
Oxidative Stress and Human Skin Connective Tissue Aging


1
Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO 63110, USA
2
Department of Dermatology, University of Michigan Medical School, Ann Arbor, MI 48109, USA
*
Author to whom correspondence should be addressed.
Cosmetics 2016, 3(3), 28; https://doi.org/10.3390/cosmetics3030028
Submission received: 14 June 2016
/
Revised: 1 August 2016
/
Accepted: 2 August 2016
/
Published: 5 August 2016
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Everyone desires healthy and beautiful-looking skin. However, as we age, our skin becomes old due to physiological changes. Reactive oxygen species (ROS) is an important pathogenic factor involved in human aging. Human skin is exposed to ROS generated from both extrinsic sources such as as ultraviolet (UV) light from the sun, and intrinsic sources such as endogenous oxidative metabolism. ROS-mediated oxidative stress damages the collagen-rich extracellular matrix (ECM), the hallmark of skin connective tissue aging. Damage to dermal collagenous ECM weakens the skin’s structural integrity and creates an aberrant tissue microenvironment that promotes age-related skin disorders, such as impaired wound healing and skin cancer development. Here, we review recent advances in our understanding of ROS/oxidative stress and skin connective tissue aging.
1. Introduction
Skin is the largest organ of the human body and changes in the skin are among the most visible signs of an aged appearance. The skin’s appearance is central to the social and visual experience, and it has significant emotional and psychological impacts on our life quality. Skin, like all human organs, undergoes progressive alterations as a consequence of the passage of time. In addition, human skin, unlike other organs, continuously experiences harmful exposure from environmental sources such as solar ultraviolet (UV) irradiation. Based on its causes, there are two types of skin aging: natural aging, also known as intrinsic aging, and photoaging, also known as extrinsic aging caused by UV irradiation from the sun [1,2,3]. Natural aging is a part of our life, observed in all individuals resulting naturally as we grow old. Photoaging refers to changes attributable to habitual UV light exposure. Both types of skin aging are cumulative and therefore photoaging is superimposed on natural skin aging. Thus, old-looking skin is a combination of intrinsic and extrinsic aging, which is the most clinically noticeable on the face, neck, and forearm [4].
The bulk of the skin is largely comprised of the collagen-rich extracellular matrix (ECM) [1]. At molecular levels, naturally aged and photoaged skin share common features of skin aging—thin and fragmented collagen [5]. Biochemical and histological studies have revealed that aged human skin is primarily characterized by a loss of collagen and damaged/disorganized collagen fibrils [6,7]. Alterations of the dermal connective tissue collagen impair the skin’s structural and mechanical integrity, and eventually result in old-looking skin, such as thin, fragile, and wrinkled skin. Age-related alterations of dermal connective tissue (loss and damaged collagen fibrils) create a tissue microenvironment for skin disorders, such as increased fragility [5,8], impaired vasculature support [9,10,11], poor wound healing [11,12], and cancer development [13,14,15,16].
Mounting evidence indicates that oxidative stress induced by reactive oxygen species (ROS) is believed to be the major driving force of the aging process; this is also known as the free radical theory of aging [17,18]. This theory proposes that ROS oxidize cellular constituents such as lipids, proteins, and nucleic acids, to yield the aged phenotype. The age-related increase in ROS generation and oxidative damage has been described in a variety of tissues including the skin [18,19,20]. The levels of protein carbonyls, a well-established biomarker of oxidative damage, are significantly elevated in the dermis of aged human skin [20]. Human skin is exposed to ROS generated from both environmental sources such as UV light from the sun [21], airborne pollutants such as ozone [22] and particulate matter [23], as well as the endogenous oxidative metabolism [20]. The elevation of ROS and oxidative damage impairs cellular functions and creates an aged-related aberrant dermal ECM microenvironment, which impairs the skin’s structure and functions [1,20,24]. As described below, oxidative stress is an important pathogenic factor involved in human skin connective tissue aging.
2. Collagen Fragmentation Collapses Dermal Fibroblasts and Increases Intracellular ROS Generation
Dermal connective tissue collagen acts as a dynamic scaffold for the attachment of cells, critically regulating their function, and is also a repository and regulator of potent biological mediators (growth factors, cytokines, chemokines, matricellular proteins, etc.). In human skin, dermal fibroblasts, the major collagen-producing cells, are responsible for the homeostasis of dermal connective tissue. Dermal fibroblasts reside in a collagenous microenvironment and intimately interact with collagen fibrils, and thus maintain normal cell shape and mechanical tension for function [20,25]. As such, collagen is a critical determinant for cell shape and mechanical tension, which is known to regulate many cellular functions [26]. In the skin dermis, fibroblast size and mechanical tension are largely regulated by cellular interactions with surrounding collagen fibrils and the intracellular cytoskeleton (Figure 1). In the young human skin dermis, the binding of fibroblasts to intact collagen fibrils allows the generation of traction forces that are necessary for spreading and maintaining normal cell size. However, in the aged human skin dermis, collagen fibril binding sites are lost and mechanical resistance to traction forces is reduced due to fragmentation. In this state, the ECM microenvironment is unable to provide sufficient mechanical stability to maintain normal cell spreading/mechanical force [7,20,27]. Therefore, age-related fragmentation of the collagen fibrils deleteriously alters fibroblast size/mechanical tension and function associated with skin connective tissue aging [7,27,28].
Therefore, reduced fibroblast size is a prominent feature of dermal fibroblasts in aged human skin [7,20,27]. As cell shape and size impact multiple cellular processes and functions [29,30,31,32], it appears that a reduced dermal fibroblast size is the major source of the constitutive elevation of ROS generation and oxidative stress [20,33]. We have previously reported that compared to fibroblasts in intact collagen gels, fibroblasts cultured in matrix metalloproteinase-1 (MMP-1), which partially degrades collagen lattices, displayed reduced cell size and three-fold increase in ROS generation [20]. This cell size-related ROS generation causes cellular oxidative damage. There are two major sources of ROS in human skin: NADPH oxidase (NOX) in cell membranes [20,21], and mitochondria [33]. In human skin, mitochondria are a significant source of ROS generation. It has been reported that the reduction of dermal fibroblast spreading and cell size can increase mitochondria ROS generation [33]. These data suggest that the reduced fibroblast cell size, as observed in aged human skin in vivo, could be one of the driving forces for the age-related elevation of ROS generation and oxidative stress in aged human skin.
Mechanistically, oxidative stress not only stimulates collagen breakdown [20], but also inhibits the production of collagen [34,35], the two primary events of human skin connective tissue aging. As described below, oxidative stress can result in the following: inducing multiple matrix metalloproteinases (MMPs), which cause fragmentation of collagen fibrils [7,20,36,37,38,39,40]; inhibiting TGF-β signaling, which causes inhibition of the collagenous ECM production [1,39,41]; and inducing multiple age-related pro-inflammatory cytokines, which create an inflammatory dermal microenvironment (inflammaging) [35,39,42]. These ROS-induced alterations of the dermal ECM microenvironment are the driving force for the most prominent clinical features of aged skin, including thinning and increased fragility of the skin [5,7,8,20,43,44,45].
3. ROS/Oxidative Stress Contributes to Damaged Dermis by Induction of Multiple MMPs
MMPs are a family of zinc-containing proteinases that are capable of degrading every type of ECM protein [46]. To date, the human MMP gene family consists of more than 20 members, with distinct structural and substrate specificities [47]. MMPs are involved in a variety of physiological and pathological processes related to ECM breakdown, including ECM remodeling after wounding, angiogenesis, and cancer development and invasion [48]. Multiple MMPs become elevated during the aging process in human skin dermis [37]. Elevated MMPs in sun-protected aged dermis can be divided into the following groups: collagenase (MMP-1), gelatinase-B (MMP-9), stromelysins (MMP-3, MMP-10, MMP-11), membrane-associated: MMP-23, MMP-24, and recently identified MMP-27. UV light from the sun is well known to transiently induce several MMPs in human skin in vivo (MMP-1, MMP-3, and MMP-9) [37,49,50]. Compared to acute UV irradiation, a larger variety of MMPs, including UV-inducible MMPs, is constitutively elevated in aged skin. Interestingly, compared to acute UV irradiation, in which the epidermis is the primary source of transient elevated MMPs [37], the dermis is the major source of elevated MMPs in naturally aged skin. These observations suggest that, although naturally aged and photoaged skin share many common molecular features, such as collagen fragmentation, the primary sources (dermis vs. epidermis) and cell types (fibroblasts vs. keratinocytes) of elevated MMPs are different.
As dermal fibroblasts are the primary source of multiple MMPs in naturally aged human skin, the reduced fibroblast size is largely responsible for multiple elevated MMPs. The primary dermal fibroblasts from aged (>80 years) or young (25–30 years) individuals are indistinguishable from each other with respect to morphology and expression of all known mammalian MMPs, when cultured in a monolayer on a plastic tissue culture plate [20,37]. In contrast, human dermal fibroblasts, obtained from individuals of any age (21–86 years of age), cultured in the conditions of reduced cell size, for example fragmented collagen lattices, resulted in the elevation of multiple MMPs [20,37]. These data suggest that the elevation of multiple MMPs in aged human skin arises, at least in part, from the reduced size of dermal fibroblasts. Further investigation indicates that reduced fibroblast size is closely associated with elevated c-Jun/c-Fos and increased transcription factor AP-1 activity, the major driving force for multiple MMPs [51,52,53]. Transcription factor AP-1, typically composed of c-Jun and c-Fos, is one of the first mammalian transcription factors to be identified [54,55]. We and others previously reported that stress-activated Mitogen-Activated Protein (MAP) kinase pathways and c-Jun mRNA and protein are increased in aged human skin as compared to young human skin in vivo [20,56,57]. AP-1 activity is regulated by a wide range of stimuli including ROS [55]. Therefore, it is postulated that elevated ROS due to reduced fibroblast size could result in elevated c-Jun/AP-1 activity, which in turn elevates multiple MMPs in aged human skin (Figure 2). This mechanism provides a foundation for understanding the cellular and molecular basis of age-related collagen fragmentation, the characteristic feature of aged human skin.
4. ROS/Oxidative Stress Contributes to Thin Dermis by Inhibition of TGF-β Signaling
TGF-β signaling is a primary regulator of ECM production and thus is critical for maintaining dermal connective tissue structural and mechanical integrity [41,58,59,60]. In human dermal fibroblasts, TGF-β not only functions as a primary regulator for collagen and other ECM synthesis, but also prevents collagen fragmentation by inhibiting MMP expression [61,62]. Therefore, impaired TGF-β signaling has a significant impact on collagen homeostasis in human skin. In mammals, three isoforms of TGF-β, TGF-β1, TGF-β2, TGF-β3, have been identified [63]. TGF-β initiates its cellular action by interacting with its cell surface receptor complex, composed of TGF-β type I receptor (TβRI) and TGF-β type II receptor (TβRII) [64]. Binding of TGF-β allows TβRII to activate TβRI, which in turn phosphorylates intracellular transcription factors, Smad2 and Smad3. Activated Smad2 or Smad3 forms heteromeric complexes with the common partner, Smad4. Activated Smad complexes translocate into the nucleus, where they interact with Smad Binding Element (SBE) in the promoter regions of TGF-β target genes, such as collagen, connective tissue growth factor (CCN2/CTGF), fibronectin, and MMP-1.
Age-related impaired TGF-β signaling due to down-regulation of TβRII in dermal fibroblasts is one of the mechanisms of the collagen loss in aged human skin [41,61,65,66,67]. The TβRII mRNA level is reduced in naturally aged [38,41], photoaged [38], and UV-irradiated human skin in vivo [67]. In contrast to TβRII, TβRI mRNA expression is not changed in naturally aged, photoaged, and UV-irradiated human skin in vivo. This suggests age-related specific down-regulation of TβRII. As described above, TGF-β initiates its cellular action by binding to TβRII. Reduced expression of TβRII causes reduced binding of TGF-β1 to the cell surface TβRII, and results in decreased cellular responsiveness to TGF-β [66].
Therefore, age-related down-regulation of TβRII is a prominent feature of aged human skin. Interestingly, both ROS and reduced dermal fibroblast size/mechanical tension specifically down-regulate TβRII, and thus impair the TGF-β/Smad signaling pathway and ECM production [25,34]. ROS/cell size-specific down-regulation of TβRII is associated with significantly decreased phosphorylation, DNA-binding, and transcriptional activity of its key down-stream effector Smad3, as well as reduced expression of Smad3-regulated type I collagen, fibronectin and CTGF/CCN2. Restoration of ROS/cell size-induced loss of TβRII expression significantly increases the TGF-β induction of Smad3 phosphorylation and stimulation of ECM production. Furthermore, ROS/cell size-induced loss of TβRII and ECM production is nearly completely reversed by inhibiting ROS generation [34] and restoring cell size [25].
In addition to the TβRII, the level of Smad3, but not Smad2, mRNA and protein is significantly reduced in aged human skin as compared to young human skin in vivo [34]. Interestingly, this reduced Smad3 is coincidentally observed with reduced type I procollagen and CTGF/CCN2 in aged human skin in vivo [34]. Furthermore, the protein level of Smad3, but not Smad2, is markedly reduced in ROS-induced senescent dermal fibroblasts, in an in vitro aging model. Smad3-specific down-regulation mediates the reduced expression of type I procollagen and CTGF/CCN2 in an in vitro aging model of senescent cells [34]. Restoration of reduced Smad3 by overexpression results in significant increases of Smad3 phosphorylation and ECM production in senescent dermal fibroblasts. These data suggest that elevated ROS causes the down-regulation of Smad3, which in turn mediates the age-related loss of collagen in aged human skin. Collectively, it is postulated that elevated ROS due to reduced fibroblast size could result in impaired TGF-β signaling by the down-regulation of TβRII and Smad3, which in turn contributes to the loss of collagen and the thin dermis in aged human skin (Figure 3).
5. CCN1 Functions as a Critical Mediator of Oxidative Stress-Induced Skin Connective Tissue Aging
CCN1, also known as cysteine-rich angiogenic inducer 61 (CYR61) is the first member of the CCN family of secreted proteins [68,69]. The CCN family of proteins comprises six distinct members: cysteine-rich protein 61 (CYR61/CCN1), connective tissue growth factor (CTGF/CCN2), nephroblastoma overexpressed (NOV/CCN3), Wnt-inducted secreted protein-1 (WISP1/CCN4), Wnt-inducted secreted protein-2 (WISP2/CCN5), and Wnt-inducted secreted protein-3 (WISP3/CCN6) [70,71,72]. All CCN proteins are secreted, ECM-associated matricellular proteins. CCN family proteins are involved in a variety of cellular functions such as regulation of cell adhesion, proliferation, migration, chemotaxis, apoptosis, motility, and ECM remodeling in wound healing [68,73]. In an in vitro tissue culture model, CCN1 regulates cell adhesion, cell migration, cell–matrix interactions and the synthesis of the extracellular matrix [74,75]. CCN1 deficiency in mice is embryonically lethal primarily due to the failure of ECM remodeling and homeostasis [76].
In human skin, CCN1 is predominantly expressed in dermal fibroblasts [38], and significantly elevated in dermal fibroblasts in aged human skin in vivo [38,39,77]. In cultured human dermal fibroblasts, the elevated expression of CCN1 not only inhibits the expression of type I procollagen, but also concurrently increases the expression of multiple MMPs and cytokines [36,38,39,77]. Mechanistically, elevated CCN1 impairs TGF-β signaling by down-regulating TβRII expression, thereby contributing to reduced type I procollagen expression. Additionally, elevated CCN1 induces transcription factor c-Jun/AP-1, which functions to stimulate the expression of multiple MMPs and cytokines.
Interestingly, CCN1 is rapidly induced by ROS in human skin dermal fibroblasts [35]. An antioxidant, N-acetyl-l-cysteine, prevented ROS-induced CCN1 expression and the loss of type I collagen in both human skin in vivo and human dermal fibroblasts in vitro [35]. ROS significantly up-regulated c-Jun, an important transcription factor of the AP-1 complex, and was able to interact with the AP-1 binding site of the CCN1 proximal promoter. Functional blocking of c-Jun significantly reduced CCN1 transcription, and thus prevented the ROS-induced aberrant collagen homeostasis. These data suggest that ROS-mediated c-Jun/CCN1 pathway plays an important role in human skin connective tissue aging, and interference of this pathway may provide clinical benefits for human skin connective tissue aging.
CCN1 is also significantly induced by acute UV irradiation in human skin in vivo, and in UV-irradiated human dermal fibroblasts in vitro [77,78]. CCN1 functions as a key mediator of UV-induced aberrant collagen homeostasis. The antioxidant N-acetyl-l-cysteine significantly reduced UV light-induced CCN1, suggesting a significant role of ROS in CCN1 induction. The functional blockade of c-Jun induction by antioxidants significantly reduced the UV irradiation-induced CCN1 expression and normalized the loss of collagen and elevated MMP-1. These data show that CCN1 is transcriptionally regulated by UV irradiation through ROS-mediated c-Jun/AP-1.
Emerging evidence indicates that CCN1 functions as a novel mediator of collagen homeostasis [1,39,79,80]. Age-related elevation of CCN1 in the human dermis alters the expression of numerous secreted proteins, which together have deleterious effects on the dermal microenvironment (Figure 3). The CCN1-induced aberrant dermal microenvironment is referred to as “Age-Associated Secretory Phenotype (AASP)” [1,39]. AASP includes: (1) reduced expression of skin ECM components by inhibiting TGF-β signaling; (2) damaged dermal collagen by induction of multiple MMPs; and (3) creation of the inflammatory dermal microenvironment (inflammaging) by induction of multiple pro-inflammatory cytokines. Importantly, CCN1-induced AASP is readily observed in the aged human dermis in vivo, and logically could account for many of the characteristic features of aged human skin. Therefore, elevated CCN1 in aged human skin induces multiple AASP-related proteins, which in turn develops an Age Associate Dermal Micronenvironment (AADM), which is a characteristic feature of dermal connective tissue aging.
It has been suggested that CCN1 proteins may represent a new class of modulator of inflammation [81]. CCN1 is markedly induced and activates a proinflammatory genetic program in response to skin wounds in both human skin [1] and mouse skin [82]. Elevated CCN1 up-regulates inflammatory cytokines and chemokines. Furthermore, evidence indicates a potential role of CCN1 in chronic inflammatory diseases such as atherosclerosis, rheumatoid arthritis, inflammatory kidney diseases and neuroinflammatory diseases [83]. Aging is associated with chronic low-grade inflammation which may promote long-term tissue damage and systemic chronic inflammation [84]. Accumulating evidence proposed the concept of “inflammaging”, which posits that low-grade chronic inflammation can be a significant risk factor for the aging progress and age-related diseases [85]. Central to this concept is that healthy aging is not an inflammatory disease, but rather sub-clinical chronic inflammation gradually damages the tissues and impairs organ function, which occurs during the aging process. For example, IL-6 is increased in the aged and has been suggested to be a marker of health status in elderly people [86]. Interestingly, IL-6 is markedly induced by CCN1 in human skin dermal fibroblasts and constitutively elevated in aged skin [78]. Nevertheless, the precise etiology of inflammaging and its potential causal role in the aging progress and age-related diseases remain largely unknown. It should be noted that aged skin does not display overt clinical inflammation. However, the role of AADM-associated cytokines in promoting long-term skin connective tissue aging and systemic inflammaging deserves further investigation.
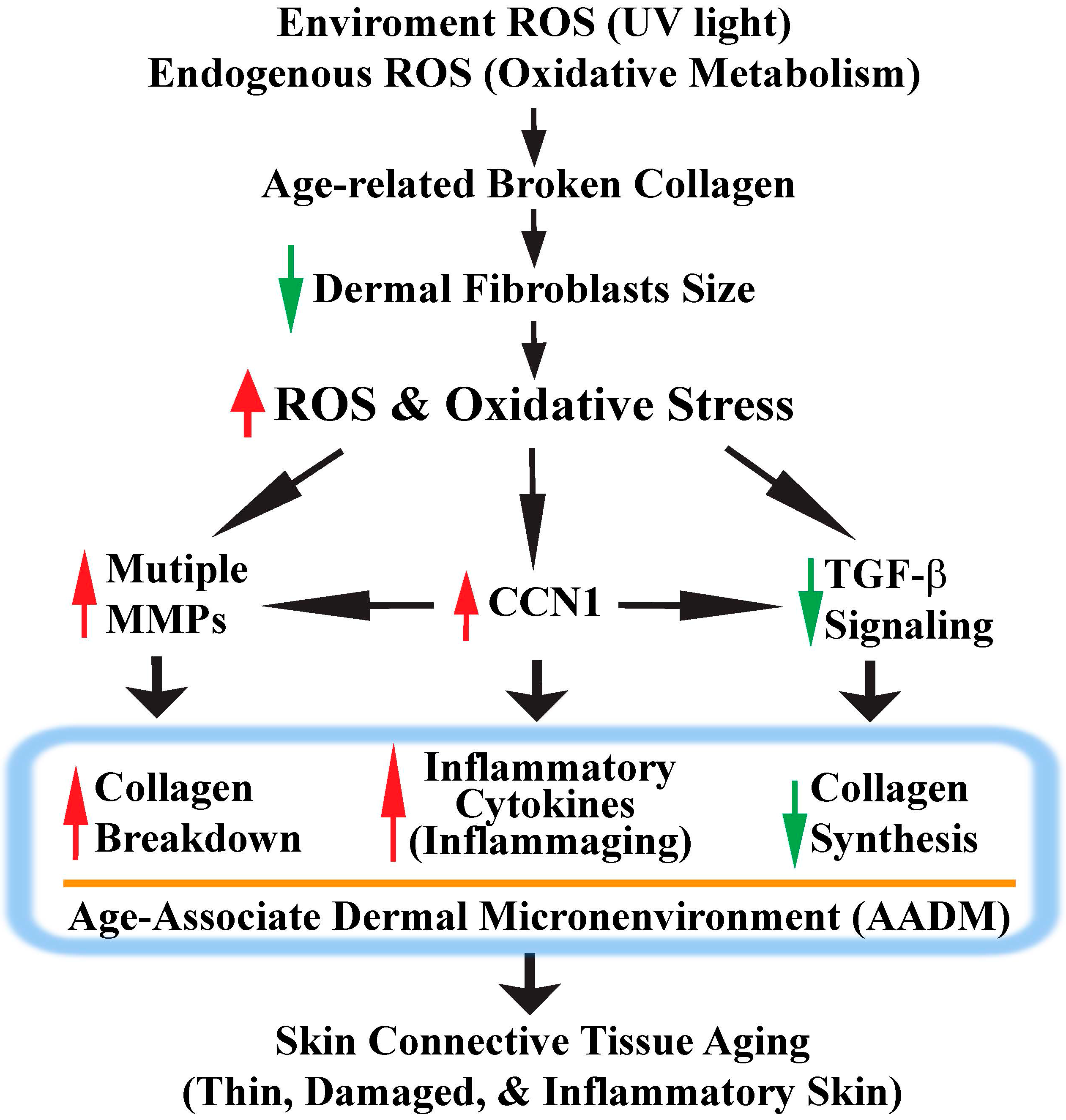
Figure 4 depicts a model in which ROS/oxidative stress contributes to human skin connective tissue aging through creating an AADM. Unlike other organs, human skin is exposed to ROS generated from both exogenous sources such as solar ultraviolet irradiation and endogenous oxidative metabolism. Chronic exposure to ROS causes collagen fragmentation, which impairs cell–matrix interactions and induces dermal fibroblast collapse. Collapsed dermal fibroblasts generate elevated ROS and induce cellular oxidative stress, which functions as a key regulator for human skin connective tissue aging. ROS/oxidative stress contributes to skin connective tissue aging through several pathways: (1) activating c-Jun/AP-1 transcription factor, which in turn elevates multiple MMPs, and thus contributes to damaged skin dermis; (2) inhibiting TβRII and Smad3, which in turn impairs TGF-β signaling, and thus contributes to thin dermis by loss of collagen production; (3) elevating CCN1, which in turn secretes multiple proteins related to AASP. Elevated CCN1 in human dermal fibroblasts acts through multiple pathways to promote AADM and thus contributes to skin connective tissue aging through the following pathways: (1) induction of multiple MMPs; (2) impairment of TGF-β signaling; (3) elevation of multiple pro-inflammatory cytokines (inflammaging).
6. Conclusions
In summary, human skin is constantly targeted by oxidative stress from ROS generated from both extrinsic and intrinsic sources. Chronic exposure to ROS eventually creates AADM, the prominent feature of human skin connective tissue aging. AADM encompasses: (1) increased production of multiple MMPs (collagen fibril fragmentation); (2) reduced production of collagens (thin skin dermis); (3) creation of a proinflammatory microenvironment (inflammaging). AADM weakens the dermal structural and mechanical integrity, and creates a tissue microenvironment that contributes to age-related skin disorders, such as delayed wound healing and epithelial skin cancer development.
Acknowledgments
This study is supported by the National Institutes of Health (Bethesda, MD, USA) Grants: ES014697, ES014697 30S1, AG019364 to Taihao Quan; Dermatology Foundation Research grant to Taihao Quan. The authors would like to thank Gary J. Fisher and John J. Voorhees for their help and support. The author also thanks Zhaoping Qian, Tianyuan He, Yuan Shao and Trupta Purohit for technical assistance, and Hehui Quan for English editing.
Author Contributions
Yidong Tu reviewed and revised this manuscript. Taihao Quan wrote and revised this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Quan, T.; Fisher, G.J. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: A mini-review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef]
- Yaar, M.; Eller, M.S.; Gilchrest, B.A. Fifty years of skin aging. J. Investig. Dermatol. Symp. Proc. 2002, 7, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H. Photoaging in Asians. Photodermatol. Photoimmunol. Photomed. 2003, 19, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Wlaschek, M.; Tantcheva-Poor, I.; Naderi, L.; Ma, W.; Schneider, L.A.; Razi-Wolf, Z.; Schuller, J.; Scharffetter-Kochanek, K. Solar UV irradiation and dermal photoaging. J. Photochem. Photobiol. B 2001, 63, 41–51. [Google Scholar] [CrossRef]
- Uitto, J.; Bernstein, E.F. Molecular mechanisms of cutaneous aging: Connective tissue alterations in the dermis. J. Investig. Dermatol. Symp. Proc. 1998, 3, 41–44. [Google Scholar] [PubMed]
- Fisher, G.J.; Wang, Z.Q.; Datta, S.C.; Varani, J.; Kang, S.; Voorhees, J.J. Pathophysiology of premature skin aging induced by ultraviolet light. N. Engl. J. Med. 1997, 337, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Varani, J.; Voorhees, J.J. Looking older: Fibroblast collapse and therapeutic implications. Arch. Dermatol. 2008, 144, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Lavker, R.M. Structural alterations in exposed and unexposed aged skin. J. Investig. Dermatol. 1979, 73, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.P. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed. Pharmacother. 2003, 57, 195–202. [Google Scholar] [CrossRef]
- Cheresh, D.A.; Stupack, D.G. Regulation of angiogenesis: Apoptotic cues from the ECM. Oncogene 2008, 27, 6285–6298. [Google Scholar] [CrossRef] [PubMed]
- Eaglstein, W.H. Wound healing and aging. Clin. Geriatr. Med. 1989, 5, 183–188. [Google Scholar] [PubMed]
- Valencia, I.C.; Falabella, A.; Kirsner, R.S.; Eaglstein, W.H. Chronic venous insufficiency and venous leg ulceration. J. Am. Acad. Dermatol. 2001, 44, 401–424. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Bader, D.A.; Robles, A.I.; Wangsa, D.; Harris, C.C.; Ried, T.; Yang, L. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-β signaling. PLoS Genet. 2013, 9, e1003251. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, V.R.; Madl, J.; Loffek, S.; Kiritsi, D.; Kern, J.S.; Romer, W.; Nystrom, A.; Bruckner-Tuderman, L. Injury-driven stiffening of the dermis expedites skin carcinoma progression. Cancer Res. 2016, 76, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The aging process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Sticozzi, C.; Pecorelli, A.; Cervellati, F.; Cervellati, C.; Maioli, E. Cutaneous responses to environmental stressors. Ann. N. Y. Acad. Sci. 2012, 1271, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Vierkotter, A.; Krutmann, J. Environmental influences on skin aging and ethnic-specific manifestations. Dermatoendocrinol 2012, 4, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Robichaud, P.; Quan, T. Oxidative stress and CCN1 protein in human skin connective tissue aging. AIMS Mol. Sci. 2016, 3, 269–279. [Google Scholar] [CrossRef]
- Fisher, G.J.; Shao, Y.; He, T.; Qin, Z.; Perry, D.; Voorhees, J.J.; Quan, T. Reduction of fibroblast size/mechanical force down-regulates TGF-β type ii receptor: Implications for human skin aging. Aging Cell 2016, 15, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Schuger, L.; Dame, M.K.; Leonard, C.; Fligiel, S.E.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Reduced fibroblast interaction with intact collagen as a mechanism for depressed collagen synthesis in photodamaged skin. J. Investig. Dermatol. 2004, 122, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Wang, F.; Shao, Y.; Rittie, L.; Xia, W.; Orringer, J.S.; Voorhees, J.J.; Fisher, G.J. Enhancing structural support of the dermal microenvironment activates fibroblasts, endothelial cells, and keratinocytes in aged human skin in vivo. J. Investig. Dermatol. 2013, 133, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, F.J.; Nauli, S.M.; Kolb, R.; Zhou, J.; Ingber, D.E. Global cytoskeletal control of mechanotransduction in kidney epithelial cells. Exp. Cell Res. 2004, 301, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Silver, F.H.; Siperko, L.M.; Seehra, G.P. Mechanobiology of force transduction in dermal tissue. Skin Res. Technol. 2003, 9, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Butler, J.P.; Ingber, D.E. Mechanotransduction across the cell surface and through the cytoskeleton. Science 1993, 260, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Cho, M.K.; Perry, D.; Quan, T. Age-associated reduction of cell spreading induces mitochondrial DNA common deletion by oxidative stress in human skin dermal fibroblasts: Implication for human skin connective tissue aging. J. Biomed. Sci. 2015, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Quan, T.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Oxidative exposure impairs TGF-β pathway via reduction of type II receptor and SMAD3 in human skin fibroblasts. Age 2014, 36, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Robichaud, P.; He, T.; Fisher, G.J.; Voorhees, J.J.; Quan, T. Oxidant exposure induces cysteine-rich protein 61 (CCN1) via c-Jun/AP-1 to reduce collagen expression in human dermal fibroblasts. PLoS ONE 2014, 9, e115402. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Fisher, G.J.; Quan, T. Cysteine-rich protein 61 (CCN1) domain-specific stimulation of matrix metalloproteinase-1 expression through αVβ3 integrin in human skin fibroblasts. J. Biol. Chem. 2013, 288, 12386–12394. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-degrading metalloproteinases in photoaging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Shao, Y.; Lin, L.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Elevated cysteine-rich 61 mediates aberrant collagen homeostasis in chronologically aged and photoaged human skin. Am. J. Pathol. 2006, 169, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Robichaud, P.; Voorhees, J.J.; Fisher, G.J. CCN1 contributes to skin connective tissue aging by inducing age-associated secretory phenotype in human skin dermal fibroblasts. J. Cell Commun. Signal. 2011, 5, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Little, E.; Quan, H.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: Impact of altered extracellular matrix microenvironment on dermal fibroblast function. J. Investig. Dermatol. 2013, 133, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Shao, Y.; He, T.; Voorhees, J.J.; Fisher, G.J. Reduced expression of connective tissue growth factor (CTGF/CCN2) mediates collagen loss in chronologically aged human skin. J. Investig. Dermatol. 2010, 130, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Okubo, T.; Voorhees, J.J.; Fisher, G.J.; Quan, T. Elevated cysteine-rich protein 61 (CCN1) promotes skin aging via upregulation of IL-1β in chronically sun-exposed human skin. Age 2014, 36, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, W.F. The aging skin. Int. J. Fertil. Women’s Med. 1997, 42, 57–66. [Google Scholar]
- Smith, J.G., Jr.; Davidson, E.A.; Sams, W.M., Jr.; Clark, R.D. Alterations in human dermal connective tissue with age and chronic sun damage. J. Investig. Dermatol. 1962, 39, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Warner, R.L.; Gharaee-Kermani, M.; Phan, S.H.; Kang, S.; Chung, J.H.; Wang, Z.Q.; Datta, S.C.; Fisher, G.J.; Voorhees, J.J. Vitamin A antagonizes decreased cell growth and elevated collagen-degrading matrix metalloproteinases and stimulates collagen accumulation in naturally aged human skin. J. Investig. Dermatol. 2000, 114, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Kohrmann, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Anacker, J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer 2009, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, L.; Cho, H.; Xie, X.; Eliceiri, G.L. Additional MDA-MB-231 breast cancer cell matrix metalloproteinases promote invasiveness. J. Cell Physiol. 2008, 216, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Brenneisen, P.; Sies, H.; Scharffetter-Kochanek, K. Ultraviolet-B irradiation and matrix metalloproteinases: From induction via signaling to initial events. Ann. N. Y. Acad. Sci. 2002, 973, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Datta, S.C.; Talwar, H.S.; Wang, Z.Q.; Varani, J.; Kang, S.; Voorhees, J.J. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature 1996, 379, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.; Mandal, M.; Das, S.; Mandal, A.; Chakraborti, T. Regulation of matrix metalloproteinases: An overview. Mol. Cell Biochem. 2003, 253, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Benbow, U.; Brinckerhoff, C.E. The AP-1 site and MMP gene regulation: What is all the fuss about? Matrix Biol. 1997, 15, 519–526. [Google Scholar] [CrossRef]
- Gutman, A.; Wasylyk, B. The collagenase gene promoter contains a TPA and oncogene-responsive unit encompassing the PEA3 and AP-1 binding sites. EMBO J. 1990, 9, 2241–2246. [Google Scholar] [PubMed]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Kang, S.; Varani, J.; Lin, J.; Fisher, G.J.; Voorhees, J.J. Decreased extracellular-signal-regulated kinase and increased stress-activated map kinase activities in aged human skin in vivo. J. Investig. Dermatol. 2000, 115, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.H.; Rhie, G.E.; Kim, Y.K.; Park, C.H.; Cho, K.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. H2O2 accumulation by catalase reduction changes map kinase signaling in aged human skin in vivo. J. Investig. Dermatol. 2005, 125, 221–229. [Google Scholar] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming growth factor β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [PubMed]
- Patil, A.S.; Sable, R.B.; Kothari, R.M. An update on transforming growth factor-β (TGF-β): Sources, types, functions and clinical applicability for cartilage/bone healing. J. Cell Physiol. 2011, 226, 3094–3103. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Rosenbloom, J.; Jimenez, S.A. Transforming growth factor β (TGF β) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem. J. 1987, 247, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Connective tissue growth factor: Expression in human skin in vivo and inhibition by ultraviolet irradiation. J. Investig. Dermatol. 2002, 118, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.C.; Young, D.A.; Waters, J.G.; Rowan, A.D.; Chantry, A.; Edwards, D.R.; Clark, I.M. The comparative role of activator protein 1 and smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-β1. J. Biol. Chem. 2003, 278, 10304–10313. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. The transforming growth factor-β family. Annu. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation alters transforming growth factor β/smad pathway in human skin in vivo. J. Investig. Dermatol. 2002, 119, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation blocks cellular responses to transforming growth factor-β by down-regulating its type-II receptor and inducing Smad7. J. Biol. Chem. 2001, 276, 26349–26356. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-β type II receptor/Smad signaling. Am. J. Pathol. 2004, 165, 741–751. [Google Scholar] [CrossRef]
- Lau, L.F.; Lam, S.C. The CCN family of angiogenic regulators: The integrin connection. Exp. Cell Res. 1999, 248, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Planque, N.; Perbal, B. A structural approach to the role of CCN (CYR61/CTGF/NOV) proteins in tumourigenesis. Cancer Cell Int. 2003, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B.; Brigstock, D.R.; Lau, L.F. Report on the second international workshop on the CCN family of genes. Mol. Pathol. 2003, 56, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Lau, L.F. Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 2009, 41, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kireeva, M.L.; Mo, F.E.; Yang, G.P.; Lau, L.F. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol. Cell Biol. 1996, 16, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chen, N.; Lau, L.F. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J. Biol. Chem. 2001, 276, 10443–10452. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.E.; Muntean, A.G.; Chen, C.C.; Stolz, D.B.; Watkins, S.C.; Lau, L.F. Cyr61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell Biol. 2002, 22, 8709–8720. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Xu, Y.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces Cyr61/CCN1, a mediator of collagen homeostasis, through activation of transcription factor AP-1 in human skin fibroblasts. J. Investig. Dermatol. 2010, 130, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Shin, S.; Qin, Z.; Fisher, G.J. Expression of CCN family of genes in human skin in vivo and alterations by solar-simulated ultraviolet irradiation. J. Cell Commun. Signal. 2009, 3, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Cysteine-rich protein 61 (CCN1) mediates replicative senescence-associated aberrant collagen homeostasis in human skin fibroblasts. J. Cell. Biochem. 2012, 113, 3011–3018. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Mo, F.E.; Lau, L.F. The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J. Biol. Chem. 2001, 276, 47329–47337. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Chen, C.C.; Lau, L.F. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J. Immunol. 2010, 184, 3223–3232. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Araneo, B.A.; Ershler, W.B.; Maloney, C.; Li, G.Z.; Ryu, S.Y. Altered regulation of IL-6 production with normal aging. Possible linkage to the age-associated decline in dehydroepiandrosterone and its sulfated derivative. J. Immunol. 1993, 150, 5219–5230. [Google Scholar] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]
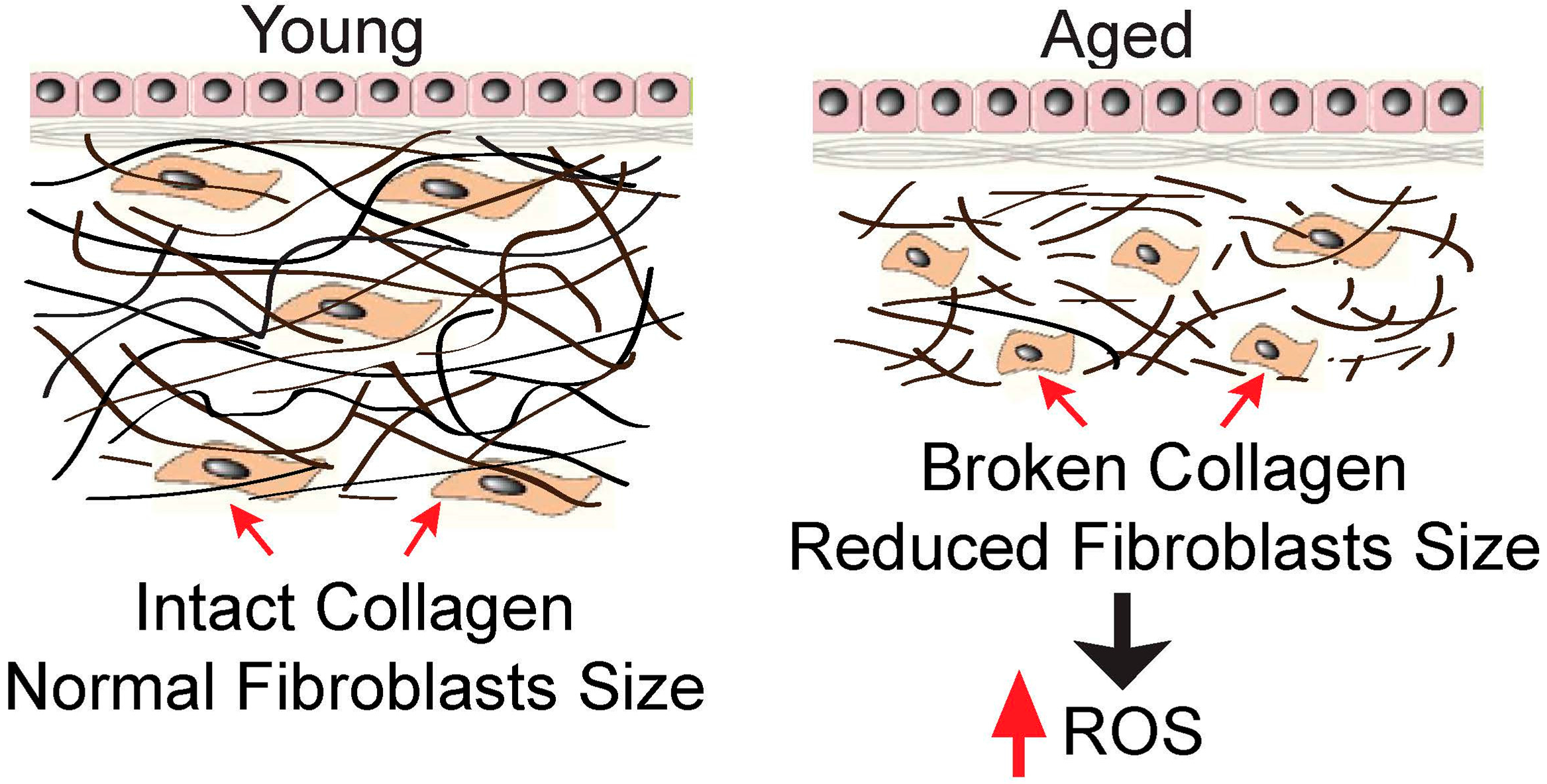
Figure 1.
Reduced fibroblasts size stimulates intracellular ROS generation. In the young human skin dermis, intact collagen fibrils interact with cells to maintain normal cell spreading and size (left). In contrast, in the aged human skin dermis (right), broken collagen fibrils are unable to support normal cell spreading, and this causes reduced cell size. One of the prominent features of the collapsed cells is the increase in intracellular ROS generation (see text for details).
Figure 1.
Reduced fibroblasts size stimulates intracellular ROS generation. In the young human skin dermis, intact collagen fibrils interact with cells to maintain normal cell spreading and size (left). In contrast, in the aged human skin dermis (right), broken collagen fibrils are unable to support normal cell spreading, and this causes reduced cell size. One of the prominent features of the collapsed cells is the increase in intracellular ROS generation (see text for details).

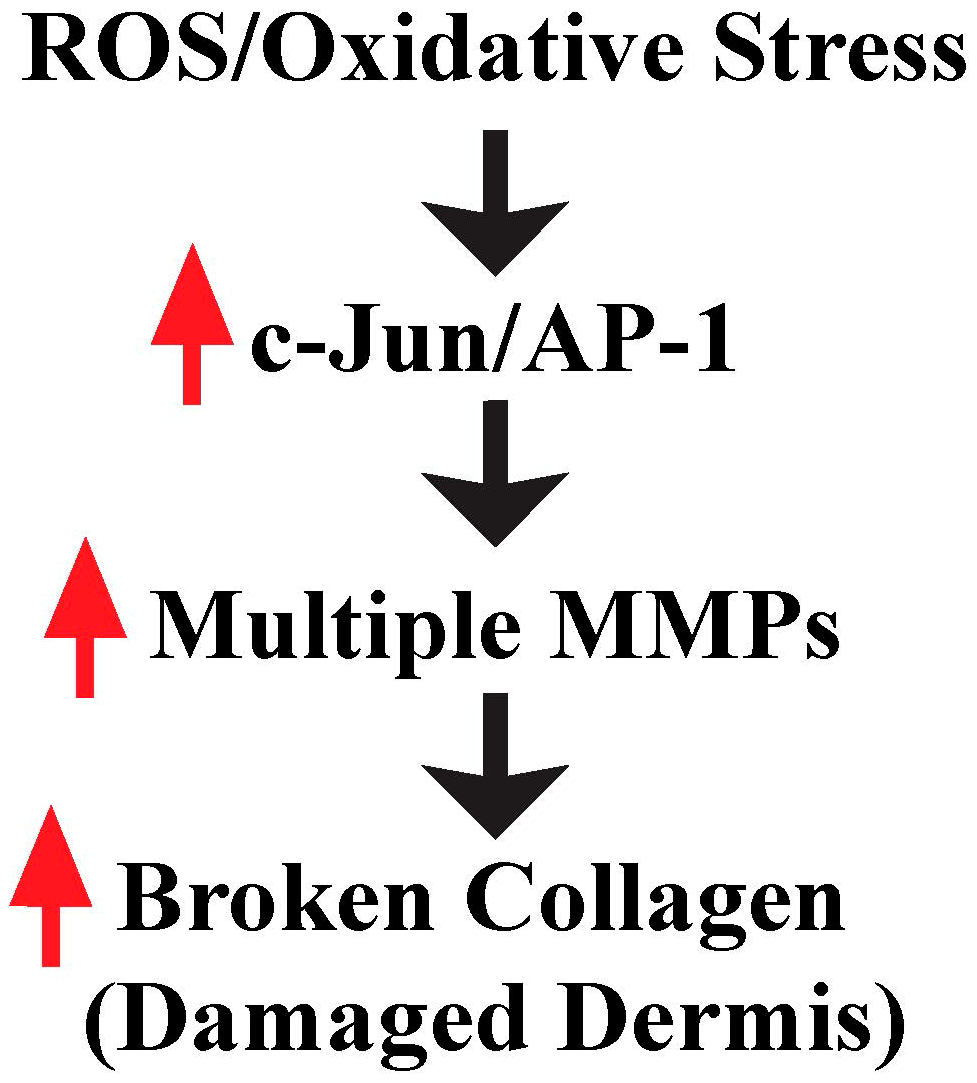
Figure 2.
ROS/oxidative stress contributes to damaged dermis by induction of multiple MMPs. Increased ROS by reduced fibroblast size activates c-Jun/AP-1, which in turn elevates multiple MMPs, and contributes to damaged skin dermis in aging skin (see text for details).
Figure 2.
ROS/oxidative stress contributes to damaged dermis by induction of multiple MMPs. Increased ROS by reduced fibroblast size activates c-Jun/AP-1, which in turn elevates multiple MMPs, and contributes to damaged skin dermis in aging skin (see text for details).

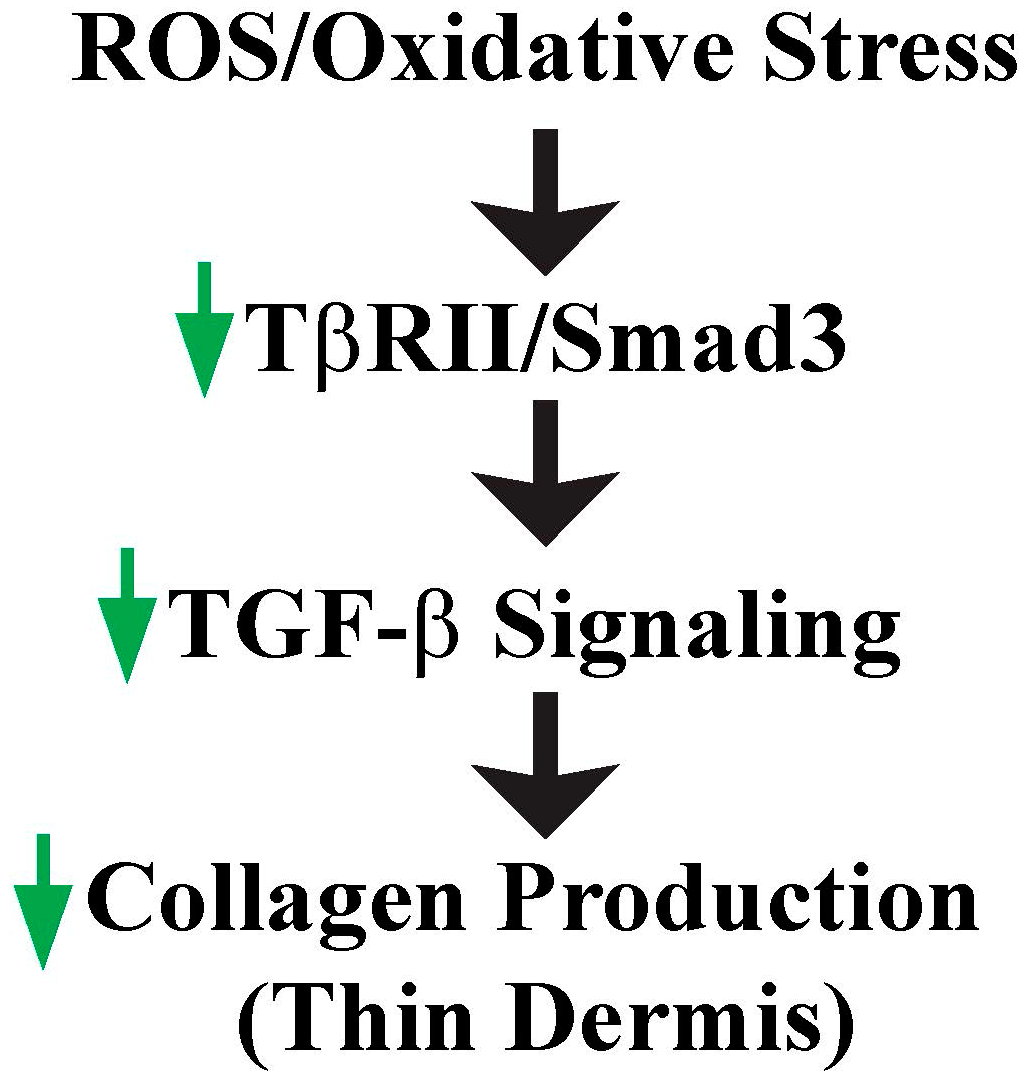
Figure 3.
ROS/oxidative stress contributes to thin dermis by inhibition of TGF-β signaling. Increased ROS due to reduced fibroblast size impairs TGF-β signaling by down-regulation of TβRII and Smad3, which in turn contributes to loss of collagen and thin dermis in aged human skin (see text for details).
Figure 3.
ROS/oxidative stress contributes to thin dermis by inhibition of TGF-β signaling. Increased ROS due to reduced fibroblast size impairs TGF-β signaling by down-regulation of TβRII and Smad3, which in turn contributes to loss of collagen and thin dermis in aged human skin (see text for details).

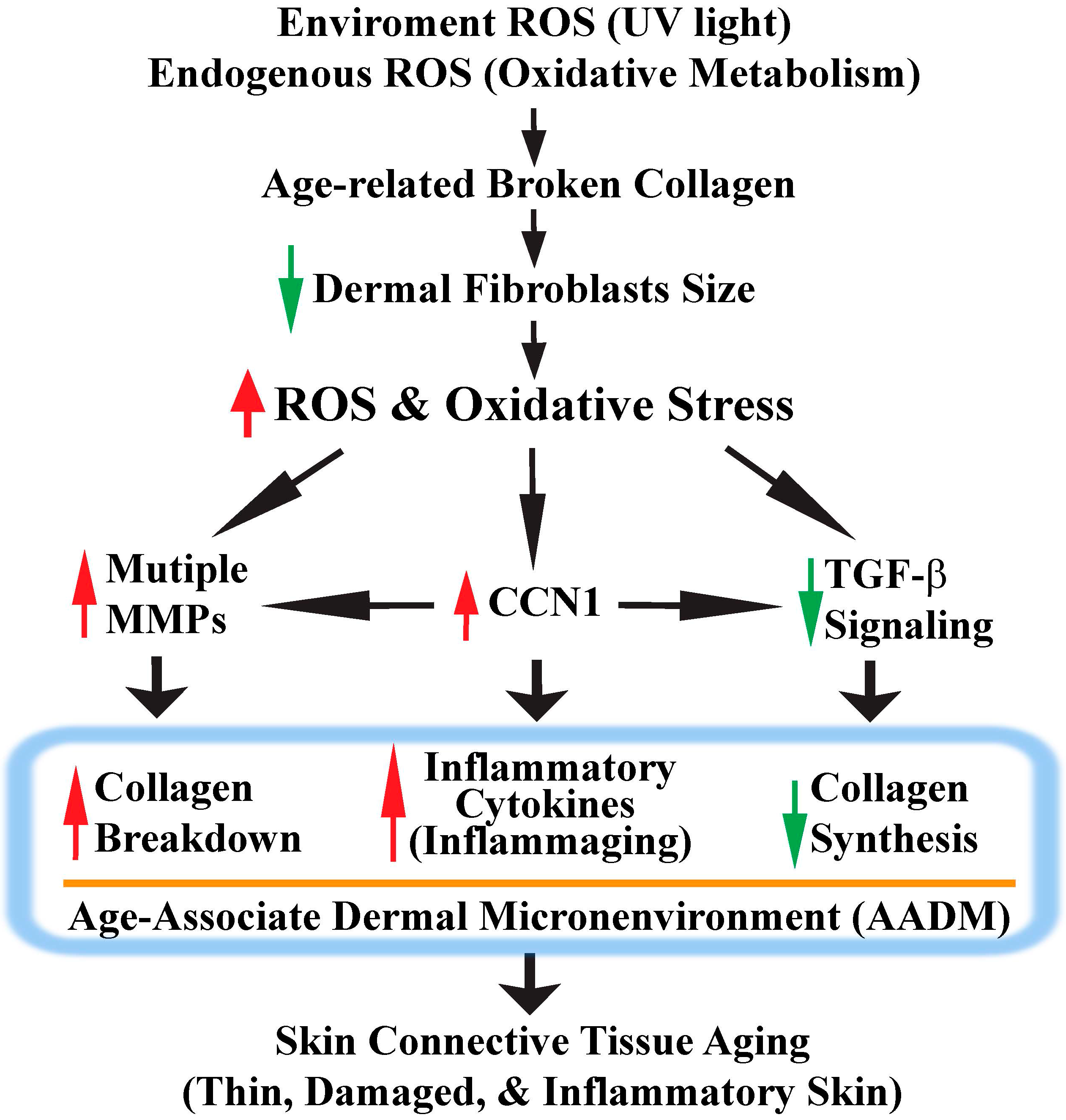
Figure 4.
Model for human skin connective tissue aging. Human skin is a main target of ROS/oxidative stress from both extrinsic (UV) and intrinsic sources (oxidative metabolism). Chronic exposure to ROS induces dermal fibroblast collapse by creating collagen fragmentation. Collapsed dermal fibroblasts generate more ROS and activate the c-Jun/AP-1 complex, which in turn up-regulates MMPs and CCN1 expression, as well as down-regulates TGF-β signaling. These ROS-mediated signaling events eventually lead to the development of an age-associated dermal microenvironment (AADM). AADM encompasses: (1) increased production of multiple MMPs, which contributes to the damaged skin dermis; (2) reduced production of collagens, which contributes to the thin skin dermis; (3) creation of a proinflammatory microenvironment (inflammaging). AADM impairs the skin’s structural and mechanical integrities, and creates a tissue microenvironment that contributes to the age-related decline of skin function (see Section 6 for details).
Figure 4.
Model for human skin connective tissue aging. Human skin is a main target of ROS/oxidative stress from both extrinsic (UV) and intrinsic sources (oxidative metabolism). Chronic exposure to ROS induces dermal fibroblast collapse by creating collagen fragmentation. Collapsed dermal fibroblasts generate more ROS and activate the c-Jun/AP-1 complex, which in turn up-regulates MMPs and CCN1 expression, as well as down-regulates TGF-β signaling. These ROS-mediated signaling events eventually lead to the development of an age-associated dermal microenvironment (AADM). AADM encompasses: (1) increased production of multiple MMPs, which contributes to the damaged skin dermis; (2) reduced production of collagens, which contributes to the thin skin dermis; (3) creation of a proinflammatory microenvironment (inflammaging). AADM impairs the skin’s structural and mechanical integrities, and creates a tissue microenvironment that contributes to the age-related decline of skin function (see Section 6 for details).
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tu, Y.; Quan, T. Oxidative Stress and Human Skin Connective Tissue Aging. Cosmetics 2016, 3, 28. https://doi.org/10.3390/cosmetics3030028
AMA Style
Tu Y, Quan T. Oxidative Stress and Human Skin Connective Tissue Aging. Cosmetics. 2016; 3(3):28. https://doi.org/10.3390/cosmetics3030028
Chicago/Turabian StyleTu, Yidong, and Taihao Quan. 2016. "Oxidative Stress and Human Skin Connective Tissue Aging" Cosmetics 3, no. 3: 28. https://doi.org/10.3390/cosmetics3030028
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.