
Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis

Abstract
:1. Introduction
2. Clinical Presentation of Inherited Glutamine Synthetase Deficiency
3. Biochemical Presentation
3.1. Physiology and Pathophysiology
3.2. Relation of Glutamine and Hyperammonemia
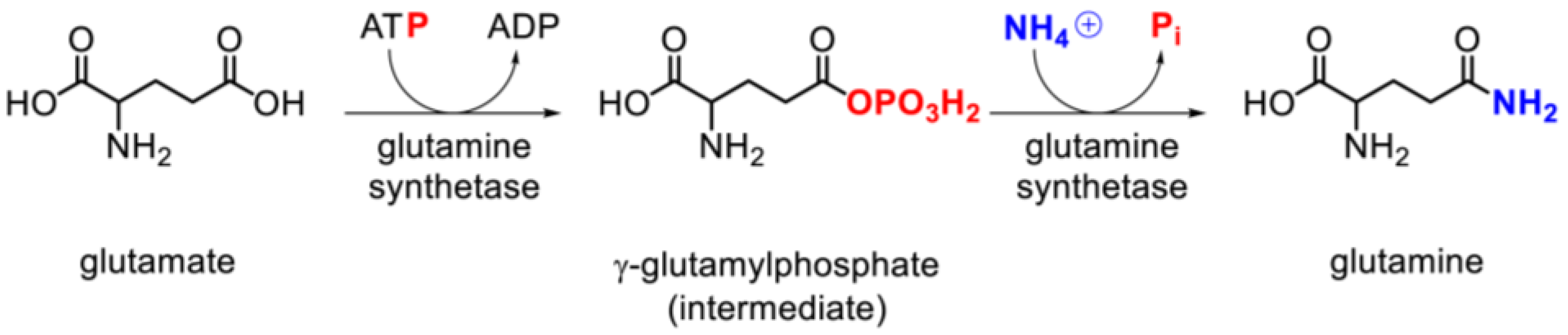
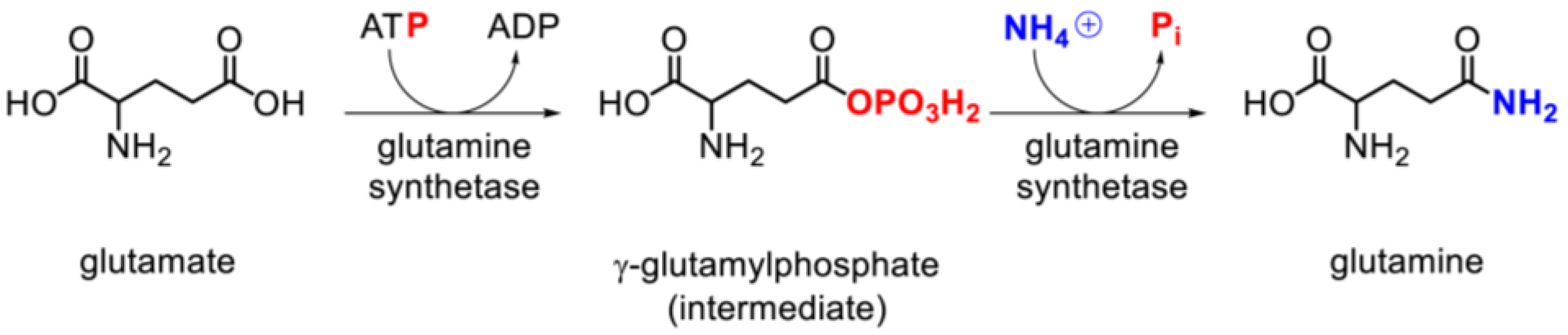
3.3. Glutamine Synthetase Activity and Expression
4. Molecular Genetics of Glutamine Synthetase Deficiency
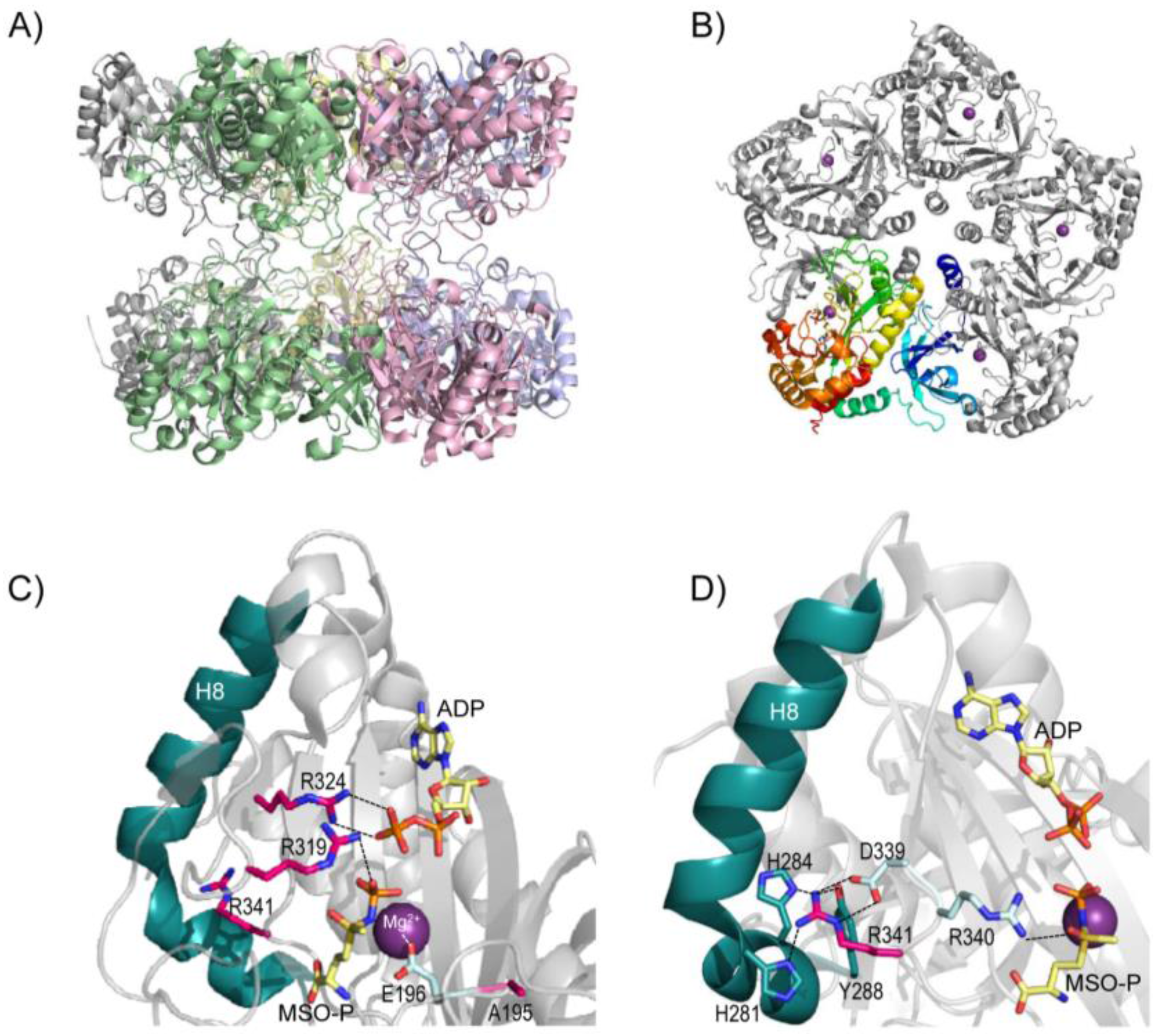
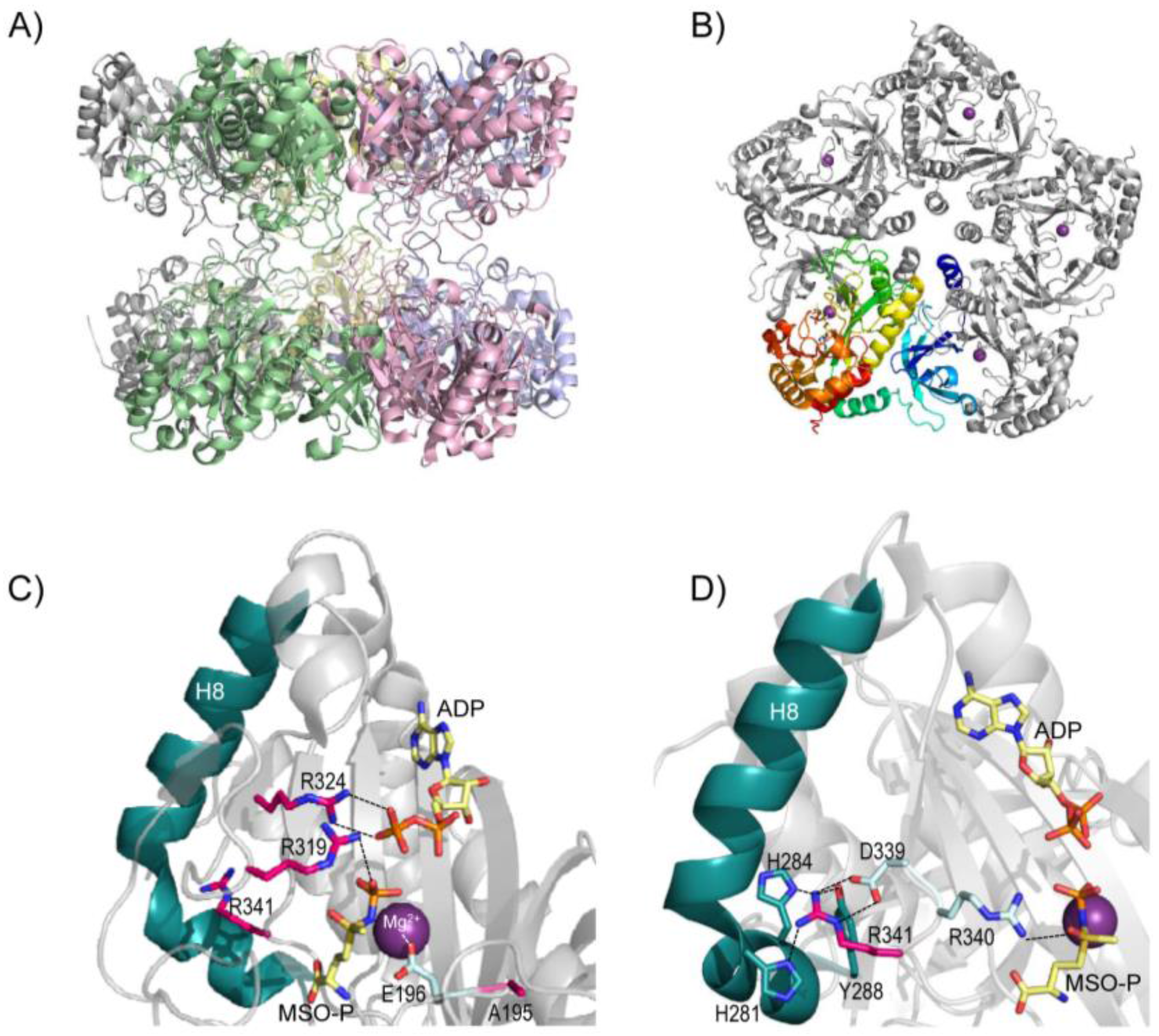
5. Structure of Glutamine Synthetase and Rationalization of the Clinical Mutations
6. Pathophysiology of Glutamine Synthetase Deficiency
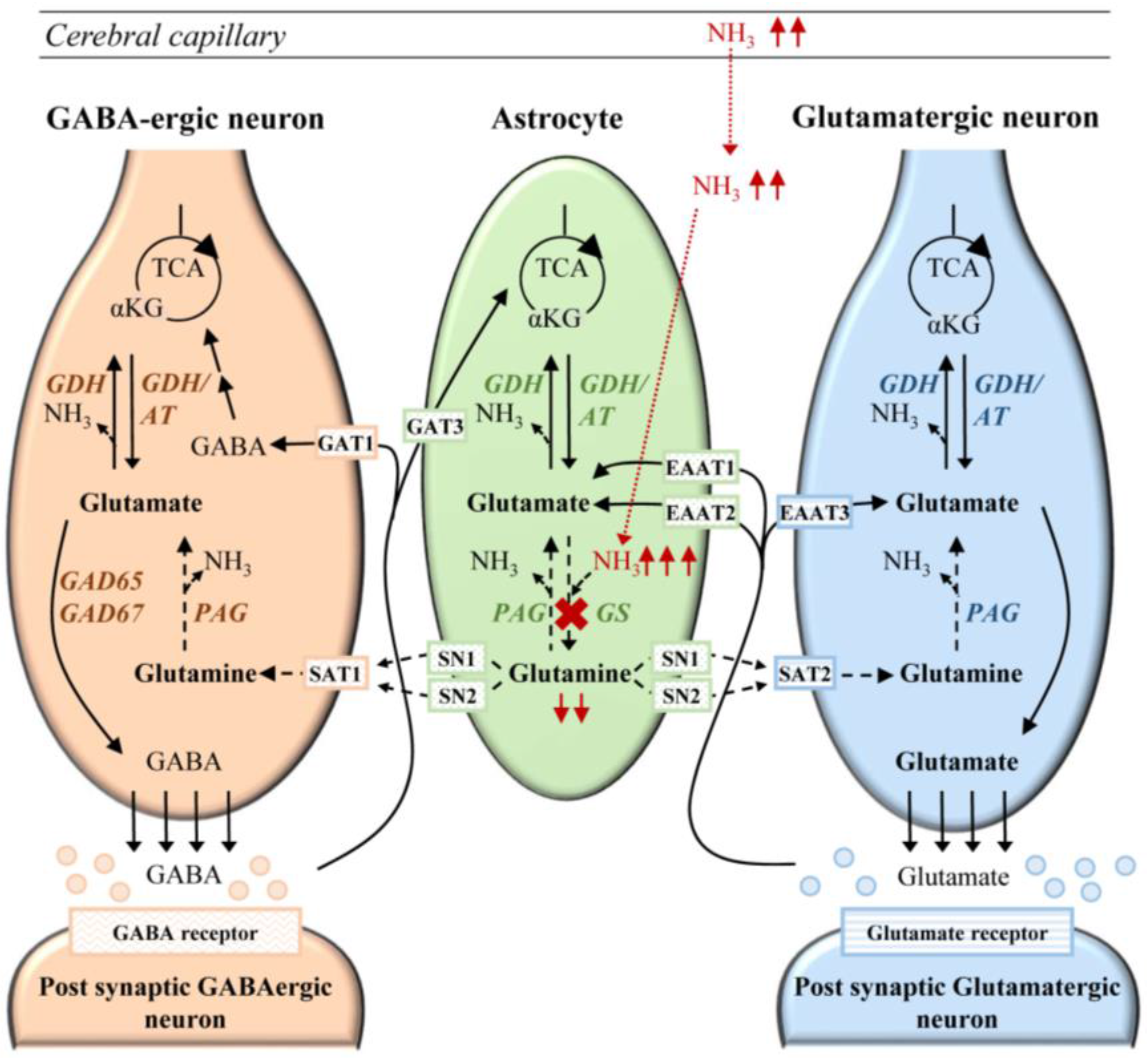
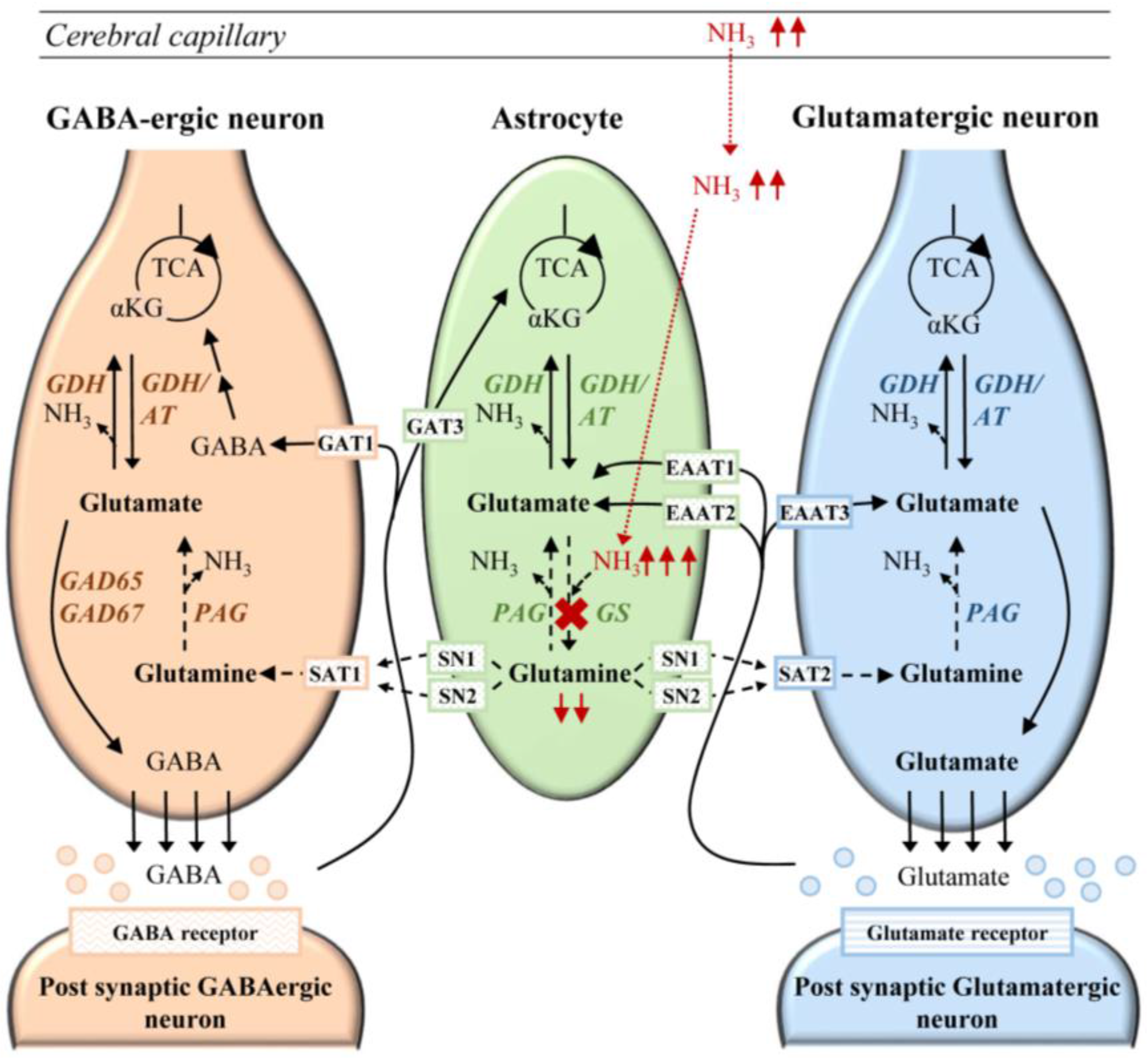
6.1. Cerebral Ammonia Toxicity in GS Deficiency
6.2. Neurotransmission in Glutamine Synthetase Deficiency
6.3. Fetal Development in Glutamine Synthetase Deficiency
6.4. NAD+ Depletion in Glutamine Synthetase Deficiency
7. Secondary Glutamine Synthetase Deficiency
8. Management of Patients with Inherited Glutamine Synthetase Deficiency
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Caggese, C.; Caizzi, R.; Barsanti, P.; Bozzetti, M.P. Mutations in the glutamine synthetase I (GSI) gene produce embryo-lethal female sterility in drosophila melanogaster. Dev. Genet. 1992, 13, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Görg, B.; Rutsch, F.; Schmidt, E.; Toutain, A.; Benoist, J.F.; Gelot, A.; Suc, A.L.; Hohne, W.; Schliess, F.; et al. Congenital glutamine deficiency with glutamine synthetase mutations. N. Engl. J. Med. 2005, 353, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Görg, B.; Toutain, A.; Rutsch, F.; Benoist, J.F.; Gelot, A.; Suc, A.L.; Koch, H.G.; Schliess, F.; Häussinger, D. Inborn error of amino acid synthesis: Human glutamine synthetase deficiency. J. Inherit. Metab. Dis. 2006, 29, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Shahbeck, N.; Ibrahim, K.; Hoffmann, G.F.; Ben-Omran, T. Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol. Genet. Metab. 2011, 103, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Shahbeck, N.; Ibrahim, K.; Schmitt, B.; Scheer, I.; O’Gorman, R.; Chaudhry, F.A.; Ben-Omran, T. Glutamine supplementation in a child with inherited GS deficiency improves the clinical status and partially corrects the peripheral and central amino acid imbalance. Orphanet J. Rare Dis. 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.J.; Jeitner, T.M. Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules 2016. [Google Scholar] [CrossRef] [PubMed]
- Solbu, T.T.; Boulland, J.L.; Zahid, W.; Lyamouri Bredahl, M.K.; Amiry-Moghaddam, M.; Storm-Mathisen, J.; Roberg, B.A.; Chaudhry, F.A. Induction and targeting of the glutamine transporter SN1 to the basolateral membranes of cortical kidney tubule cells during chronic metabolic acidosis suggest a role in ph regulation. J. Am. Soc. Nephrol. 2005, 16, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Soeters, P.B.; Grecu, I. Have we enough glutamine and how does it work? A clinician’s view. Ann. Nutr. Metab. 2012, 60, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D.; Schliess, F. Glutamine metabolism and signaling in the liver. Front. Biosci. 2007, 12, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Curi, R.; Lagranha, C.J.; Doi, S.Q.; Sellitti, D.F.; Procopio, J.; Pithon-Curi, T.C.; Corless, M.; Newsholme, P. Molecular mechanisms of glutamine action. J. Cell. Physiol. 2005, 204, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Sidoryk-Wegrzynowicz, M.; Zielinska, M.; Aschner, M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010, 6, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P. Why is l-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J. Nutr. 2001, 131, 2515S–2522S. [Google Scholar] [PubMed]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Watford, M. Glutamine metabolism and function in relation to proline synthesis and the safety of glutamine and proline supplementation. J. Nutr. 2008, 138, 2003S–2007S. [Google Scholar] [PubMed]
- Cooper, A.J. The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem. Res. 2012, 37, 2439–2455. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J. The role of glutamine transaminase K (GTK) in sulfur and alpha-keto acid metabolism in the brain, and in the possible bioactivation of neurotoxicants. Neurochem. Int. 2004, 44, 557–577. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Gill, H.S.; Pfluegl, G.M.; Rotstein, S.H. Structure-function relationships of glutamine synthetases. Biochim. Biophys. Acta 2000, 1477, 122–145. [Google Scholar] [CrossRef]
- Windmueller, H.G. Metabolism of vascular and luminal glutamine by intestinal mucosa in vivo. In Glutamine Metabolism in Mamalian Tissues; Häussinger, D., Sies, H., Eds.; Springer: Berlin, Germany, 1884; pp. 61–77. [Google Scholar]
- Watford, M.; Reeds, P.J. Glutamate metabolism in the gut. Forum Nutr. 2003, 56, 81–82. [Google Scholar] [PubMed]
- Gianotti, L.; Oldani, M.; Coppola, S.; Nespoli, L.; Zanello, M.; Braga, M. Glutamine supplementation in major surgery and intensive care. In Glutamine in Clinical Nutrition; Rajendram, R., Preedy, V.R., Patel, V.B., Eds.; Springer: New York, NY, USA, 2015; pp. 153–168. [Google Scholar]
- Ekmark, L.; Rooyackers, O.; Wernerman, J.; Flaring, U. Plasma glutamine deficiency is associated with multiple organ failure in critically ill children. Amino Acids 2015, 47, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D. Hepatocyte heterogeneity in glutamine and ammonia metabolism and the role of an intercellular glutamine cycle during ureogenesis in perfused rat liver. Eur. J. Biochem. FEBS 1983, 133, 269–275. [Google Scholar] [CrossRef]
- Häussinger, D. Liver glutamine metabolism. JPEN 1990, 14, 56s–62s. [Google Scholar] [CrossRef]
- Gaunitz, F.; Deichsel, D.; Heise, K.; Werth, M.; Anderegg, U.; Gebhardt, R. An intronic silencer element is responsible for specific zonal expression of glutamine synthetase in the rat liver. Hepatology 2005, 41, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; McDonald, J.M.; Gelbard, A.S.; Gledhill, R.F.; Duffy, T.E. The metabolic fate of 13N-labeled ammonia in rat brain. J. Biol. Chem. 1979, 254, 4982–4992. [Google Scholar] [PubMed]
- Häussinger, D.; Gerok, W. Hepatocyte heterogeneity in glutamate uptake by isolated perfused rat liver. Eur. J. Biochem. FEBS 1983, 136, 421–425. [Google Scholar] [CrossRef]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Qvartskhava, N.; Lang, P.A.; Görg, B.; Pozdeev, V.I.; Ortiz, M.P.; Lang, K.S.; Bidmon, H.J.; Lang, E.; Leibrock, C.B.; Herebian, D.; et al. Hyperammonemia in gene-targeted mice lacking functional hepatic glutamine synthetase. Proc. Natl. Acad. Sci. USA 2015, 112, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D. Nitrogen metabolism in liver: Structural and functional organization and physiological relevance. Biochem. J. 1990, 267, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Ibrahim, K.; Stucki, M.; Frapolli, M.; Shahbeck, N.; Chaudhry, F.A.; Görg, B.; Häussinger, D.; Penberthy, W.T.; Ben-Omran, T.; et al. Secondary NAD+ deficiency in the inherited defect of glutamine synthetase. J. Inherit. Metab. Dis. 2015, 38, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, F.C. Glutamine and glutamate exchange between the fetal liver and the placenta. J. Nutr. 2000, 130, 974s–977s. [Google Scholar] [PubMed]
- Day, P.E.; Cleal, J.K.; Lofthouse, E.M.; Goss, V.; Koster, G.; Postle, A.; Jackson, J.M.; Hanson, M.A.; Jackson, A.A.; Lewis, R.M. Partitioning of glutamine synthesised by the isolated perfused human placenta between the maternal and fetal circulations. Placenta 2013, 34, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, T.; Görg, B.; Vogl, T.; Wolf, M.; Varga, G.; Toutain, A.; Paul, R.; Schliess, F.; Häussinger, D.; Häberle, J. Glutamine synthetase is essential for proliferation of fetal skin fibroblasts. Arch. Biochem. Biophys. 2008, 478, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Schliess, F.; Görg, B.; Fischer, R.; Desjardins, P.; Bidmon, H.J.; Herrmann, A.; Butterworth, R.F.; Zilles, K.; Häussinger, D. Ammonia induces MK-801-sensitive nitration and phosphorylation of protein tyrosine residues in rat astrocytes. FASEB J. 2002, 16, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Görg, B.; Qvartskhava, N.; Voss, P.; Grune, T.; Häussinger, D.; Schliess, F. Reversible inhibition of mammalian glutamine synthetase by tyrosine nitration. FEBS Lett. 2007, 581, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; McLin, V.A.; Cudalbu, C. Ammonia toxicity to the brain. J. Inherit. Metab. Dis. 2013, 36, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Görg, B.; Wettstein, M.; Metzger, S.; Schliess, F.; Häussinger, D. Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology 2005, 41, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Albrecht, J. Oxidative and nitrosative stress in ammonia neurotoxicity. Neurochem. Int. 2013, 62, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Görg, B.; Qvartskhava, N.; Keitel, V.; Bidmon, H.J.; Selbach, O.; Schliess, F.; Häussinger, D. Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology 2008, 48, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.; Llansola, M.; Montoliu, C.; Monfort, P.; Rodrigo, R.; Hernandez-Viadel, M.; Erceg, S.; Sanchez-Perez, A.M.; Felipo, V. Glutamine synthetase activity and glutamine content in brain: Modulation by NMDA receptors and nitric oxide. Neurochem. Int. 2003, 43, 493–499. [Google Scholar] [CrossRef]
- Rodrigo, R.; Felipo, V. Control of brain glutamine synthesis by NMDA receptors. Front. Biosci. 2007, 12, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Lee, J.E.; Sweredoski, M.J.; Yang, S.J.; Jeon, S.J.; Harrison, J.S.; Yim, J.H.; Lee, S.G.; Handa, H.; Kuhlman, B.; et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell 2016, 61, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Lan, B.Y.; Bedolli, M.; Feng, S.; Hebrok, M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 2006, 43, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Suarez, I.; Bodega, G.; Fernandez, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Hansson, E. Regulation of glutamine synthetase synthesis and activity by glucocorticoids and adrenoceptor activation in astroglial cells. Neurochem. Res. 1989, 14, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Abcouwer, S.F.; Bode, B.P.; Souba, W.W. Glucocorticoids regulate rat glutamine synthetase expression in a tissue-specific manner. J. Surg. Res. 1995, 59, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Watford, M. Glutamine, insulin and glucocorticoids regulate glutamine synthetase expression in C2C12 myotubes, Hep G2 hepatoma cells and 3T3 L1 adipocytes. Biochim. Biophys. Acta 2007, 1770, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Hunt, A.; Tahourdin, C.S. Regulation of in vivo glutamine synthetase activity by glucocorticoids in the developing rat brain. Brain Res. 1983, 312, 83–91. [Google Scholar] [CrossRef]
- Nissim, I. Newer aspects of glutamine/glutamate metabolism: The role of acute pH changes. Am. J. Physiol. 1999, 277, F493–F497. [Google Scholar] [PubMed]
- Saitoh, F.; Araki, T. Proteasomal degradation of glutamine synthetase regulates Schwann cell differentiation. J. Neurosci. 2010, 30, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Rosier, F.; Lambert, D.; Mertens-Strijthagen, M. Effect of glucose deprivation on rat glutamine synthetase in cultured astrocytes. Biochem. J. 1996, 315, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K.; Chung, M.C.; Verkhovsky, L.M.; Cooper, A.J. Brain glutamine synthetase increases following cerebral ischemia in the rat. Brain Res. 1992, 569, 275–280. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Battaile, K.; Cooper, A.J. Critical evaluation of the changes in glutamine synthetase activity in models of cerebral stroke. Neurochem. Res. 2015, 40, 2544–2556. [Google Scholar] [CrossRef] [PubMed]
- Labow, B.I.; Souba, W.W.; Abcouwer, S.F. Mechanisms governing the expression of the enzymes of glutamine metabolism—Glutaminase and glutamine synthetase. J. Nutr. 2001, 131, 2467S–2474S. [Google Scholar] [PubMed]
- Kumada, Y.; Benson, D.R.; Hillemann, D.; Hosted, T.J.; Rochefort, D.A.; Thompson, C.J.; Wohlleben, W.; Tateno, Y. Evolution of the glutamine synthetase gene, one of the oldest existing and functioning genes. Proc. Natl. Acad. Sci. USA 1993, 90, 3009–3013. [Google Scholar] [CrossRef] [PubMed]
- Helou, K.; Das, A.T.; Lamers, W.H.; Hoovers, J.M.; Szpirer, C.; Szpirer, J.; Klinga-Levan, K.; Levan, G. FISH mapping of three ammonia metabolism genes (Glul, Cps1, Glud1) in rat, and the chromosomal localization of Glul in human and Cps1 in mouse. Mamm. Genome 1997, 8, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kudoh, J.; Kubota, R.; Asakawa, S.; Minoshima, S.; Shimizu, N. Chromosomal mapping of a family of human glutamine synthetase genes: Functional gene (GLUL) on 1q25, pseudogene (GLULP) on 9p13, and three related genes (GLULL1, GLULL2, GLULL3) on 5q33, 11p15, and 11q24. Genomics 1996, 37, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, W.W.; Collins, R.; Holmberg-Schiavone, L.; Jones, T.A.; Karlberg, T.; Mowbray, S.L. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 2008, 375, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Denman, R.B.; Wedler, F.C. Association-dissociation of mammalian brain glutamine synthetase: Effects of metal ions and other ligands. Arch. Biochem. Biophys. 1984, 232, 427–440. [Google Scholar] [CrossRef]
- Frieg, B.; Görg, B.; Homeyer, N.; Keitel, V.; Häussinger, D.; Gohlke, H. Molecular mechanisms of glutamine synthetase mutations that lead to clinically relevant pathologies. PLoS Comp. Biol. 2016, 12, e1004693. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J. Glutamine supplementation in glutamine synthetase deficiency. In Glutamine in Clinical Nutrition; Rajendram, R., Preedy, V.R., Patel, V.B., Eds.; Springer: New York, NY, USA, 2015; pp. 427–436. [Google Scholar]
- Martinez-Hernandez, A.; Bell, K.P.; Norenberg, M.D. Glutamine synthetase: Glial localization in brain. Science 1977, 195, 1356–1358. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- Yamamoto, H.; Konno, H.; Yamamoto, T.; Ito, K.; Mizugaki, M.; Iwasaki, Y. Glutamine synthetase of the human brain: Purification and characterization. J. Neurochem. 1987, 49, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Riepe, R.E.; Norenburg, M.D. Müller cell localisation of glutamine synthetase in rat retina. Nature 1977, 268, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Bannier, J.; Meyer-Lotz, G.; Steiner, J.; Keilhoff, G.; Dobrowolny, H.; Walter, M.; Bogerts, B. Distribution of immunoreactive glutamine synthetase in the adult human and mouse brain. Qualitative and quantitative observations with special emphasis on extra-astroglial protein localization. J. Chem. Neuroanat. 2014, 61–62, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J. Quantitative analysis of neurotransmitter pathways under steady state conditions—A perspective. Front. Endocrinol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The glutamine-glutamate/GABA cycle: Function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.F.; Verkhratsky, A.; Parpura, V. Astrocyte glutamine synthetase: Pivotal in health and disease. Biochem. Soc. Trans. 2013, 41, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. A tribute to Mary C. Mckenna: Glutamate as energy substrate and neurotransmitter-functional interaction between neurons and astrocytes. Neurochem. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O. Ammonia toxicity to the brain: Effects on creatine metabolism and transport and protective roles of creatine. Mol. Genet. Metab. 2010, 100, S53–S58. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Song, D.; Peng, L.; Chen, Y. Multifactorial effects on different types of brain cells contribute to ammonia toxicity. Neurochem. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rangroo Thrane, V.; Thrane, A.S.; Wang, F.; Cotrina, M.L.; Smith, N.A.; Chen, M.; Xu, Q.; Kang, N.; Fujita, T.; Nagelhus, E.A.; et al. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 2013, 19, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D.; Schliess, F. Pathogenetic mechanisms of hepatic encephalopathy. Gut 2008, 57, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Zieminska, E.; Dolinska, M.; Lazarewicz, J.W.; Albrecht, J. Induction of permeability transition and swelling of rat brain mitochondria by glutamine. Neurotoxicology 2000, 21, 295–300. [Google Scholar] [PubMed]
- Albrecht, J.; Zielinska, M.; Norenberg, M.D. Glutamine as a mediator of ammonia neurotoxicity: A critical appraisal. Biochem. Pharmacol. 2010, 80, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Norenberg, M.D. Glutamine in the pathogenesis of hepatic encephalopathy: The Trojan horse hypothesis revisited. Neurochem. Res. 2014, 39, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Rama Rao, K.V.; Jayakumar, A.R. Ammonia neurotoxicity and the mitochondrial permeability transition. J. Bioenerg. Biomembr. 2004, 36, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Eid, T.; Behar, K.; Dhaher, R.; Bumanglag, A.V.; Lee, T.S. Roles of glutamine synthetase inhibition in epilepsy. Neurochem. Res. 2012, 37, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Westergaard, N.; Petersen, S.B.; Unsgard, G.; Schousboe, A. Metabolism of [U-13C]glutamate in astrocytes studied by 13C NMR spectroscopy: Incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J. Neurochem. 1993, 61, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Rothman, D.L.; Cline, G.W.; Behar, K.L. Glutamine is the major precursor for GABA synthesis in rat neocortex in vivo following acute GABA-transaminase inhibition. Brain Res. 2001, 919, 207–220. [Google Scholar] [CrossRef]
- Patel, A.B.; de Graaf, R.A.; Mason, G.F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 5588–5593. [Google Scholar] [CrossRef] [PubMed]
- Laake, J.H.; Slyngstad, T.A.; Haug, F.M.; Ottersen, O.P. Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: Immunogold evidence from hippocampal slice cultures. J. Neurochem. 1995, 65, 871–881. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hakvoort, T.B.; Vermeulen, J.L.; Labruyere, W.T.; De Waart, D.R.; Van Der Hel, W.S.; Ruijter, J.M.; Uylings, H.B.; Lamers, W.H. Glutamine synthetase deficiency in murine astrocytes results in neonatal death. Glia 2010, 58, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Duncan, N.W.; Wiebking, C.; Northoff, G. Associations of regional GABA and glutamate with intrinsic and extrinsic neural activity in humans—A review of multimodal imaging studies. Neurosci. Biobehav. Rev. 2014, 47, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Petroff, O.A.; Errante, L.D.; Rothman, D.L.; Kim, J.H.; Spencer, D.D. Neuronal and glial metabolite content of the epileptogenic human hippocampus. Ann. Neurol. 2002, 52, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Bacci, A.; Sancini, G.; Verderio, C.; Armano, S.; Pravettoni, E.; Fesce, R.; Franceschetti, S.; Matteoli, M. Block of glutamate-glutamine cycle between astrocytes and neurons inhibits epileptiform activity in hippocampus. J. Neurophysiol. 2002, 88, 2302–2310. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Sontheimer, H. Glia as drivers of abnormal neuronal activity. Nat. Neurosci. 2016, 19, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Ortinski, P.I.; Dong, J.; Mungenast, A.; Yue, C.; Takano, H.; Watson, D.J.; Haydon, P.G.; Coulter, D.A. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 2010, 13, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Self, J.T.; Spencer, T.E.; Johnson, G.A.; Hu, J.; Bazer, F.W.; Wu, G. Glutamine synthesis in the developing porcine placenta. Biol. Reprod. 2004, 70, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Schlett, K. Glutamate as a modulator of embryonic and adult neurogenesis. Curr. Top. Med. Chem. 2006, 6, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L. The glutamate-glutamine (GABA) cycle: Importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front. Endocrinol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J. Practical Skin Pathology: A Diagnostic Approach; Leslie, K.O., Wick, M.R., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2013. [Google Scholar]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bieganowski, P.; Brenner, C. The reported human NADsyn2 is ammonia-dependent NAD synthetase from a pseudomonad. J. Biol. Chem. 2003, 278, 33056–33059. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Conforti, L. Diversification of NAD biological role: The importance of location. FEBS J. 2013, 280, 4711–4728. [Google Scholar] [CrossRef] [PubMed]
- Hara, N.; Yamada, K.; Terashima, M.; Osago, H.; Shimoyama, M.; Tsuchiya, M. Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J. Biol. Chem. 2003, 278, 10914–10921. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.R. Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 2000, 36, 471–482. [Google Scholar] [CrossRef]
- Fluteau, A.; Ince, P.G.; Minett, T.; Matthews, F.E.; Brayne, C.; Garwood, C.J.; Ratcliffe, L.E.; Morgan, S.; Heath, P.R.; Shaw, P.J.; et al. The nuclear retention of transcription factor FOXO3A correlates with a DNA damage response and increased glutamine synthetase expression by astrocytes suggesting a neuroprotective role in the ageing brain. Neurosci. Lett. 2015, 609, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Coffer, P.J. Glutamine metabolism links growth factor signaling to the regulation of autophagy. Autophagy 2012, 8, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Kulijewicz-Nawrot, M.; Sykova, E.; Chvatal, A.; Verkhratsky, A.; Rodriguez, J.J. Astrocytes and glutamate homoeostasis in Alzheimer’s disease: A decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro 2013, 5, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, M.; Lichtenstein, G.R.; Rajagopal, B.S.; McCann, M.T.; Furth, E.E.; Bavaria, J.; Kaplan, P.B.; Gibson, J.B.; Berry, G.T. Hepatic glutamine synthetase deficiency in fatal hyperammonemia after lung transplantation. Ann. Intern. Med. 1997, 127, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, G.R.; Yang, Y.X.; Nunes, F.A.; Lewis, J.D.; Tuchman, M.; Tino, G.; Kaiser, L.R.; Palevsky, H.I.; Kotloff, R.M.; Furth, E.E.; et al. Fatal hyperammonemia after orthotopic lung transplantation. Ann. Intern. Med. 2000, 132, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rosati, A.; Poliani, P.L.; Todeschini, A.; Cominelli, M.; Medicina, D.; Cenzato, M.; Simoncini, E.L.; Magrini, S.M.; Buglione, M.; Grisanti, S.; et al. Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme. Neuro-Oncology 2013, 15, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Nagashima, I.; Tsuno, N.H.; Kitayama, J.; Nagawa, H. Prognostic significance of glutamine synthetase expression in unifocal advanced hepatocellular carcinoma. J. Hepatol. 2000, 33, 247–253. [Google Scholar] [CrossRef]
- De Vitto, H.; Perez-Valencia, J.; Radosevich, J.A. Glutamine at focus: Versatile roles in cancer. Tumor Biol. 2016, 37, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- De Kieviet, J.F.; Oosterlaan, J.; Vermeulen, R.J.; Pouwels, P.J.; Lafeber, H.N.; van Elburg, R.M. Effects of glutamine on brain development in very preterm children at school age. Pediatrics 2012, 130, e1121–e1127. [Google Scholar] [CrossRef] [PubMed]
- Irimia, R.; Stanciu, C.; Cojocariu, C.; Sfarti, C.; Trifan, A. Oral glutamine challenge improves the performance of psychometric tests for the diagnosis of minimal hepatic encephalopathy in patients with liver cirrhosis. J. Gastrointest. Liver Dis. 2013, 22, 277–281. [Google Scholar]
- Albrecht, J.; Norenberg, M.D. Glutamine: A Trojan horse in ammonia neurotoxicity. Hepatology 2006, 44, 788–794. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Variable | Patient 1 [2] | Patient 2 [2] | Patient 3 [4] |
|---|---|---|---|
| Ethnicity/Gender | Turkish boy | Turkish girl | Sudanese boy |
| Prenatal ultrasonography | Polyhydramnios, large lateral brain ventricles with a left frontal paraventricular cyst, micromelia | Dilatation of cerebral posterior fossa | Delayed gyration, marked white matter changes, subependymal cysts |
| Presentation at birth | Spontaneously at 354/7 week | Spontaneously at 386/7 week | Term |
| Birth weight Length Head circumfer. | 2220 g (10th percentile) 44 cm (3rd–10th percentile) 34 cm (75th–90th percentile) | 2460 g (3rd–10th percentile) 42 cm (<3rd percentile) 28.5 cm (<3rd percentile) | 3245 g (50th percentile) 53 cm (90th percentile) 34 cm (50th percentile) |
| Clinical course | Epileptic encephalopathy Multiple organ failure | Epileptic encephalopathy Multiple organ failure Necrolytic erythema Diarrhea | Epileptic encephalopathy Severe developmental delay Necrolytic skin erythema (one episode) |
| Onset | Immediately after birth | Immediately after birth | Day 1 |
| Life Time | 2 days | 28 days | 6 years |
| Dysmorphic features | Flat nasal root, low-set ears, flexion contractures at elbows and knees, shortness of limbs | Broad nasal root, low-set ears | None |
| EEG | Generalized seizures, Outbursts of theta waves | Multifocal seizures | Generalized tonic-clonic seizures |
| Brain MRI | Cerebral and cerebellar atrophy, almost complete agyria, immature white matter, multiple paraventricular cysts in frontal and temporal lobes, enlarged lateral ventricles | Small frontal lobe and cerebellum, delayed gyration, marked white matter changes, subependymal cysts | Mild degree of brain atrophy with prominent cortical sulci and sylvian fissures, hypomyelination of white matter, thinning of the corpus callosum |
| Glutamine | |||
| Serum | 2 μmol/L (normal range: 433–619) | 6 μmol/L (normal range: 300–800) | First test: 126 μmol/L, range: 8–354 μmol/L (normal range: 376–709) |
| Urine | Not detectable (normal range: 52–205) | 8 μmol/g creatinine (normal range: 640–3230) | |
| Cerebrospinal fluid | 11 μmol/L (normal range: 352–885) | 12 μmol/L (normal range: 520–1280) | Range: 50–238 μmol/L (normal range: 373–556) |
| Glutamate | |||
| Serum | 45 μmol/L (normal range: 36–82) | 80 μmol/L (normal range: 70–220) | Range: 20–143 μmol/L (normal range: 62–260) |
| Urine | 2 mmol/mol creatinine (normal range: 0–30) | 34 μmol/g creatinine (normal range: 0–250) | |
| Cerebrospinal fluid | Not detectable (normal range: 1–48) | 2 μmol/L (normal range: 2–51) | |
| Ammonia | |||
| Plasma | 140 μmol/L (normal < 110) | Mean: 94 μmol/L, range: 38–424 μmol/L (normal < 50) | |
| Mutation | |||
| c.970C > T (p.R324C) | c.1021C > T (p.R341C) | c.970C > A (p.R324S) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spodenkiewicz, M.; Diez-Fernandez, C.; Rüfenacht, V.; Gemperle-Britschgi, C.; Häberle, J. Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis. Biology 2016, 5, 40. https://doi.org/10.3390/biology5040040
Spodenkiewicz M, Diez-Fernandez C, Rüfenacht V, Gemperle-Britschgi C, Häberle J. Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis. Biology. 2016; 5(4):40. https://doi.org/10.3390/biology5040040
Chicago/Turabian StyleSpodenkiewicz, Marta, Carmen Diez-Fernandez, Véronique Rüfenacht, Corinne Gemperle-Britschgi, and Johannes Häberle. 2016. "Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis" Biology 5, no. 4: 40. https://doi.org/10.3390/biology5040040