
Skin Redox Balance Maintenance: The Need for an Nrf2-Activator Delivery System

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidation Processes in Skin
2.1. Reactive Oxygen Species (ROS) and Oxidative Stress in Skin
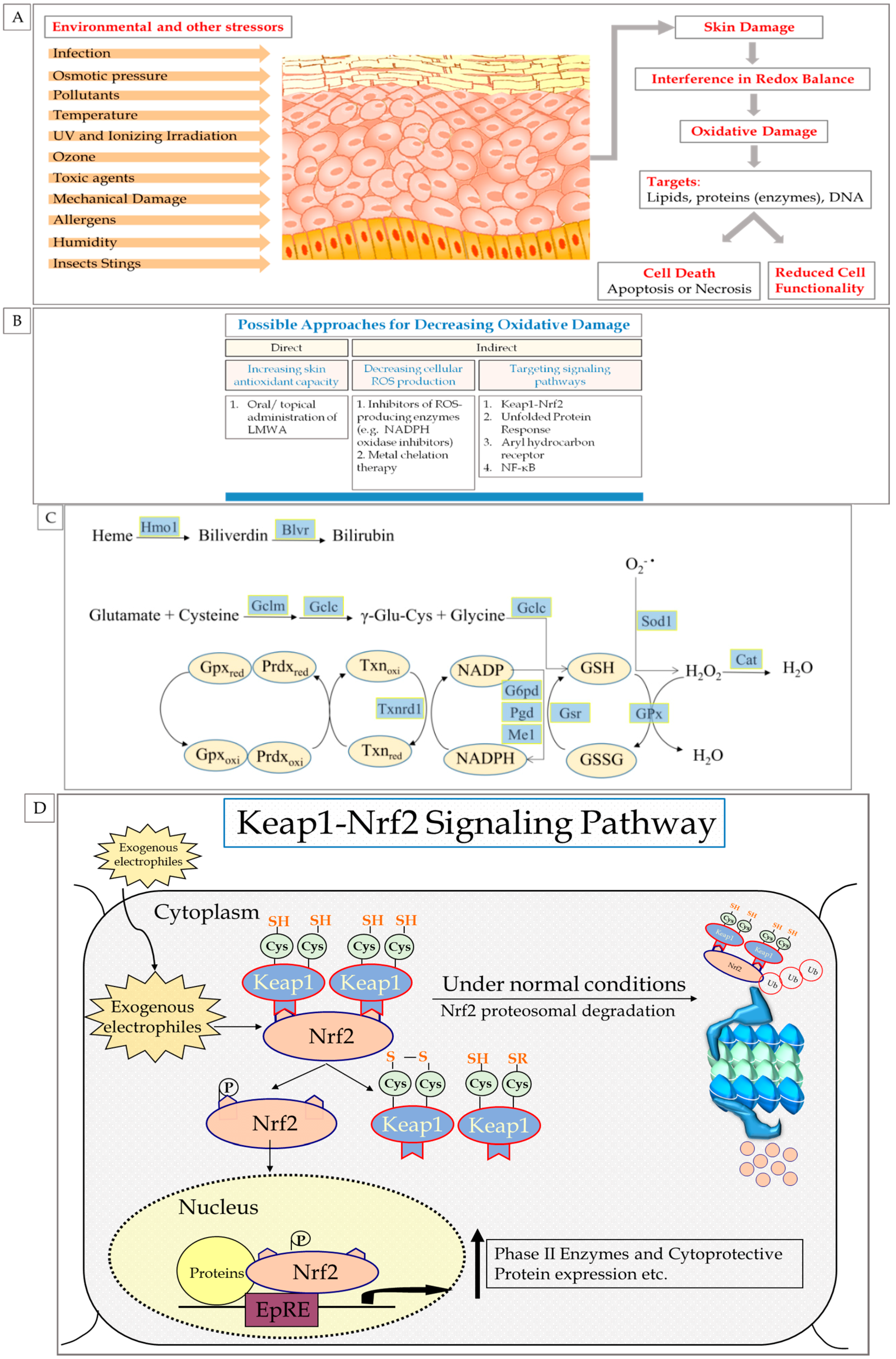
2.2. Skin Exposure to Oxidative Stress: Exogenous and Endogenous Sources
2.3. ROS: Beneficial and Detrimental Consequences to Skin
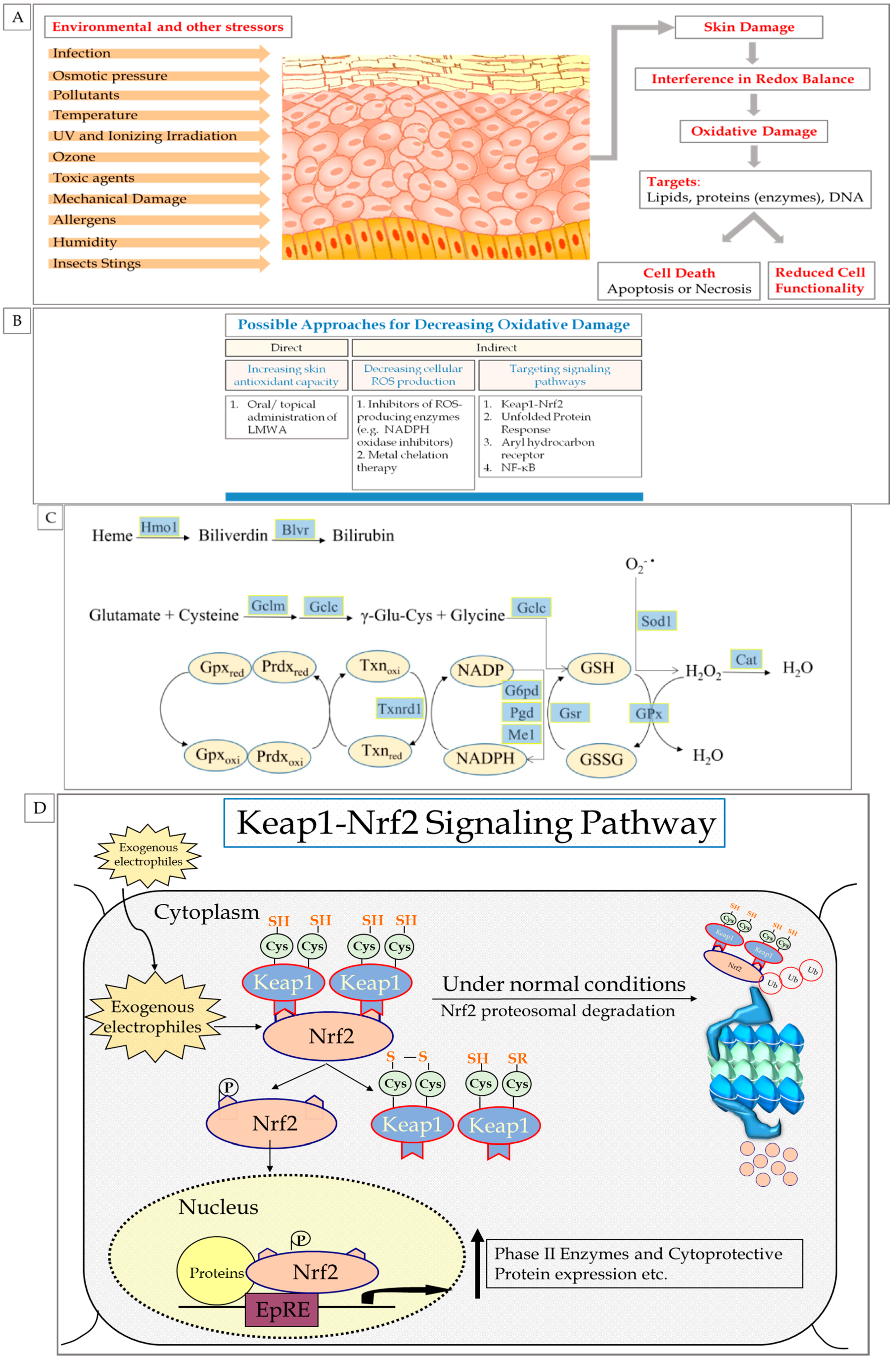
3. Endogenous Cellular Defense Systems in the Skin
3.1. Antioxidant Defense System
3.1.1. Low Molecular Weight Antioxidants (LMWA)
3.1.2. Antioxidant Enzymes
4. Can Skin Redox Balance Be Efficiently Affected by Exogenous Intervention?
4.1. Is Topical Application of LMWA Useful?
4.2. Can Activation of Cytoprotective Signaling Pathways in Skin Be Effective?
5. The Role of Nrf2 in Skin Redox Balance
6. Nrf2 Pathway
6.1. Nrf2 Mechanism of Action
6.2. Nrf2-Activating Agents
7. Enhanced Protection against Oxidative Damage by Nrf2-Activating Agents Using Delivery Systems
7.1. The Need for Transient and Controlled Nrf2 Activation
7.2. Is the Usage of Topical Delivery Systems Suitable?
7.3. Desired Features of an Nrf2-Activator Delivery System

Examples of Nrf2-Activator Delivery Systems
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Trouba, K.J.; Hamadeh, H.K.; Amin, R.P.; Germolec, D.R. Oxidative stress and its role in skin disease. Antioxid. Redox Signal. 2002, 4, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R.; Gati, I. Skin low molecular weight antioxidants and their role in aging and in oxidative stress. Toxicology 2000, 148, 149–157. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: Differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review). Int. J. Mol. Med. 1999, 4, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Lavrovsky, Y.; Chatterjee, B.; Clark, R.A.; Roy, A.K. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000, 35, 521–532. [Google Scholar] [CrossRef]
- Kohen, R. Skin antioxidants: Their role in aging and in oxidative stress-new approaches for their evaluation. Biomed. Pharmacother. 1999, 53, 181–192. [Google Scholar] [CrossRef]
- Wölfle, U.; Seelinger, G.; Bauer, G.; Meinke, M.C.; Lademann, J.; Schempp, C.M. Reactive molecule species and antioxidative mechanisms in normal skin and skin aging. Skin Pharmacol. Physiol. 2014, 27, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Bickers, D.R.; Athar, M. Oxidative stress in the pathogenesis of skin disease. J. Investig. Dermatol. 2006, 126, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.; Carels, C.E.; Lundvig, D. Targeting the redox balance in inflammatory skin conditions. Int. J. Mol. Sci. 2013, 14, 9126–9167. [Google Scholar] [PubMed]
- Schäfer, M.; Werner, S. NRF2—A regulator of keratinocyte redox signaling. Free Radic. Biol. Med. 2015, 88, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P.; Fahey, J.W.; Healy, Z.R.; Wehage, S.L.; Benedict, A.L.; Min, C.; Dinkova-Kostova, A.T. Sulforaphane mobilizes cellular defenses that protect skin against damage by UV radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 17500–17505. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods of their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: Oxford, UK, 1999; Volume 3. [Google Scholar]
- Blanpain, C.D.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar]
- Rinnerthaler, M.; Streubel, M.K.; Bischof, J.; Richter, K. Skin aging, gene expression and calcium. Exp. Gerontol. 2015, 68, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Wood, J.M. Free radical reduction in the human epidermis. Free Radic. Biol. Med. 1989, 6, 519–532. [Google Scholar] [CrossRef]
- Krutmann, J.; Schruder, P.; Morita, A. Molekulare mechanismen der hautalterung durch UV-strahlung und andere exogene noxen. In Hautalterung; Springer: Berlin, Germany, 2008; pp. 23–36. (In German) [Google Scholar]
- Poljšak, B.; Dahmane, R. Free radicals and extrinsic skin aging. Dermatol. Res. Pract. 2012, 2012, 135206. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Gogvadze, V.; Laffranchi, R.; Schlapbach, R.; Schweizer, M.; Suter, M.; Walter, P.; Yaffee, M. Oxidants in mitochondria: From physiology to diseases. Biochim. Biophys. Acta Mol. Basis Dis. 1995, 1271, 67–74. [Google Scholar] [CrossRef]
- Kandola, K.; Bowman, A.; Birch-Machin, M.A. Oxidative stress—A key emerging impact factor in health, ageing, lifestyle and aesthetics. Int. J. Cosmet. Sci. 2015, 37 (Suppl. 2), 1–8. [Google Scholar] [CrossRef]
- Deliconstantinos, G.; Villiotou, V. Gas phase oxidants of cigarette smoke increase nitric oxide synthase and xanthine oxidase activities of rabbit brain synaptosomes. Neurochem. Res. 2000, 25, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Kadiiska, M.B.; Jiang, J.J.; Stadler, K.; Mason, R.P. Free radical production requires both inducible nitric oxide synthase and xanthine oxidase in LPS-treated skin. Proc. Natl. Acad. Sci. USA 2006, 103, 4616–4621. [Google Scholar] [CrossRef] [PubMed]
- Portugal-Cohen, M.; Kohen, R. Exposure of human keratinocytes to ischemia, hyperglycemia and their combination induces oxidative stress via the enzymes inducible nitric oxide synthase and xanthine oxidase. J. Dermatol. Sci. 2009, 55, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Portugal-Cohen, M.; Numa, R.; Yaka, R.; Kohen, R. Cocaine induces oxidative damage to skin via xanthine oxidase and nitric oxide synthase. J. Dermatol. Sci. 2010, 58, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Kammeyer, A.; Luiten, R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Lipid peroxidation-DNA damage by malondialdehyde. Mutat. Res. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Briganti, S.; Picardo, M. Antioxidant activity, lipid peroxidation and skin diseases. What’s new. J. Eur. Acad. Dermatol. Venereol. 2003, 17, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M. 11th Hour: Introduction to Immunology; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Pillai, S.; Oresajo, C.; Hayward, J. Ultraviolet radiation and skin aging: Roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation—A review. Int. J. Cosmet. Sci. 2005, 27, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Wlaschek, M.; Tantcheva-Poor, I.; Naderi, L.; Ma, W.; Schneider, L.A.; Razi-Wolf, Z.; Schuller, J.; Scharffetter-Kochanek, K. Solar UV irradiation and dermal photoaging. J. Photochem. Photobiol. B Biol. 2001, 63, 41–51. [Google Scholar] [CrossRef]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Drew, J.E. Cellular defense system gene expression profiling of human whole blood: Opportunities to predict health benefits in response to diet. Adv. Nutr. Int. Rev. J. 2012, 3, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Birch-Machin, M.A.; Russell, E.V.; Latimer, J.A. Mitochondrial DNA damage as a biomarker for ultraviolet radiation exposure and oxidative stress. Br. J. Dermatol. 2013, 169 (Suppl. 2), 9–14. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Chua, P.P.S.; Chong, M.T.; Sim, A.B.T.; Nikaido, O.; Tron, V.A. Nucleotide excision repair genes are upregulated by low-dose artificial ultraviolet B: Evidence of a photoprotective SOS response? J. Investig. Dermatol. 2001, 117, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative Stress in Aging Human Skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S. Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxid. Med. Cell. Longev. 2009, 2, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Packer, J.E.; Slater, T.; Willson, R.L. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Martensson, J.; Meister, A.; Mrtensson, J. Glutathione deficiency decreases tissue ascorbate levels in newborn rats: Ascorbate spares glutathione and protects. Proc. Natl. Acad. Sci. USA 1991, 88, 4656–4660. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Torres, M.; Thiele, J.J.; Shindo, Y.; Han, D.; Packer, L. Topical application of alpha-tocopherol modulates the antioxidant network and diminishes ultraviolet-induced oxidative damage in murine skin. Br. J. Dermatol. 1998, 138, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, W.P.; Alia, A.; Backendorf, C. ROS quenching potential of the epidermal cornified cell envelope. J. Investig. Dermatol. 2011, 131, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Stojiljković, D.; Pavlović, D.I.; Arsić, I. Oxidative Stress, Skin Aging and Antioxidant Therapy/Oksidacioni Stres, Starenje Kooe I Antioksidaciona Terapija. Acta Fac. Med. Naissensis 2014, 31, 207–217. [Google Scholar]
- Rigo, A.; Stevanato, R.; Finazzi-Agro, A.; Rotilio, G. An attempt to evaluate the rate of the haber-weiss reaction by using OH radical scavengers. FEBS Lett. 1977, 80, 130–132. [Google Scholar] [CrossRef]
- Yang, H.; Wang, X.; Liu, X.; Wu, J.; Liu, C.; Gong, W.; Zhao, Z.; Hong, J.; Lin, D.; Wang, Y.; et al. Antioxidant peptidomics reveals novel skin antioxidant system. Mol. Cell. Proteomics 2009, 8, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Weinraub, D.; Levy, P.; Faraggi, M. Chemical properties of water-soluble porphyrins. 5. Reactions of some manganese (III) porphyrins with the superoxide and other reducing radicals. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1986, 50, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A. Does carnosine possess direct antioxidant activity? Int. J. Biochem. 1993, 25, 1101–1107. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; de Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Kohen, R.; Yamamoto, Y.; Cundy, K.C.; Ames, B.N. Antioxidant activity of carnosine, homocarnosine, and anserine present in muscle and brain. Proc. Natl. Acad. Sci. USA 1988, 85, 3175–3179. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Kraemer, K.; Rimbach, G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition 2001, 17, 888–895. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Myriam, M.; Sabatier, M.; Steiling, H.; Williamson, G. Skin bioavailability of dietary vitamin E, carotenoids, polyphenols, vitamin C, zinc and selenium. Br. J. Nutr. 2006, 96, 227–238. [Google Scholar] [CrossRef]
- Ratnam, D.V.; Ankola, D.D.; Bhardwaj, V.; Sahana, D.K.; Kumar, M.N. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Release 2006, 113, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Fernández-García, E. Skin protection against UV light by dietary antioxidants. Food Funct. 2014, 5, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Boddupalli, S.; Mein, J.R.; James, D.R.; Lakkanna, S. Induction of phase 2 antioxidant enzymes by broccoli sulforaphane: Perspectives in maintaining the antioxidant activity of vitamins A, C, and E. Front. Genet. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Auf dem Keller, U.; Kumin, A.; Braun, S.; Werner, S. Reactive oxygen species and their detoxification in healing skin wounds. J. Investig. Dermatol. Symp. Proc. 2006, 11, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Shindo, Y.; Witt, E.; Packer, L. Antioxidant defense mechanisms in murine epidermis and dermis and their responses to ultraviolet light. J. Investig. Dermatol. 1993, 100, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Haines, D.D.; Lekli, I.; Teissier, P.; Bak, I.; Tosaki, A. Role of haeme oxygenase-1 in resolution of oxidative stress-related pathologies: Focus on cardiovascular, lung, neurological and kidney disorders. Acta Physiol. 2012, 204, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, R.M. Solar ultraviolet A radiation: An oxidizing skin carcinogen that activates heme oxygenase-1. Antioxid. Redox Signal. 2004, 6, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Saw, C.L.; Yang, A.Y.; Huang, M.-T.; Liu, Y.; Lee, J.H.; Khor, T.O.; Su, Z.-Y.; Shu, L.; Lu, Y.; Conney, A.H.; et al. Nrf2 null enhances UVB-induced skin inflammation and extracellular matrix damages. Cell Biosci. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Gul, M.; Kutay, F.Z.; Temocin, S.; Hanninen, O. Cellular and clinical implications of glutathione. Indian J. Exp. Biol. 2000, 38, 625–634. [Google Scholar] [PubMed]
- Peres, P.S.; Terra, V.A.; Guarnier, F.A.; Cecchini, R.; Cecchini, A.L. Photoaging and chronological aging profile: Understanding oxidation of the skin. J. Photochem. Photobiol. B Biol. 2011, 103, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.-S.; Kundu, J.; Kundu, J.K.; Surh, Y.-J. Targeting Nrf2-Keap1 signaling for chemoprevention of skin carcinogenesis with bioactive phytochemicals. Toxicol. Lett. 2014, 229, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Engin, A. Glutathione content of human skin carcinomas. Arch. Dermatol. Res. 1976, 257, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Matsumoto, M.; Iizuka, H.; Suzuki, K.; Taniguchi, N. Superoxide dismutase in psoriasis, squamous cell carcinoma and basal cell epithelioma: An immunohistochemical study. Br. J. Dermatol. 1991, 124, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Kartasova, T.; Segre, J.A. Mouse Sprr locus: A tandem array of coordinately regulated genes. Mamm. Genome 2003, 14, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Duschl, J.; Steinbacher, P.; Salzmann, M.; Bischof, J.; Schuller, M.; Wimmer, H.; Peer, T.; Bauer, J.W.; Richter, K. Age-related changes in the composition of the cornified envelope in human skin. Exp. Dermatol. 2013, 22, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Baroni, A.; Buommino, E.; de Gregorio, V.; Ruocco, E.; Ruocco, V.; Wolf, R. Structure and function of the epidermis related to barrier properties. Clin. Dermatol. 2012, 30, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.J.; Hsieh, S.N.; Briviba, K.; Sies, H. Protein oxidation in human stratum corneum: Susceptibility of keratins to oxidation in vitro and presence of a keratin oxidation gradient in vivo. J. Investig. Dermatol. 1999, 113, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.-L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Black, H.S. ROS: A step closer to elucidating their role in the etiology of light-induced skin disorders. J. Investig. Dermatol. 2004, 122, xiii–xiv. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Sticozzi, C.; Pecorelli, A.; Cervellati, F.; Cervellati, C.; Maioli, E. Cutaneous responses to environmental stressors. Ann. N. Y. Acad. Sci. 2012, 1271, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Krause, K.-H.; Clark, R.A. NOX enzymes as novel targets for drug development. Semin. Immunopathol. 2008, 30, 339–363. [Google Scholar] [CrossRef] [PubMed]
- Jaquet, V.; Scapozza, L.; Clark, R.A.; Krause, K.-H.; Lambeth, J.D. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal. 2009, 11, 2535–2552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-J.; Wei, H.; Frei, B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic. Biol. Med. 2009, 46, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Marriott, H.M.; Jackson, L.E.; Wilkinson, T.S.; Simpson, A.J.; Mitchell, T.J.; Buttle, D.J.; Cross, S.S.; Ince, P.G.; Hellewell, P.G.; Whyte, M.K.B.; et al. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 2008, 177, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Bharathi, P.; Suram, A.; Venugopal, C.; Jagannathan, R.; Poddar, P.; Srinivas, P.; Sambamurti, K.; Rao, K.J.; Scancar, J.; et al. Challenges associated with metal chelation therapy in Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 457–468. [Google Scholar] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 377, 1019–1031. [Google Scholar] [CrossRef]
- Dong, J.; Atwood, C.S.; Anderson, V.E.; Siedlak, S.L.; Smith, M.A.; Perry, G.; Carey, P.R. Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 2003, 42, 2768–2773. [Google Scholar] [CrossRef] [PubMed]
- Finefrock, A.E.; Bush, A.I.; Doraiswamy, P.M. Current status of metals as therapeutic targets in Alzheimer’s disease. J. Am. Geriatr. Soc. 2003, 51, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ashcroft, A.E.; Korchazhkina, O.V.; Exley, C. Metal-mediated formation of fibrillar ABri amyloid. J. Inorg. Biochem. 2004, 98, 2006–2010. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Ricchelli, F.; Drago, D.; Filippi, B.; Tognon, G.; Zatta, P. Aluminum-triggered structural modifications and aggregation of β-amyloids. Cell. Mol. Life Sci. 2005, 62, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 2007, 82, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.-Y.; Kim, Y.; Ryoo, Z.Y.; Kim, T.-Y. Inhibition of the TPA-induced cutaneous inflammation and hyperplasia by EC-SOD. Biochem. Biophys. Res. Commun. 2006, 348, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, B.H.; Lee, H.; Jeon, B.; Lee, Y.S.; Kwon, M.-J.; Kim, T.-Y. Regulation of skin inflammation and angiogenesis by EC-SOD via HIF-1α and NF-κB pathways. Free Radic. Biol. Med. 2011, 51, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J.; Canada, F.J.; Diaz, J.C.; Kroon, P.A.; McLauchlan, R.; Faulds, C.B.; Plumb, G.W.; Morgan, M.R.A.; Williamson, G. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett. 2000, 468, 166–170. [Google Scholar] [CrossRef]
- Nemeth, K.; Plumb, G.W.; Berrin, J.-G.; Juge, N.; Jacob, R.; Naim, H.Y.; Williamson, G.; Swallow, D.M.; Kroon, P.A. Deglycosylation by small intestinal epithelial cell β-glucosidases is a critical step in the absorption and metabolism of dietary flavonoid glycosides in humans. Eur. J. Nutr. 2003, 42, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Rim, E.; Muralidhara, K.S. Mechanism and site of small intestinal absorption of alpha-tocopherol in the rat. Gastroenterology 1975, 68, 1492–1499. [Google Scholar] [PubMed]
- Hollander, D.; Ruble, P.E. Beta-carotene intestinal absorption: Bile, fatty acid, pH, and flow rate effects on transport. Am. J. Physiol. 1978, 235, G686–G691. [Google Scholar]
- During, A.; Hussain, M.M.; Morel, D.W.; Harrison, E.H. Carotenoid uptake and secretion by CaCo-2 cells: β-carotene isomer selectivity and carotenoid interactions. J. Lipid Res. 2002, 43, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- During, A.; Dawson, H.D.; Harrison, E.H. Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is downregulated in Caco-2 cells treated with ezetimibe. J. Nutr. 2005, 135, 2305–2312. [Google Scholar] [PubMed]
- Reboul, E.; Abou, L.; Mikail, C.L.; Ghiringhelli, O.; Andre, M.; Portugal, H.; Jourdheuil-Rahmani, D.; Amiot, M.; Lairon, D.; Borel, P. Lutein transport by Caco-2 TC-7 cells occurs partly by a facilitated process involving the scavenger receptor class B type I (SR-BI). Biochem. J. 2005, 387, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Kwak, D.H.; Choi, I.S.; Park, H.K.; Kang, S.J.; Yoo, H.S.; Lee, M.S.; Oh, K.-W.; Hong, J.T. Inhibitory effect of proanthocyanidin on ultraviolet B irradiation-induced melanogenesis. J. Toxicol. Environ. Health A 2009, 72, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Elmets, C.A.; Katiyar, S.K. Dietary feeding of proanthocyanidins from grape seeds prevents photocarcinogenesis in SKH-1 hairless mice: Relationship to decreased fat and lipid peroxidation. Carcinogenesis 2003, 24, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Vaid, M.; Singh, T.; Katiyar, S.K. Grape seed proanthocyanidins inhibit melanoma cell invasiveness by reduction of PGE2 synthesis and reversal of epithelial-to-mesenchymal transition. PLoS ONE 2011, 6, e21539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Huang, M.-T.; Ho, C.-T.; Chang, R.; Ma, W.; Ferraro, T.; Reuhl, K.R.; Yang, C.S.; Conney, A.H. Inhibitory effect of green tea on the growth of established skin papillomas in mice. Cancer Res. 1992, 52, 6657–6665. [Google Scholar] [PubMed]
- Tong, L.X.; Young, L.C. Nutrition: The future of melanoma prevention? J. Am. Acad. Dermatol. 2014, 71, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Vayalil, P.K.; Mittal, A.; Hara, Y.; Elmets, C.A.; Katiyar, S.K. Green tea polyphenols prevent ultraviolet light-induced oxidative damage and matrix metalloproteinases expression in mouse skin. J. Investig. Dermatol. 2004, 122, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Stahl, W.; Heinrich, U.; Jungmann, H.; Sies, H.; Tronnier, H. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am. J. Clin. Nutr. 2000, 71, 795–798. [Google Scholar] [PubMed]
- Lee, J.; Jiang, S.; Levine, N.; Watson, R.R. Carotenoid supplementation reduces erythema in human skin after simulated solar radiation exposure. Proc. Soc. Exp. Biol. Med. 2000, 223, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, U.; Gartner, C.; Wiebusch, M.; Eichler, O.; Sies, H.; Tronnier, H.; Stahl, W. Supplementation with β-carotene or a similar amount of mixed carotenoids protects humans from UV-induced erythema. J. Nutr. 2003, 133, 98–101. [Google Scholar] [PubMed]
- Eicker, J.R.; Kurten, V.; Wild, S.; Riss, G.; Goralczyk, R.; Krutmann, J.; Berneburg, M. Betacarotene supplementation protects from photoaging-associated mitochondrial DNA mutation. Photochem. Photobiol. Sci. 2003, 2, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Asgari, M.M.; Maruti, S.S.; Kushi, L.H.; White, E. A cohort study of vitamin D intake and melanoma risk. J. Investig. Dermatol. 2009, 129, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, R.; Calvanese, M.; Murino, P.; Manzo, R.; Guida, C.; di Gennaro, D.; Anania, C.; Ravo, V. Skin toxicity from external beam radiation therapy in breast cancer patients: Protective effects of Resveratrol, Lycopene, Vitamin C and anthocianin (Ixor®). Radiat. Oncol. 2012, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- USDA. USDA-NCC Carotenoid Database for U.S. Foods—1998. Available online: http://healthybusiness.co.za/blog/wp-content/uploads/USDA_carotenoids-in-foods.pdf (accessed on 9 August 2013).
- Mathews-Roth, M.M.; Pathak, M.A.; Parrish, J.; Fitzpatrick, T.B.; Kass, E.H.; Toda, K.; Clemens, W. A clinical trial of the effects of oral beta-carotene on the responses of human skin to solar radiation. J. Investig. Dermatol. 1972, 59, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, H.P.M.; Hopfenmller, W.; Hemmes, C.; Chun, S.C.; Schmid, C.; Sundermeier, K.; Biesalski, H.K. Systemic beta carotene plus topical UV-sunscreen are an optimal protection against harmful effects of natural UV-sunlight: Results of the Berlin-Eilath study. Eur. J. Dermatol. 1996, 6, 200–205. [Google Scholar]
- Stahl, W.; Heinrich, U.; Wiseman, S.; Eichler, O.; Sies, H.; Tronnier, H. Dietary tomato paste protects against ultraviolet light-induced erythema in humans. J. Nutr. 2001, 131, 1449–1451. [Google Scholar] [PubMed]
- Wolf, C.; Steiner, A.; Hunigsmann, H. Do oral carotenoids protect human skin against ultraviolet erythema, psoralen phototoxicity, and ultraviolet-induced DNA damage? J. Investig. Dermatol. 1988, 90, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Garmyn, M.; Ribaya-Mercado, J.D.; Russel, R.M.; Bhawan, J.; Gilchrest, B.A. Effect of beta-carotene supplementation on the human sunburn reaction. Exp. Dermatol. 1995, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Placzek, M.; Gaube, S.; Kerkmann, U.; Gilbertz, K.-P.; Herzinger, T.; Haen, E.; Przybilla, B. Ultraviolet B-induced DNA damage in human epidermis is modified by the antioxidants ascorbic acid and d-α-tocopherol. J. Investig. Dermatol. 2005, 124, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Eberlein-Konig, B.; Ring, J. Relevance of vitamins C and E in cutaneous photoprotection. J. Cosmet. Dermatol. 2005, 4, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Makrantonaki, E.; Zouboulis, C.C. Skin alterations and diseases in advanced age. Drug Discov. Today Dis. Mech. 2008, 5, e153–e162. [Google Scholar] [CrossRef]
- Elmets, C.A.; Singh, D.; Tubesing, K.; Matsui, M.; Katiyar, S.; Mukhtar, H. Cutaneous photoprotection from ultraviolet injury by green tea polyphenols. J. Am. Acad. Dermatol. 2001, 44, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Bickers, D.R.; Athar, M. Novel approaches to chemoprevention of skin cancer. J. Dermatol. 2000, 27, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K. Skin photoprotection by green tea: Antioxidant and immunomodulatory effects. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2003, 3, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Matsui, M.S.; Elmets, C.A.; Mukhtar, H. Polyphenolic antioxidant (−)-epigallocatechin-3-gallate from green tea reduces UVB-induced inflammatory responses and infiltration of leukocytes in human skin. Photochem. Photobiol. 1999, 69, 148–153. [Google Scholar] [CrossRef]
- Mittal, A.; Piyathilake, C.; Hara, Y.; Katiyar, S.K. Exceptionally high protection of photocarcinogenesis by topical application of (−)-epigallocatechin-3-gallate in hydrophilic cream in SKH-1 Hairless Mouse Model: Relationship to inhibition of UVB-induced global DNA hypomethylation. Neoplasia 2003, 5, 555–565. [Google Scholar] [CrossRef]
- Gensler, H.L.; Timmermann, B.N.; Valcic, S.; Wachter, G.A.; Dorr, R.; Dvorakova, K.; Alberts, D.S. Prevention of photocarcinogenesis by topical administration of pure epigallocatechin gallate isolated from green tea. 1996, 26, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-Y.; Liu, W.; Hao, J.-C.; Gu, W.-J.; Zhao, Y.-S. Topical grape seed proanthocyandin extract reduces sunburn cells and mutant p53 positive epidermal cell formation, and prevents depletion of Langerhans cells in an acute sunburn model. Photomed. Laser Surg. 2012, 30, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Oyewole, A.O.; Wilmot, M.-C.; Fowler, M.; Birch-Machin, M.A. Comparing the effects of mitochondrial targeted and localized antioxidants with cellular antioxidants in human skin cells exposed to UVA and hydrogen peroxide. FASEB J. 2014, 28, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2014, 29, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Spearman, D.; Perone, P.; Fligiel, S.E.G.; Datta, S.C.; Wang, Z.Q.; Shao, Y.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Inhibition of type I procollagen synthesis by damaged collagen in photoaged skin and by collagenase-degraded collagen in vitro. Am. J. Pathol. 2001, 158, 931–942. [Google Scholar] [CrossRef]
- Humbert, P.G.; Haftek, M.; Creidi, P.; Lapiere, C.; Nusgens, B.; Richard, A.; Schmitt, D.; Rougier, A.; Zahouani, H. Topical ascorbic acid on photoaged skin. Clinical, topographical and ultrastructural evaluation: Double-blind study vs. placebo. Exp. Dermatol. 2003, 12, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Maroni, P.; Özer, N.; Zingg, J.-M.; Azzi, A. Age-dependent increase of collagenase expression can be reduced by α-tocopherol via protein kinase C inhibition. Free Radic. Biol. Med. 1999, 27, 729–737. [Google Scholar] [CrossRef]
- Chen, W.; Barthelman, M.; Martinez, J.; Alberts, D.; Gensler, H.L. Inhibition of cyclobutane pyrimidine dimer formation in epidermal p53 gene of UV-irradiated mice by alpha-tocopherol. Nutr. Cancer 1997, 29, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Zussman, J.; Ahdout, J.; Kim, J. Vitamins and photoaging: Do scientific data support their use? J. Am. Acad. Dermatol. 2010, 63, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.C.; Burch, J.A.; Streilein, R.D.; Iannacchione, M.A.; Hall, R.P.; Pinnell, S.R. A topical antioxidant solution containing vitamins C and E stabilized by ferulic acid provides protection for human skin against damage caused by ultraviolet irradiation. J. Am. Acad. Dermatol. 2008, 59, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Barnes, P.J.; Roberts, L.J. Isoprostanes: Markers and mediators of oxidative stress. FASEB J. 2004, 18, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G. Does vitamin E decrease heart attack risk? Summary and implications with respect to dietary recommendations. J. Nutr. 2001, 131, 395S–397S. [Google Scholar] [PubMed]
- Morrow, J.D. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.J.; Oates, J.A.; Linton, M.F.; Fazio, S.; Meador, B.P.; Gross, M.D.; Shyr, Y.; Morrow, J.D. The relationship between dose of vitamin E and suppression of oxidative stress in humans. Free Radic. Biol. Med. 2007, 43, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Caballero, B.; Chang, S.; Alberg, A.J.; Semba, R.D.; Schneyer, C.R.; Wilson, R.F.; Cheng, T.Y.; Vassy, J.; Prokopowicz, G.; et al. The efficacy and safety of multivitamin and mineral supplement use to prevent cancer and chronic Disease in adults: A systematic review for a national institutes of health state-of-the-science conference. Ann. Intern. Med. 2006, 145, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Gey, K.F. Prospects for the prevention of free radical disease, regarding cancer and cardiovascular disease. Br. Med. Bull. 1993, 49, 679–699. [Google Scholar] [PubMed]
- Wright, M.E.; Lawson, K.A.; Weinstein, S.J.; Pietinen, P.; Taylor, P.R.; Virtamo, J.; Albanes, D. Higher baseline serum concentrations of vitamin E are associated with lower total and cause-specific mortality in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Am. J. Clin. Nutr. 2006, 84, 1200–1207. [Google Scholar] [PubMed]
- Marchioli, R.; Levantesi, G.; Macchia, A.; Marfisi, R.M.; Nicolosi, G.L.; Tavazzi, L.; Tognoni, G.; Valagussa, F.; Investigators, G.I.P. Vitamin E increases the risk of developing heart failure after myocardial infarction: Results from the GISSI-Prevenzione trial. J. Cardiovasc. Med. 2006, 7, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Frei, B.; Beckman, J.S. Vitamin E revisited: Do new data validate benefits for chronic disease prevention? Curr. Opin. Lipidol. 2008, 19, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, 3. [Google Scholar] [CrossRef]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z. Window Period for Oxidative Stress Attenuating Intervention (WPOS Theory). Am. J. Biomed. Sci. 2009, 1, 250–259. [Google Scholar] [CrossRef]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346, f10. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Terentis, A.C.; Thomas, S.R.; Burr, J.A.; Liebler, D.C.; Stocker, R. Vitamin E oxidation in human atherosclerotic lesions. Circ. Res. 2002, 90, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P. Chemoprotection against cancer by induction of phase 2 enzymes. Biofactors 2000, 12, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Birringer, M. Hormetics: Dietary triggers of an adaptive stress response. Pharm. Res. 2011, 28, 2680–2694. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Okamoto, S.I.; Cui, J.; Watanabe, Y.; Furuta, K.; Suzuki, M.; Tohyama, K.; Lipton, S.A. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophillic phase II inducers. Proc. Natl. Acad. Sci. USA 2006, 103, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Lipton, S.A. Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 2007, 30, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kosaka, K.; Itoh, K.; Kobayashi, A.; Yamamoto, M.; Shimojo, Y.; Kitajima, C.; Cui, J.; Kamins, J.; Okamoto, S.I.; et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J. Neurochem. 2008, 104, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Rezaie, T.; Seki, M.; Sunico, C.R.; Tabuchi, T.; Kitagawa, T.; Yanagitai, M.; Senzaki, M.; Kosegawa, C.; Taira, H.; et al. Dual neuroprotective pathways of a pro-electrophilic compound via HSF-1-activated heat-shock proteins and Nrf2-activated phase 2 antioxidant response enzymes. J. Neurochem. 2011, 119, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Akhtar, M.W.; Lipton, S.A. Combating oxidative/nitrosative stress with electrophilic counterattack strategies. In Oxidative Stress and Redox Regulation; Springer: Berlin, Germany, 2013; pp. 277–307. [Google Scholar]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Patel, R.P.; Brookes, P.S.; Darley-Usmar, V.M. Nanotransducers in cellular redox signaling: Modification of thiols by reactive oxygen and nitrogen species. Trends Biochem. Sci. 2002, 27, 489–492. [Google Scholar] [CrossRef]
- Higdon, A.N.; Landar, A.; Barnes, S.; Darley-Usmar, V.M. The electrophile responsive proteome: Integrating proteomics and lipidomics with cellular function. Antioxid. Redox Signal. 2012, 17, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, K.; Perez-Sala, D. Prostanoids with cyclopentenone structure as tools for the characterization of electrophilic lipid-protein interactomes. Ann. NY Acad. Sci. 2006, 1091, 548–570. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap‘n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar] [PubMed]
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; auf dem Keller, U.; Born-Berclaz, C.; Chan, K.; Kan, Y.W.; Werner, S. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol. Cell. Biol. 2002, 22, 5492–5505. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Auf dem Keller, U.; Braun, S.; Schafer, M.; Werner, S. Roles and mechanisms of action of the Nrf2 transcription factor in skin morphogenesis, wound repair and skin cancer. Cell Death Differ. 2007, 14, 1250–1254. [Google Scholar] [CrossRef] [PubMed]
- Huebner, A.J.; Dai, D.; Morasso, M.; Schmidt, E.E.; Schafer, M.; Werner, S.; Roop, D.R. Amniotic fluid activates the Nrf2/keap1 pathway to repair an epidermal barrier defect in utero. Dev. Cell 2012, 23, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Hirota, A.; Kawachi, Y.; Yamamoto, M.; Koga, T.; Hamada, K.; Otsuka, F. Acceleration of UVB-induced photoageing in Nrf2 gene-deficient mice. Exp. Dermatol. 2011, 20, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Jaiswal, A.K. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H: Quinone oxidoreductasel gene. Oncogene 2001, 20, 3906–3917. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [PubMed]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Maher, J.; Yamamoto, M. Select heterozygous Keap1 mutations have a dominant-negative effect on wild-type Keap1 in vivo. Cancer Res. 2011, 71, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Yan, X.X.; Freeman, M.L. Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene 2002, 21, 6829–6834. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Ma, Q. Critical cysteine residues of Kelch-like ECH-associated protein 1 in arsenic sensing and suppression of nuclear factor erythroid 2-related factor 2. J. Pharmacol. Exp. Ther. 2010, 332, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Yang, C.S.; Pickett, C.B. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic. Biol. Med. 2004, 37, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, V.O.; Menshchikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Moscow) 2011, 76, 407–422. [Google Scholar] [CrossRef]
- Giudice, A.; Arra, C.; Turco, M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. In Transcription Factors; Springer: Berlin, Germany, 2010; pp. 37–74. [Google Scholar]
- Hur, W.; Gray, N.S. Small molecule modulators of antioxidant response pathway. Curr. Opin. Chem. Biol. 2011, 15, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap-n-collar transcription factors in aging and human disease. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS ONE 2009, 4, e6588. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.G.; Jain, A.D.; Speltz, T.E.; Moore, T.W. Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorg. Med. Chem. Lett. 2015, 25, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Llores, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kivela, A.M.; Levonen, A.L. Regulation of Nrf2-dependent gene expression by 15-deoxy-Δ12,14-prostaglandin J2. Free Radic. Biol. Med. 2009, 47, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.; Landar, A.; Ramachandran, A.; Ceaser, E.; Dickinson, D.; Zanoni, G.; Morrow, J.; Darley-Usmar, V. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; LaHair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Owuor, E.D.; Kong, A.N.T. Antioxidants and oxidants regulated signal transduction pathways. Biochem. Pharmacol. 2002, 64, 765–770. [Google Scholar] [CrossRef]
- Numazawa, S.; Ishikawa, M.; Yoshida, A.; Tanaka, S.; Yoshida, T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am. J. Physiol. Cell Physiol. 2003, 285, C334–C342. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H: Quinone oxidoreductase-1 gene expression. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar] [PubMed]
- Guo, Y.; Yu, S.; Zhang, C.; Kong, A.N.T. Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Prestera, T.; Holtzclaw, W.D.; Zhang, Y.; Talalay, P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc. Natl. Acad. Sci. 1993, 90, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.S.; Jun, M.; Kong, A.N.T. Nrf2: A potential molecular target for cancer chemoprevention by natural compounds. Antioxid. Redox Signal. 2006, 8, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N. Nrf2: Friend or foe for chemoprevention? Carcinogenesis 2010, 31, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Ben Yehuda Greenwald, M.; Anzi, S.; Ben Sasson, S.; Bianco-Peled, H.; Kohen, R. Can nitroxides evoke the Keap1-Nrf2-ARE pathway in skin? Free Radic. Biol. Med. 2014, 77, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.S.; Matos, M.F.; Li, B.; Hronowski, X.; Gao, B.; Juhasz, P.; Rhodes, K.J.; Scannevin, R.H. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS ONE 2015, 10, e0120254. [Google Scholar]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Farwanah, H.; Willrodt, A.H.; Huebner, A.J.; Sandhoff, K.; Roop, D.; Hohl, D.; Bloch, W.; Werner, S. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol. Med. 2012, 4, 364–379. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharm. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Lieder, F.; Reisen, F.; Geppert, T.; Sollberger, G.; Beer, H.D.; auf dem Keller, U.; Schafer, M.; Detmar, M.; Schneider, G.; Werner, S. Identification of UV-protective activators of nuclear factor erythroid-derived 2-related factor 2 (Nrf2) by combining a chemical library screen with computer-based virtual screening. J. Biol. Chem. 2012, 287, 33001–33013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, B.W. Novel mechanisms and devices to enable successful transdermal drug delivery. Eur. J. Pharm. Sci. 2001, 14, 101–114. [Google Scholar] [CrossRef]
- Raza, K.; Kumar, M.; Kumar, P.; Malik, R.; Sharma, G.; Kaur, M.; Katare, O.P. Topical delivery of aceclofenac: Challenges and promises of novel drug delivery systems. BioMed Res. Int. 2014, 2014, 406731. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shin, J.M.; Jang, S.; Choi, D.K.; Seo, M.S.; Kim, H.R.; Sohn, K.C.; Im, M.; Seo, Y.J.; Lee, J.H.; et al. Role of nuclear factor E2-related factor 2 (Nrf2) in epidermal differentiation. Arch. Dermatol. Res. 2014, 306, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Dutsch, S.; auf dem Keller, U.; Werner, S. Nrf2: A central regulator of UV protection in the epidermis. Cell Cycle 2010, 9, 2917–2918. [Google Scholar] [CrossRef] [PubMed]
- Gašperlin, M.; Gosenca, M. Main approaches for delivering antioxidant vitamins through the skin to prevent skin ageing. Expert Opin. Drug Deliv. 2011, 8, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.C. Conventional topical delivery systems. Dermatol. Ther. 2011, 24, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Ben Yehuda Greenwald, M.; Frusic-Zlotkin, M.; Soroka, Y.; Sasson, S.B.; Bianco-Peled, H.; Bitton, R.; Kohen, R. Nitroxide delivery system for Nrf2 activation and skin protection. Eur. J. Pharm. Biopharm. 2015, 94, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Csiszar, A.; Pinto, J.T.; Gautam, T.; Kleusch, C.; Hoffmann, B.; Tucsek, Z.; Toth, P.; Sonntag, W.E.; Ungvari, Z. Resveratrol encapsulated in novel fusogenic liposomes activates Nrf2 and attenuates oxidative stress in cerebromicrovascular endothelial cells from aged rats. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 70, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sabzichi, M.; Hamishehkar, H.; Ramezani, F.; Sharifi, S.; Tabasinezhad, M.; Pirouzpanah, M.; Ghanbari, P.; Samadi, N. Luteolin-loaded phytosomes sensitize human breast carcinoma MDA-MB 231 cells to doxorubicin by suppressing Nrf2 mediated signalling. Asian Pac. J. Cancer Prev. 2014, 15, 5311–5316. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben-Yehuda Greenwald, M.; Ben-Sasson, S.; Bianco-Peled, H.; Kohen, R. Skin Redox Balance Maintenance: The Need for an Nrf2-Activator Delivery System. Cosmetics 2016, 3, 1. https://doi.org/10.3390/cosmetics3010001
Ben-Yehuda Greenwald M, Ben-Sasson S, Bianco-Peled H, Kohen R. Skin Redox Balance Maintenance: The Need for an Nrf2-Activator Delivery System. Cosmetics. 2016; 3(1):1. https://doi.org/10.3390/cosmetics3010001
Chicago/Turabian StyleBen-Yehuda Greenwald, Maya, Shmuel Ben-Sasson, Havazelet Bianco-Peled, and Ron Kohen. 2016. "Skin Redox Balance Maintenance: The Need for an Nrf2-Activator Delivery System" Cosmetics 3, no. 1: 1. https://doi.org/10.3390/cosmetics3010001