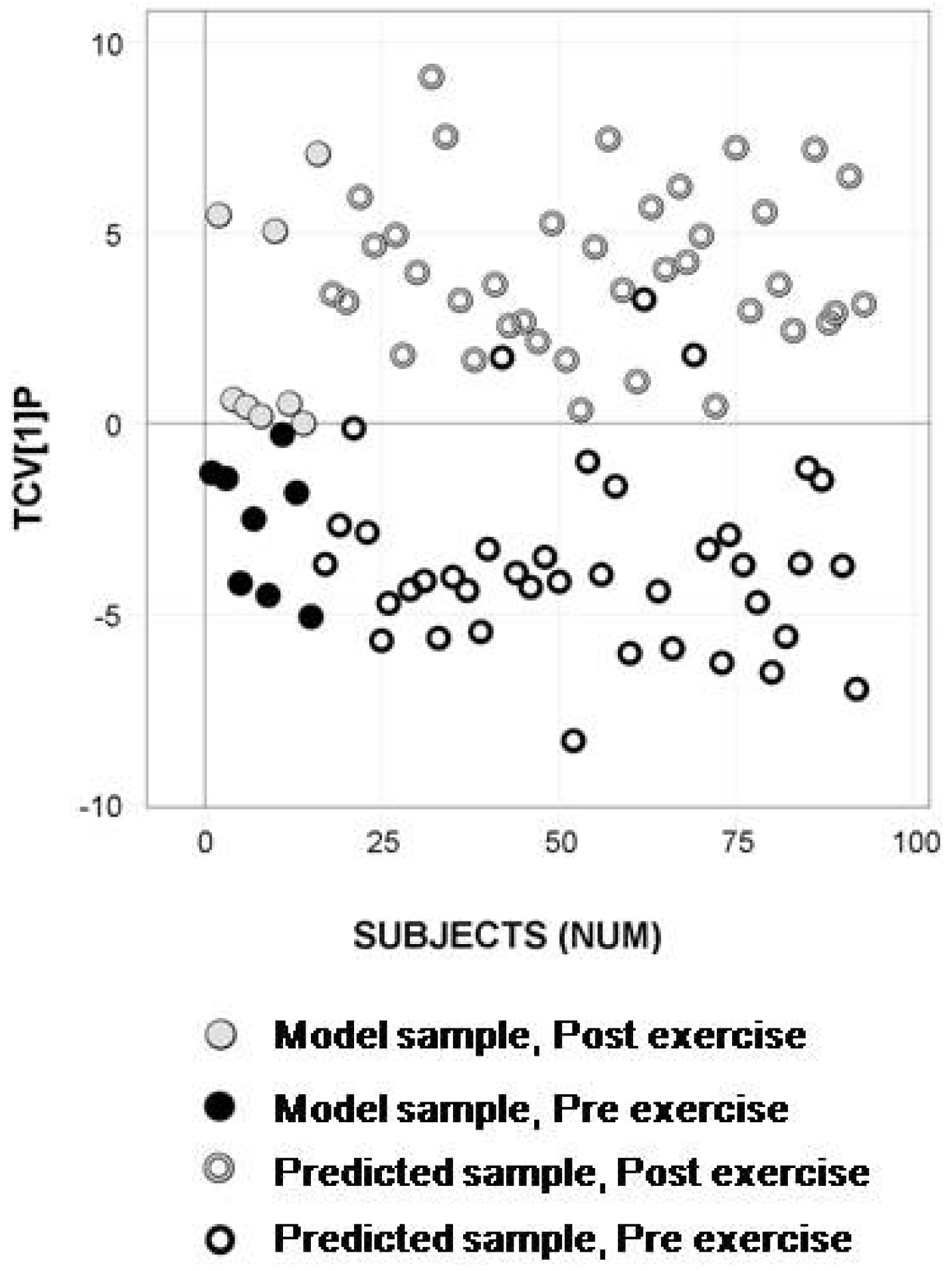
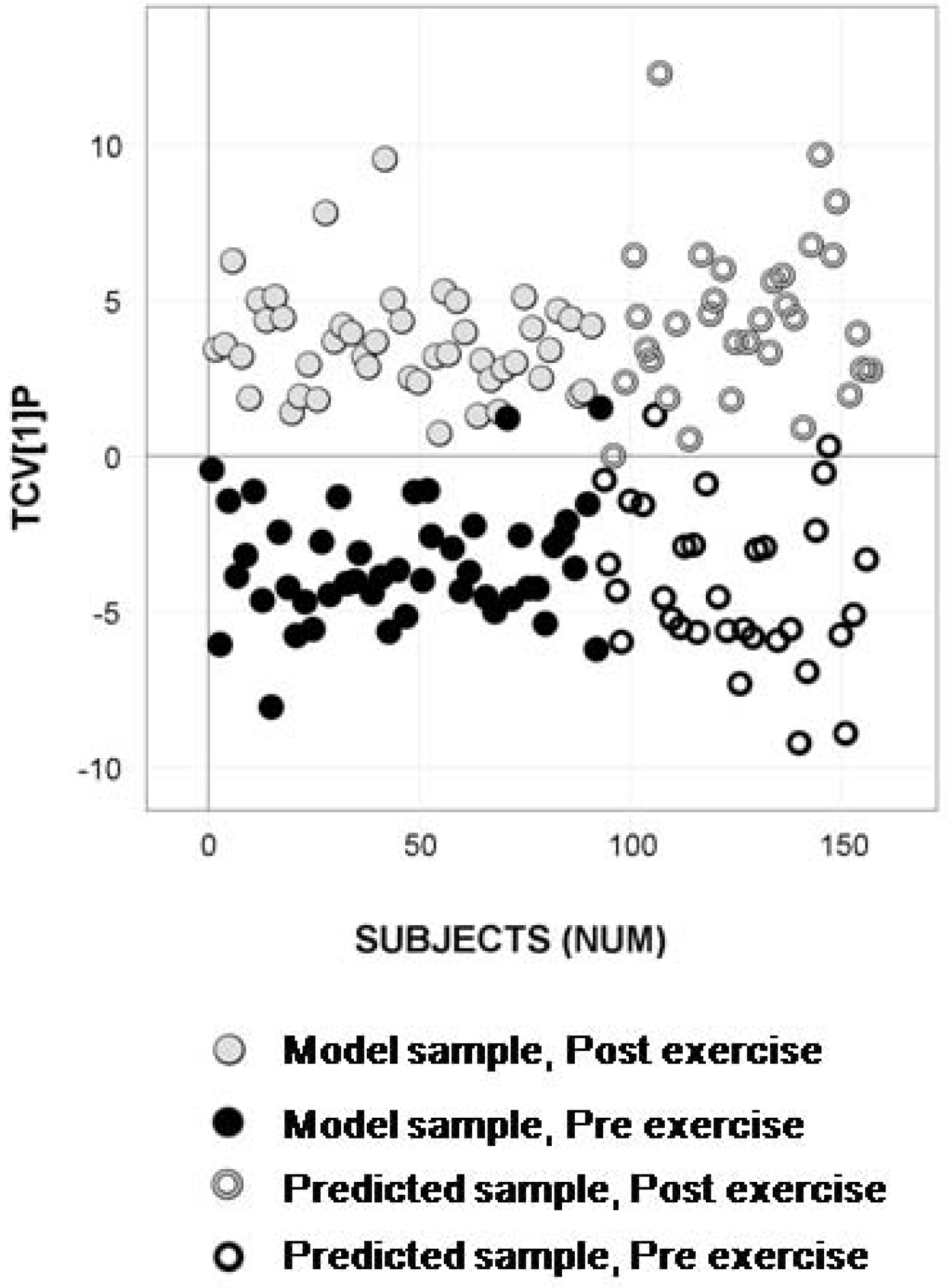
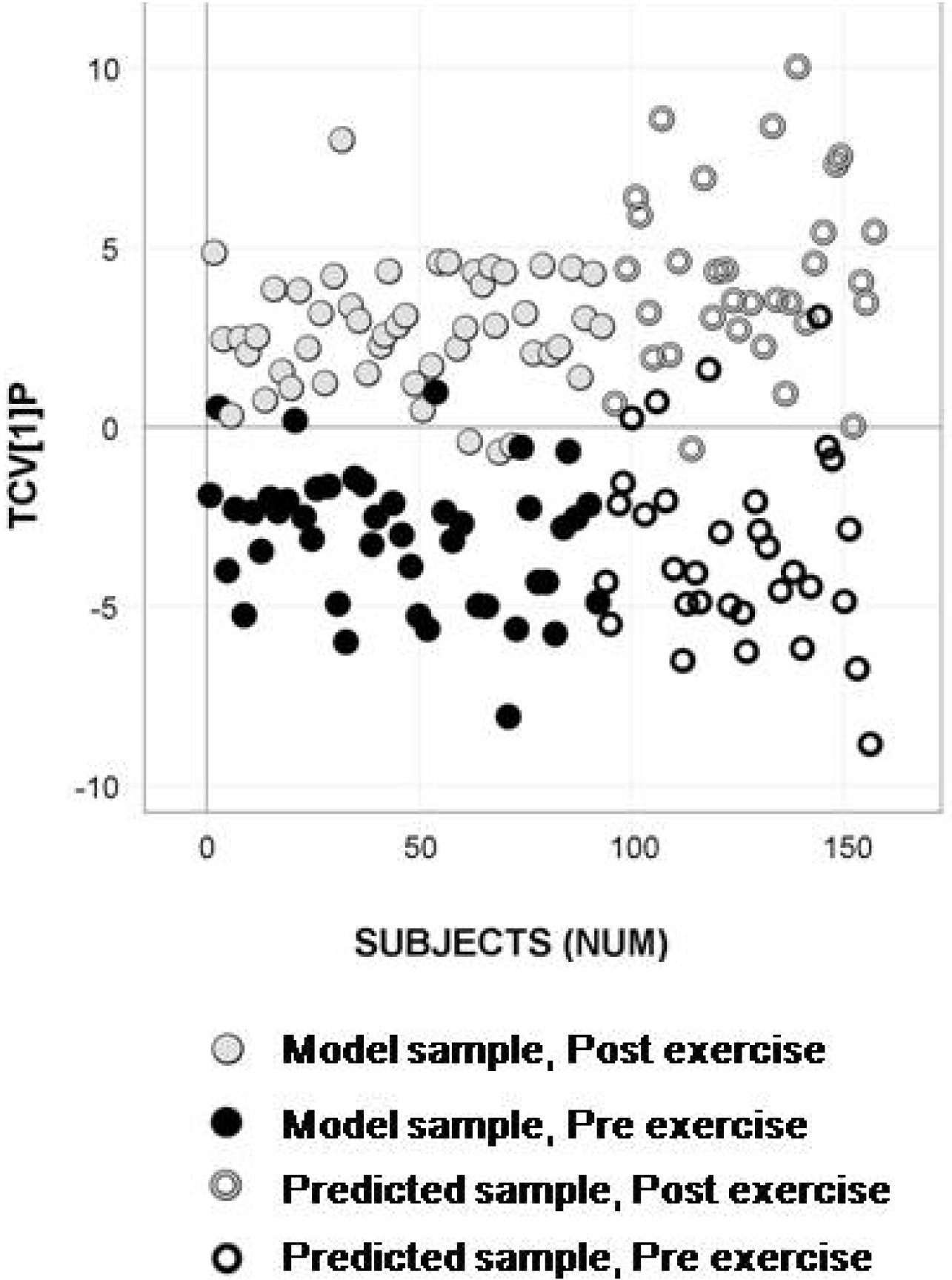
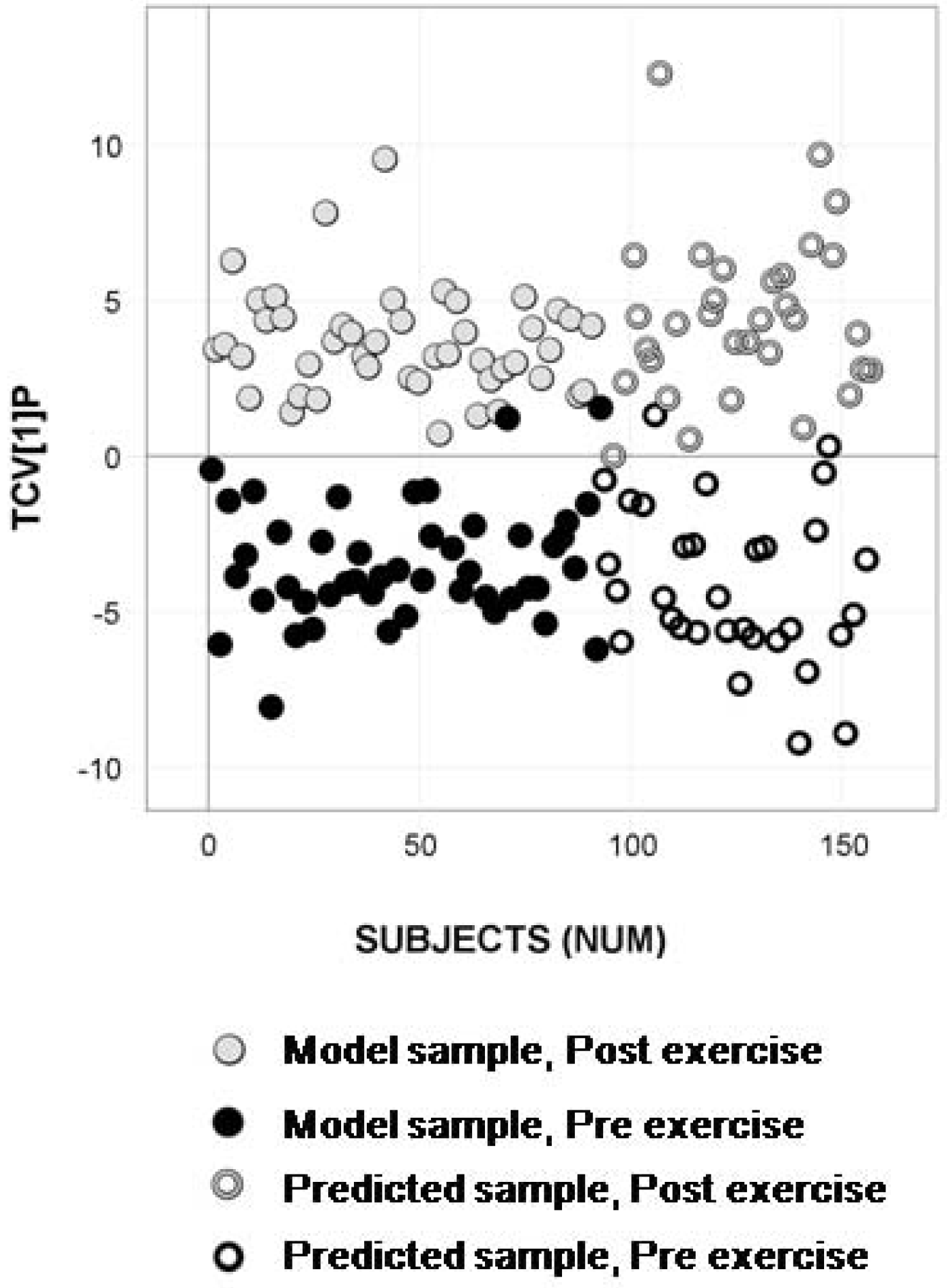
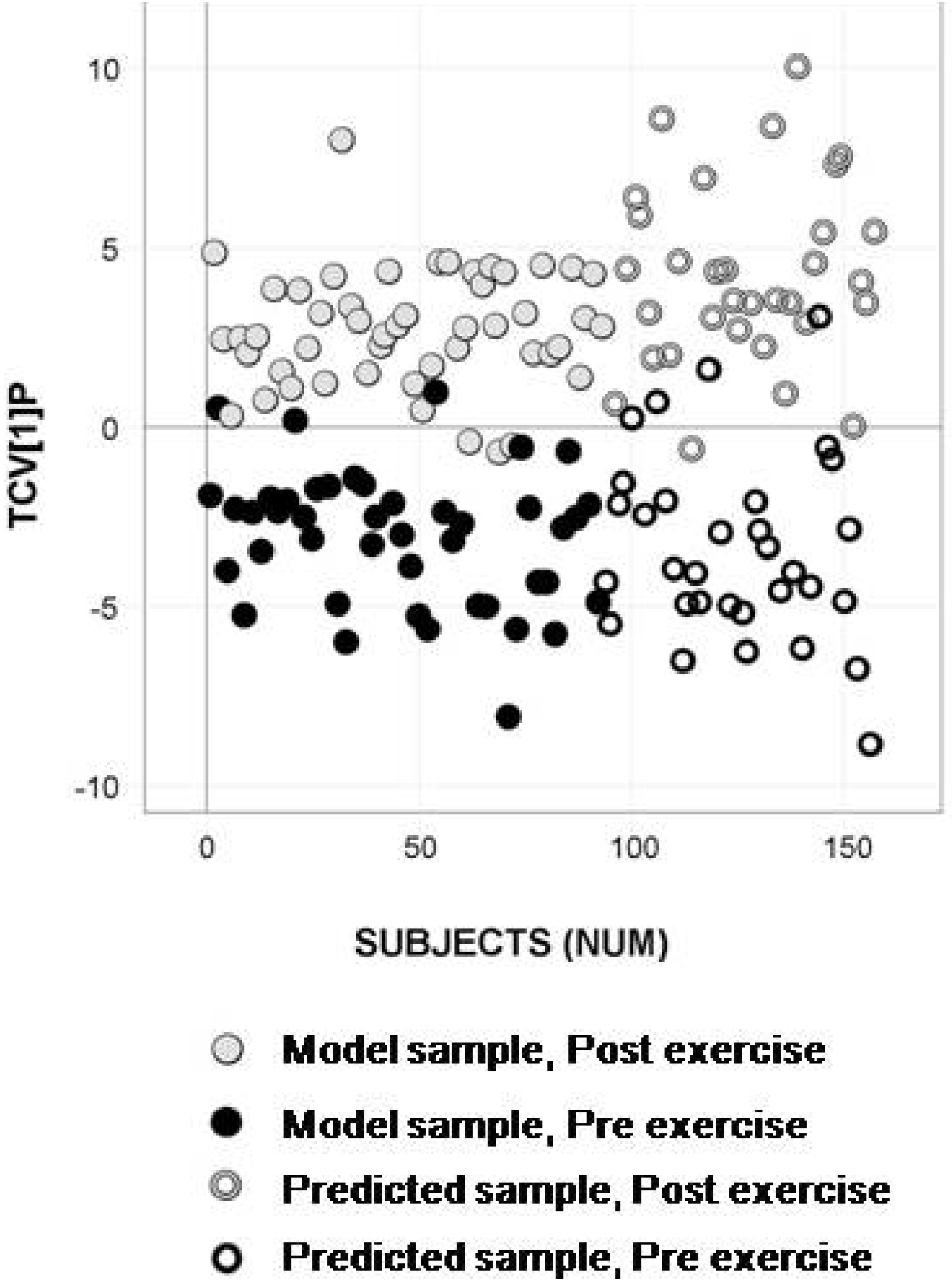
2.4. Longitudinal Sample Predictions

3. Discussion
3.1. Data Processing and Analysis
3.2. Biological Relevance
4. Experimental Section
4.1. Dataset
4.1.1. Pre-Experimental Procedures
4.1.2.Experimental Procedure
4.2.Selection of Representative Samples
4.2.1. Subset Selection 1— Metadata
4.2.2. Subset Selection 2—Analytical data
4.3. Generation, Processing and Modeling of Representative Data
4.3.1. Data Processing and Analysis
4.3.2. Hierarchical Multivariate Curve Resolution (H-MCR)
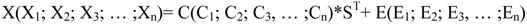
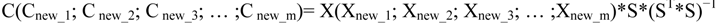
4.3.3. Multivariate Classification and Prediction
4.4. Evaluation of Data Processing and Modeling
4.4.1. Multiple Sample Comparisons
4.4.2. Metabolic information content
4.4.3. Sample Predictions
4.4.4. Longitudinal Sample Predictions
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. 'Metabonomics': understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics - the link between genotypes and phenotypes. Plant. Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Bijlsma, S.; Bobeldijk, L.; Verheij, E.R.; Ramaker, R.; Kochhar, S.; Macdonald, I.A.; van Ommen, B.; Smilde, A.K. Large-scale human metabolomics studies: A strategy for data (pre-) processing and validation. Anal. Chem. 2006, 78, 567–574. [Google Scholar]
- Yap, I.K.S.; Brown, I.J.; Chan, Q.; Wijeyesekera, A.; Garcia-Perez, I.; Bictash, M.; Loo, R.L.; Chadeau-Hyam, M.; Ebbeis, T.; De Iorio, M.; Maibaum, E.; Zhao, L.C.; Kesteloot, H.; Daviglus, M.L.; Stamler, J.; Nicholson, J.K.; Elliott, P.; Holmes, E. Metabolome-Wide Association Study Identifies Multiple Biomarkers that Discriminate North and South Chinese Populations at Differing Risks of Cardiovascular Disease: INTERMAP Study. J.Proteome Res. 2010, 9, 6647–6654. [Google Scholar]
- Holmes, E.; Loo, R.L.; Stamler, J.; Bictash, M.; Yap, I.K.S.; Chan, Q.; Ebbels, T.; De Iorio, M.; Brown, I.J.; Veselkov, K.A.; Daviglus, M.L.; Kesteloot, H.; Ueshima, H.; Zhao, L.C.; Nicholson, J.K.; Elliott, P. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008, 453, 396–400. [Google Scholar]
- Bictash, M.; Ebbels, T.M.; Chan, Q.; Loo, R.L.; Yap, I.K.S.; Brown, I.J.; de Iorio, M.; Daviglus, M.L.; Holmes, E.; Stamler, J.; et al. Opening up the "Black Box": Metabolic phenotyping and metabolome-wide association studies in epidemiology. J. Clin. Epidemiol. 2010, 63, 970–979. [Google Scholar] [CrossRef]
- Suhre, K.; Wallaschofski, H.; Raffler, J.; Friedrich, N.; Haring, R.; Michael, K.; Wasner, C.; Krebs, A.; Kronenberg, F.; Chang, D.; et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 2011, 43, 565–569. [Google Scholar]
- Dumas, M.E.; Maibaum, E.C.; Teague, C.; Ueshima, H.; Zhou, B.F.; Lindon, J.C.; Nicholson, J.K.; Stamler, J.; Elliott, P.; Chan, Q.; Holmes, E. Assessment of analytical reproducibility of H-1 NMR spectroscopy based metabonomics for large-scale epidemiological research: the INTERMAP study. Anal. Chem. 2006, 78, 2199–2208. [Google Scholar]
- Begley, P.; Francis-McIntyre, S.; Dunn, W.B.; Broadhurst, D.I.; Halsall, A.; Tseng, A.; Knowles, J.; Goodacre, R.; Kell, D.B.; Consortium, H. Development and Performance of a Gas Chromatography-Time-of-Flight Mass Spectrometry Analysis for Large-Scale Nontargeted Metabolomic Studies of Human Serum. Anal. Chem. 2009, 81, 7038–7046. [Google Scholar]
- Zelena, E.; Dunn, W.B.; Broadhurst, D.; Francis-McIntyre, S.; Carroll, K.M.; Begley, P.; O'Hagan, S.; Knowles, J.D.; Halsall, A.; Wilson, I.D.; Kell, D.B.; Consortium, H. Development of a Robust and Repeatable UPLC-MS Method for the Long-Term Metabolomic Study of Human Serum. Anal. Chem. 2009, 81, 1357–1364. [Google Scholar]
- Romisch-Margl, W.; Prehn, C.; Bogumil, R.; Rohring, C.; Suhre, K.; Adamski, J. Procedure for tissue sample preparation and metabolite extraction for high-throughput targeted metabolomics. Metabolomics 2012, 8, 133–142. [Google Scholar] [CrossRef]
- Illig, T.; Gieger, C.; Zhai, G.J.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmuller, G.; Kato, B.S.; Mewes, H.W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar]
- Nicholson, G.; Rantalainen, M.; Li, J.V.; Maher, A.D.; Malmodin, D.; Ahmadi, K.R.; Faber, J.H.; Barrett, A.; Min, J.L.; Rayner, N.W.; et al. A Genome-Wide Metabolic QTL Analysis in Europeans Implicates Two Loci Shaped by Recent Positive Selection. PLoS Genet. 2011, 7, e100227. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.L.; Grung, B.; Kvalheim, O.M.; Eide, I. Automated curve resolution applied to data from multi-detection instruments. Anal. Chim. Acta 2001, 446, 313–328. [Google Scholar]
- Liang, Y.Z.; Kvalheim, O.M. Resolution of two-way data: theoretical background and practical problem-solving - Part 1: Theoretical background and methodology. Fresenius J. Anal.Chem. 2001, 370, 694–704. [Google Scholar] [CrossRef]
- Tauler, R. Multivariate curve resolution applied to second order data. Chemom. Intell. Lab. Syst. 1995, 30, 133–146. [Google Scholar] [CrossRef]
- Fiehn, O. Extending the breadth of metabolite profiling by gas chromatography coupled to mass spectrometry. Trac-Trends Anal. Chem. 2008, 27, 261–269. [Google Scholar] [CrossRef]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.D.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar]
- Wishart, D.S.; Lewis, M.J.; Morrissey, J.A.; Flegel, M.D.; Jeroncic, K.; Xiong, Y.P.; Cheng, D.; Eisner, R.; Gautam, B.; Tzur, D.; et al. The human cerebrospinal fluid metabolome. J.Chromatogr. B 2008, 871, 164–173. [Google Scholar] [CrossRef]
- Chan, E.C.Y.; Pasikanti, K.K.; Nicholson, J.K. Global urinary metabolic profiling procedures using gas chromatography-mass spectrometry. Nature Prot. 2011, 6, 1483–1499. [Google Scholar] [CrossRef]
- Jonsson, P.; Johansson, A.I.; Gullberg, J.; Trygg, J.; A, J.; Grung, B.; Marklund, S.; Sjostrom, M.; Antti, H.; Moritz, T. High-throughput data analysis for detecting and identifying differences between samples in GC/MS-based metabolomic analyses. Anal. Chem 2005, 77, 5635–5642. [Google Scholar] [CrossRef]
- Jonsson, P.; Johansson, E.S.; Wuolikainen, A.; Lindberg, J.; Schuppe-Koistinen, I.; Kusano, M.; Sjostrom, M.; Trygg, J.; Moritz, T.; Antti, H. Predictive metabolite profiling applying hierarchical multivariate curve resolution to GC-MS datas - A potential tool for multi-parametric diagnosis. J. Proteome Res. 2006, 5, 1407–1414. [Google Scholar] [CrossRef]
- Thysell, E.; Surowiec, I.; Hornberg, E.; Crnalic, S.; Widmark, A.; Johansson, A.I.; Stattin, P.; Bergh, A.; Moritz, T.; Antti, H.; et al. Metabolomic Characterization of Human Prostate Cancer Bone Metastases Reveals Increased Levels of Cholesterol. Plos One 5, e14175. [CrossRef]
- Chorell, E.; Moritz, T.; Branth, S.; Antti, H.; Svensson, M.B. Predictive Metabolomics Evaluation of Nutrition-Modulated Metabolic Stress Responses in Human Blood Serum During the Early Recovery Phase of Strenuous Physical Exercise. J.Proteome Res. 2009, 8, 2966–2977. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Andersen, P.M.; Moritz, T.; Marklund, S.L.; Antti, H. ALS patients with mutations in the SOD1 gene have an unique metabolomic profile in the cerebrospinal fluid compared with ALS patients without mutations. Mol. Genet. Metab. 2012, 105, 472–478. [Google Scholar] [CrossRef]
- Stein, S.E. An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry data. J. Am. Soc. Mass Spectrom. 1999, 10, 770–781. [Google Scholar] [CrossRef]
- Luedemann, A.; Strassburg, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography - mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinf. 2008, 24, 732–737. [Google Scholar] [CrossRef]
- Jiang, W.X.; Qiu, Y.P.; Ni, Y.; Su, M.M.; Jia, W.; Du, X.X. An Automated Data Analysis Pipeline for GC-TOF-MS Metabonomics Studies. J. Proteome Res. 2010, 9, 5974–5981. [Google Scholar] [CrossRef]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemometr. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Bylesjo, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J.K.; Holmes, E.; Trygg, J. OPLS discriminant analysis: combining the strengths of PLS-DA and SIMCA classification. J. Chemometr. 2006, 20, 341–351. [Google Scholar] [CrossRef]
- Mcardle, D.W; Katch, I.F.; Katch, V.L. Exercise Physiology : Energy, Nutrition, and Human Performance, 6th ed; Lippincott Williams and Wilkins: Philadelphia, FL, USA, 2006. [Google Scholar]
- Jonsson, P.; Gullberg, J.; Nordstrom, A.; Kusano, M.; Kowalczyk, M.; Sjostrom, M.; Moritz, T. A strategy for identifying differences in large series of metabolomic samples analyzed by GC/MS. Anal. Chem. 2004, 76, 1738–1745. [Google Scholar] [CrossRef]
- Chadeau-Hyam, M.; Ebbels, T.M.D.; Brown, I.J.; Chan, Q.; Stemler, J.; Huang, C.C.; Daviglus, M.L.; Ueshima, H.; Zhao, L.C.; Holmes, E.; Nicholson, J.K.; Elliott, P.; De Iorio, M. Metabolic Profiling and the Metabolome-Wide Association Study: Significance Level For Biomarker Identification. J. Proteome Res. 2010, 9, 4620–4627. [Google Scholar]
- Nicholson, J.K.; Holmes, E.; Elliott, P. The metabolome-wide association study: A new look at human disease risk factors. J. Proteome Res. 2008, 7, 3637–3638. [Google Scholar] [CrossRef]
- Trygg, J.; Holmes, E.; Lundstedt, T. Chemometrics in metabonomics. J. Proteome Res. 2007, 6, 469–479. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Hedenstrom, M.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Optimization of procedures for collecting and storing of CSF for studying the metabolome in ALS. Amyotroph. Lateral Scler. 2009, 10, 229–236. [Google Scholar] [CrossRef]
- Thysell, E.; Pohjanen, E.; Lindberg, J.; Schuppe-Koistinen, I.; Sjöström, M.; Moritz, T.; Jonsson, P.; Antti, H. Reliable profile detection in comparative metabolomics. OMICS 2007, 11, 209–224. [Google Scholar] [CrossRef]
- Hodson, M.P.; Dear, G.J.; Griffin, J.L.; Haselden, J.N. An approach for the development and selection of chromatographic methods for high-throughput metabolomic screening of urine by ultra pressure LC-ESI-ToF-MS. Metabolomics 2009, 5, 166–182. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; Nicholls, A.W.; Wilson, I.D.; Kell, D.B.; Goodacre, R.; Human Serum Metabolome, H.C. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protocol. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Ranallo, R.F.; Rhodes, E.C. Lipid metabolism during exercise. Sports Med. 1998, 26, 29–42. [Google Scholar] [CrossRef]
- Donsmark, M.; Langfort, J.; Holm, C.; Ploug, T.; Galbo, H. Hormone-sensitive lipase as mediator of lipolysis in contracting skeletal muscle. Exerc Sport Sci Rev. 2005, 33, 127–133. [Google Scholar] [CrossRef]
- Romijn, J.A.; Coyle, E.F.; Sidossis, L.S.; Gastaldelli, A.; Horowitz, J.F.; Endert, E.; Wolfe, R.R. Regulation of Endogenous Fat and Carbohydrate-Metabolism in Relation to Exercise Intensity and Duration. Am. J. Physiol 1993, 265, E380–E391. [Google Scholar]
- Weltan, S.M.; Bosch, A.N.; Dennis, S.C.; Noakes, T.D. Preexercise muscle glycogen content affects metabolism during exercise despite maintenance of hyperglycemia. Am. J. Physiol-Endoc. M. 1998, 274, E83–E88. [Google Scholar]
- Friedlander, A.L.; Jacobs, K.A.; Fattor, J.A.; Horning, M.A.; Hagobian, T.A.; Bauer, T.A.; Wolfel, E.E.; Brooks, G.A. Contributions of working muscle to whole body lipid metabolism are altered by exercise intensity and training. Am. J. Physiol-Endoc M. 2007, 292, E107–E116. [Google Scholar]
- Wahren, J.; Ekberg, K. Splanchnic regulation of glucose production. Annu Rev. Nutr 2007, 27, 329–345. [Google Scholar] [CrossRef]
- Rennie, M.J.; Tipton, K.D. Protein and amino acid metabolism during and after exercise and the effects of nutrition. Annu Rev. Nutr 2000, 20, 457–483. [Google Scholar] [CrossRef]
- Gibala, M.J. Protein metabolism and endurance exercise. Sports Med. 2007, 37, 337–340. [Google Scholar] [CrossRef]
- Hellsten, Y.; Svensson, M.; Sjodin, B.; Smith, S.; Christensen, A.; Richter, E.A.; Bangsbo, J. Allantoin formation and urate and glutathione exchange in human muscle during submaximal exercise. Free Radical Bio.Med. 2001, 31, 1313–1322. [Google Scholar] [CrossRef]
- Svensson, M.B.; Ekblom, B.; Cotgreave, I.A.; Norman, B.; Sjoberg, B.; Ekblom, O.; Sjodin, B.; Sjodin, A. Adaptive stress response of glutathione and uric acid metabolism in man following controlled exercise and diet. Acta Physiol. Scand. 2002, 176, 43–56. [Google Scholar] [CrossRef]
- Hellsten, Y.; Richter, E.A.; Kiens, B.; Bangsbo, J. AMP deamination and purine exchange in human skeletal muscle during and after intense exercise. J. Physiol. (London) 1999, 520, 909–920. [Google Scholar] [CrossRef]
- Pohjanen, E.; Thysell, E.; Jonsson, P.; Eklund, C.; Silfver, A.; Carlsson, I.B.; Lundgren, K.; Moritz, T.; Svensson, M.B.; Antti, H. A multivariate screening strategy for investigating metabolic effects of strenuous physical exercise in human serum. J. Proteome Res. 2007, 6, 2113–2120. [Google Scholar] [CrossRef]
- Larsson, P.U.; Wadell, K.M.E.; Jakobsson, E.J.I.; Burlin, L.U.; Henriksson-Larsen, K.B. Validation of the MetaMax II portable metabolic measurement system. Int. J. Sports Med. 2004, 25, 115–123. [Google Scholar] [CrossRef]
- Jiye, A.; Trygg, J.; Gullberg, J.; Johansson, A.I.; Jonsson, P.; Antti, H.; Marklund, S.L.; Moritz, T. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal. Chem. 2005, 77, 8086–8094. [Google Scholar]
- Wold, S.; Sjostrom, M.; Carlson, R.; Lundstedt, T.; Hellberg, S.; Skagerberg, B.; Wikstrom, C.; Ohman, J. Multivariate Design. Anal. Chim. Acta 1986, 191, 17–32. [Google Scholar] [CrossRef]
- Lundstedt, T.; Seifert, E.; Abramo, L.; Thelin, B.; Nystrom, A.; Pettersen, J.; Bergman, R. Experimental design and optimization. Chemom. Intell. Lab.Syst. 1998, 42, 3–40. [Google Scholar] [CrossRef]
- Linusson, A.; Wold, S.; Norden, B. Statistical molecular design of peptoid libraries. Mol. Diversity 1998, 4, 103–114. [Google Scholar] [CrossRef]
- Marengo, E.; Todeschini, R. A New Algorithm for Optimal, Distance-Based Experimental-Design. Chemom. Intell. Lab.Syst. 1992, 16, 37–44. [Google Scholar] [CrossRef]
- Stone, M. Cross-Validatory Choice and Assessment of Statistical Predictions. J. Royal Statist. Soc. Ser. B (Methodol.) 1974, 36, 111–147. [Google Scholar]
- Wold, S.; Sjostrom, M.; Eriksson, L. PLS-regression: a basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Wold, S.; Antti, H.; Lindgren, F.; Ohman, J. Orthogonal signal correction of near-infrared spectra. Chemom. Intell. Lab.Syst. 1998, 44, 175–185. [Google Scholar] [CrossRef]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thysell, E.; Chorell, E.; Svensson, M.B.; Jonsson, P.; Antti, H. Validated and Predictive Processing of Gas Chromatography-Mass Spectrometry Based Metabolomics Data for Large Scale Screening Studies, Diagnostics and Metabolite Pattern Verification. Metabolites 2012, 2, 796-817. https://doi.org/10.3390/metabo2040796
Thysell E, Chorell E, Svensson MB, Jonsson P, Antti H. Validated and Predictive Processing of Gas Chromatography-Mass Spectrometry Based Metabolomics Data for Large Scale Screening Studies, Diagnostics and Metabolite Pattern Verification. Metabolites. 2012; 2(4):796-817. https://doi.org/10.3390/metabo2040796
Chicago/Turabian StyleThysell, Elin, Elin Chorell, Michael B. Svensson, Pär Jonsson, and Henrik Antti. 2012. "Validated and Predictive Processing of Gas Chromatography-Mass Spectrometry Based Metabolomics Data for Large Scale Screening Studies, Diagnostics and Metabolite Pattern Verification" Metabolites 2, no. 4: 796-817. https://doi.org/10.3390/metabo2040796