Cellular Localization and Trafficking of the Human ABCG1 Transporter


Abstract
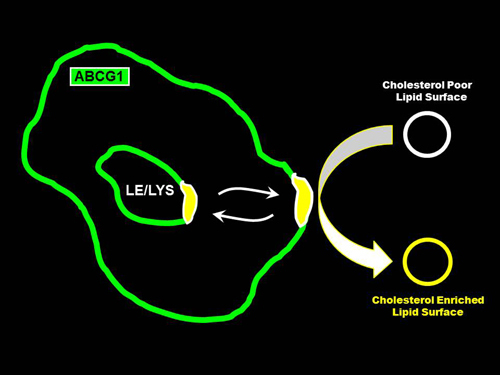
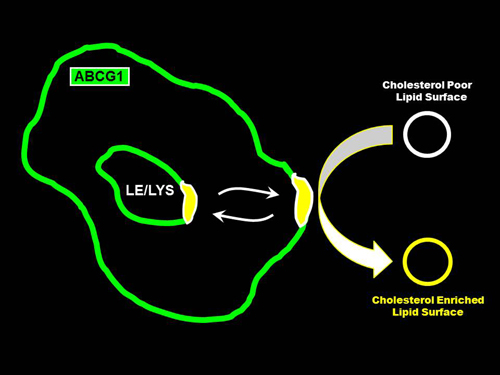
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
3. Results and Discussion
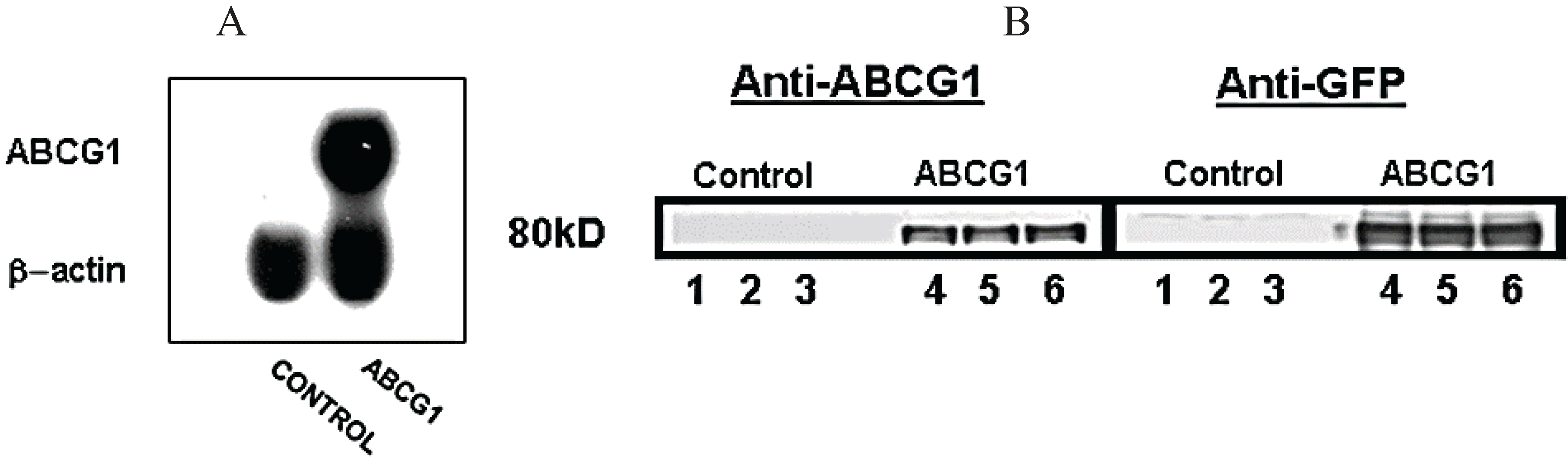
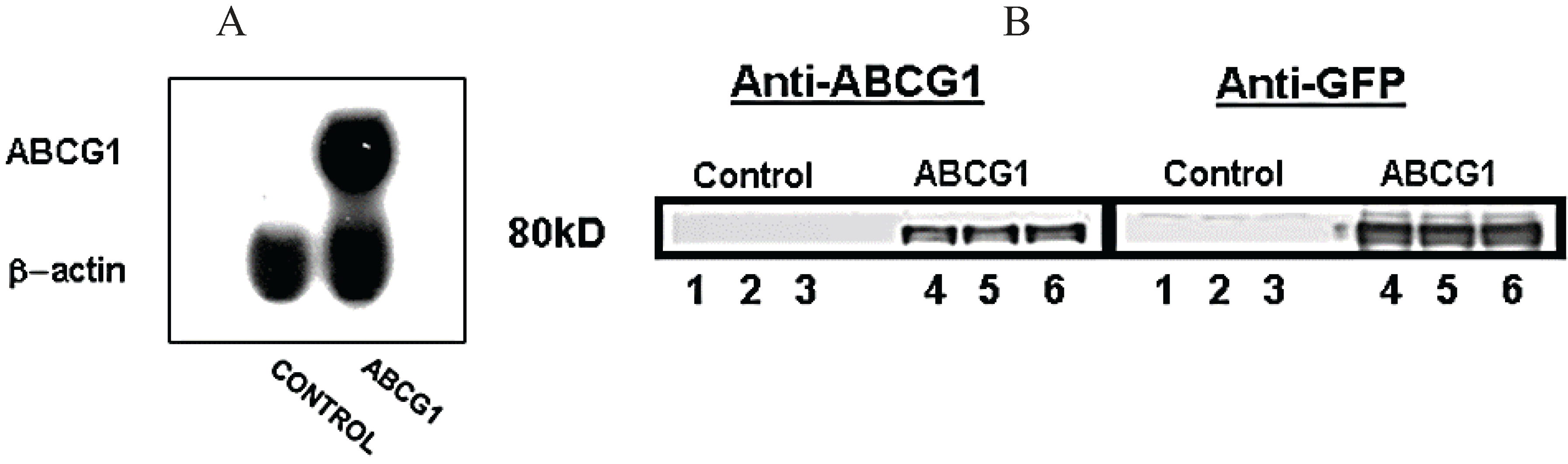
3.1. Stable Expression of GFP-Tagged Human ABCG1
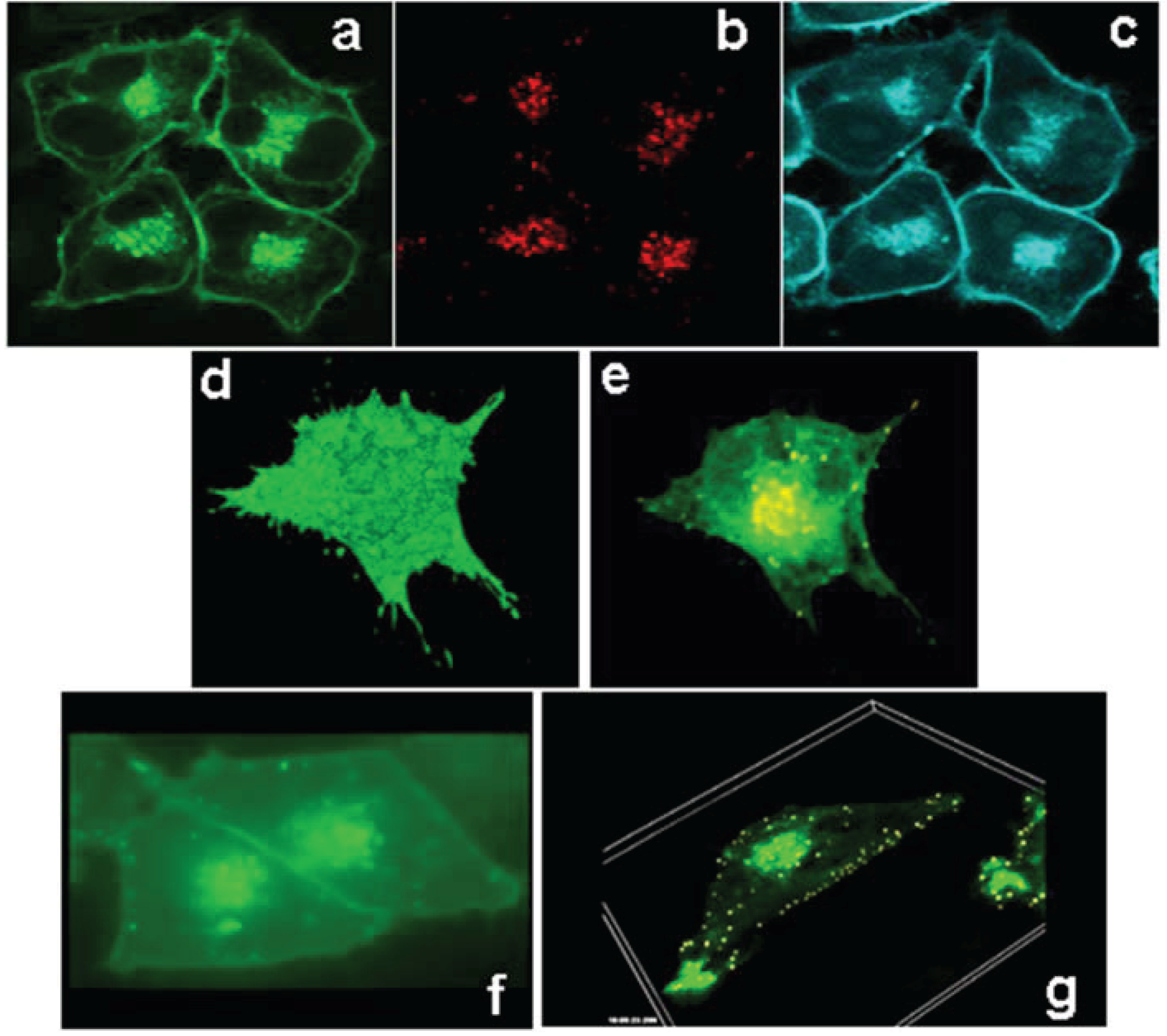
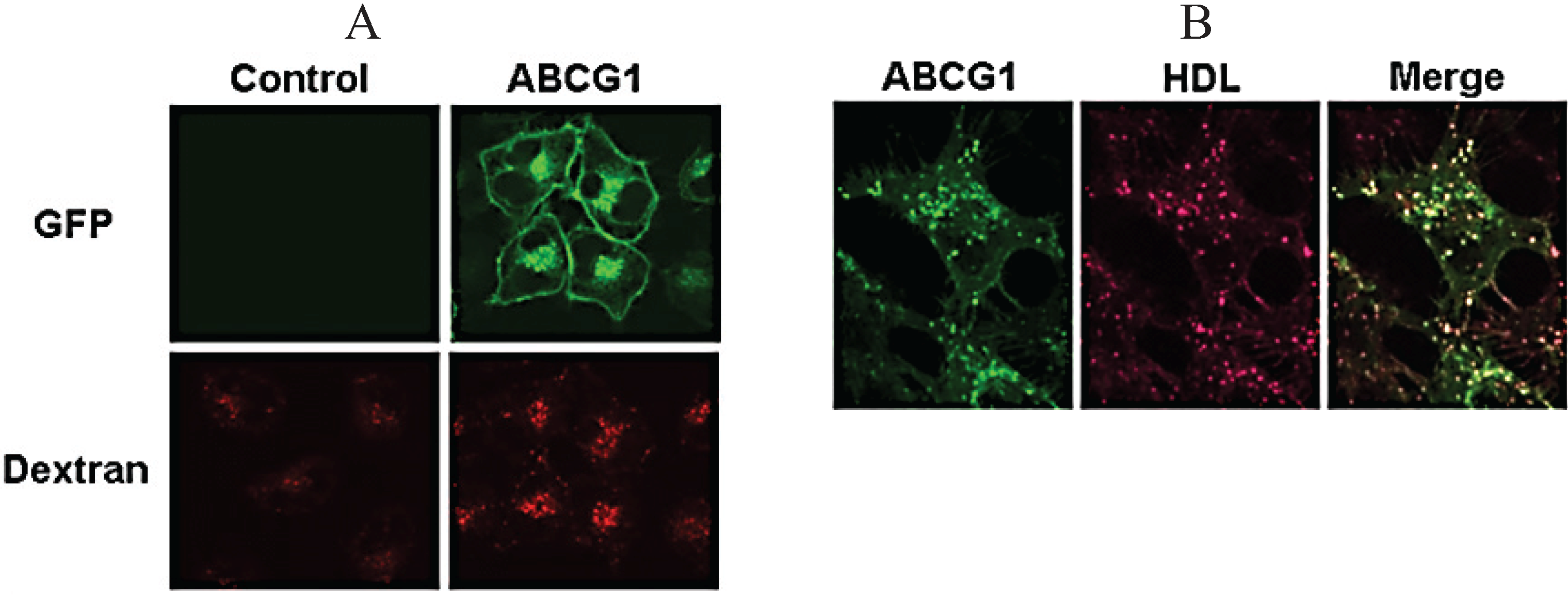
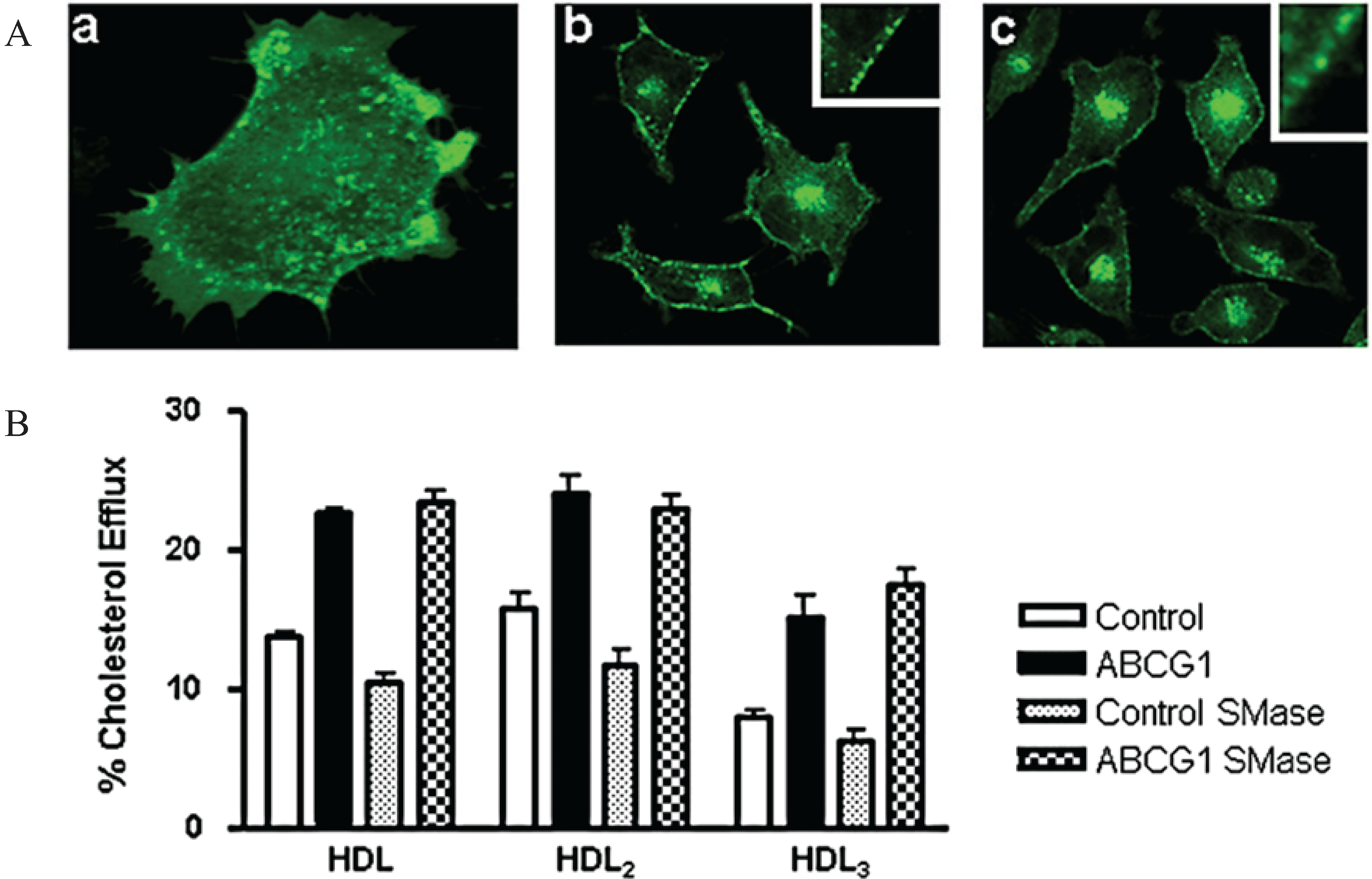
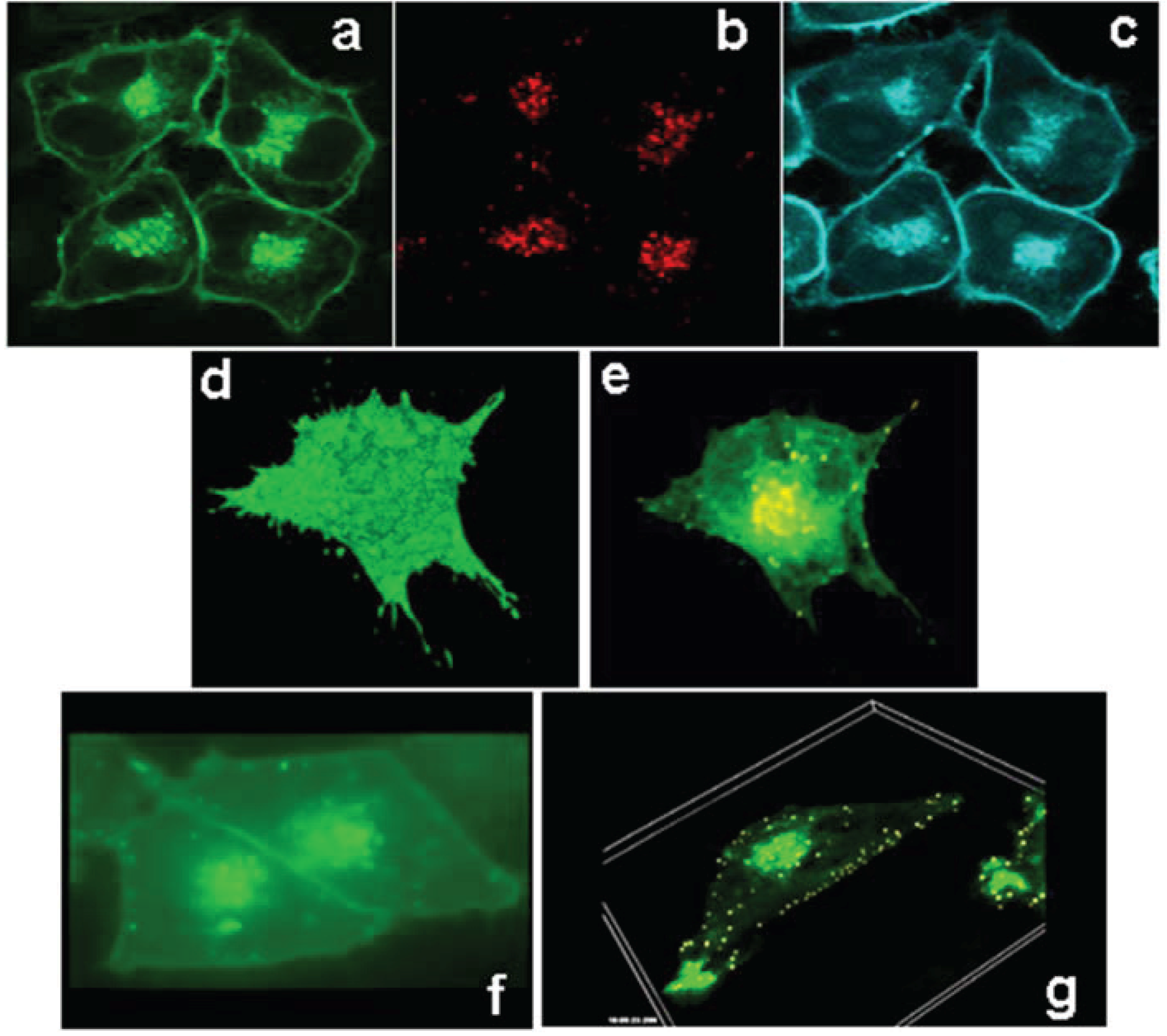
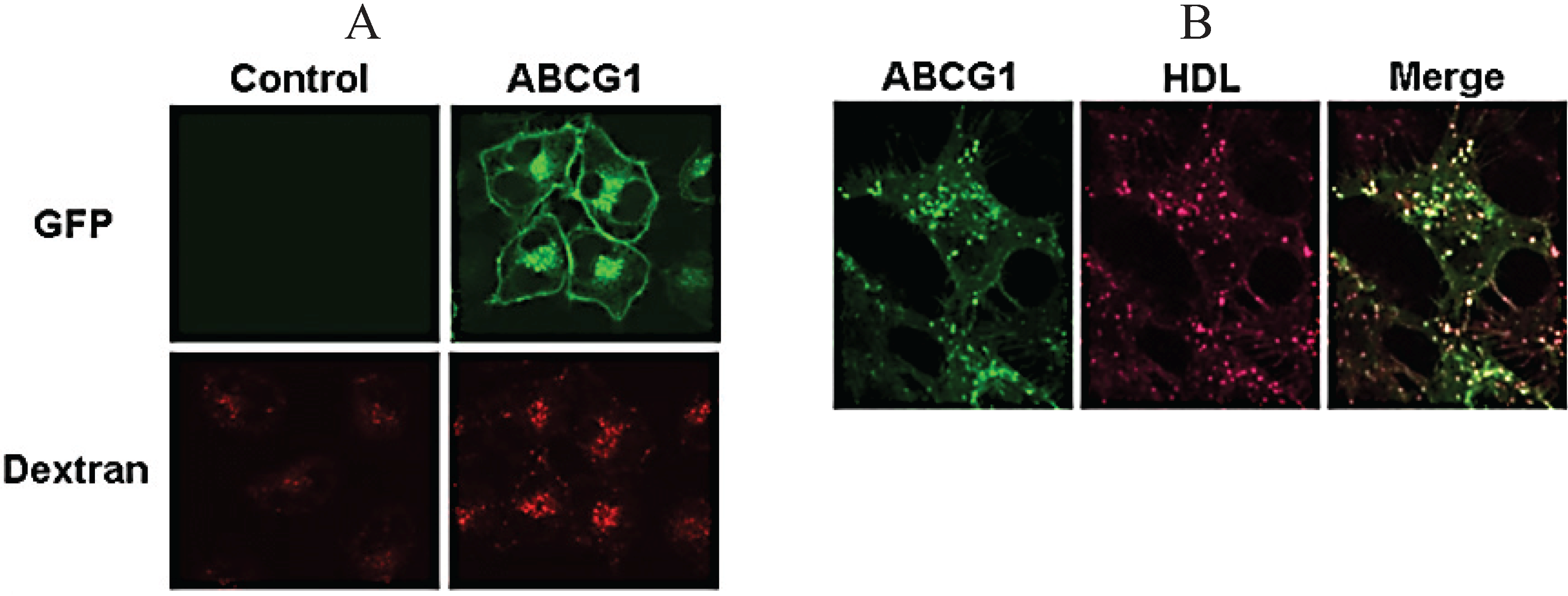
3.2. ABCG1 Resides on the Cell Surface and Late Endosomes
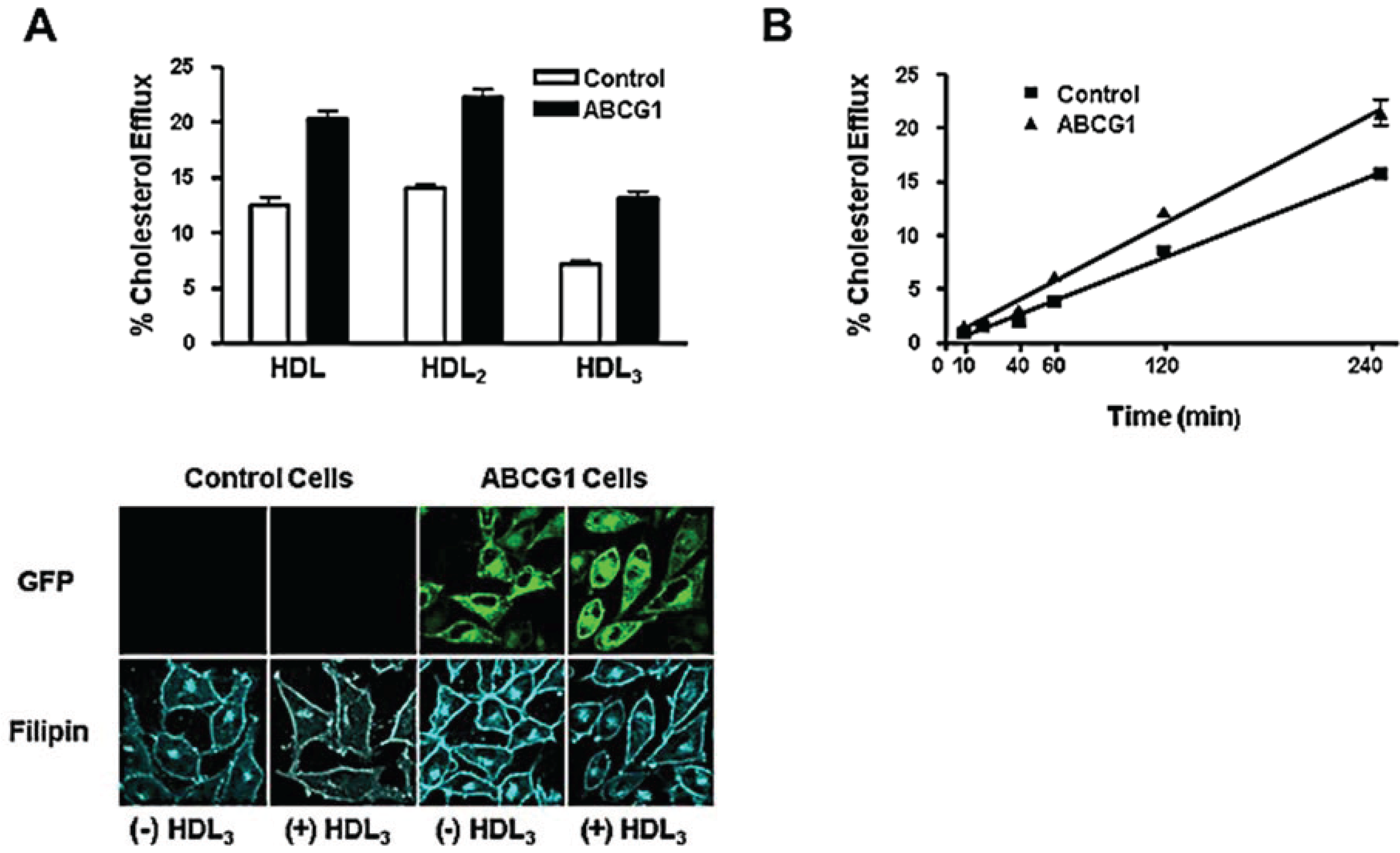
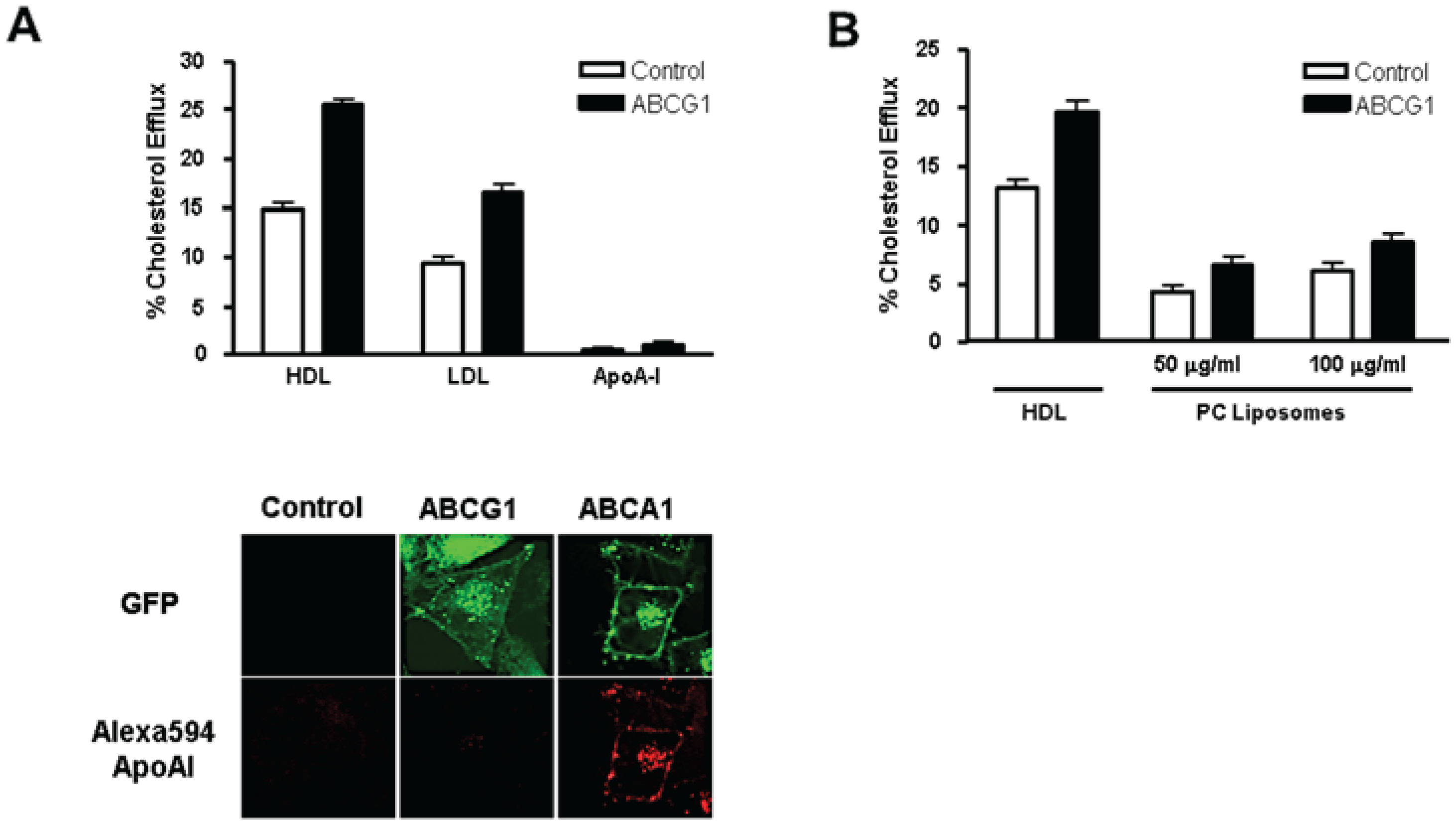
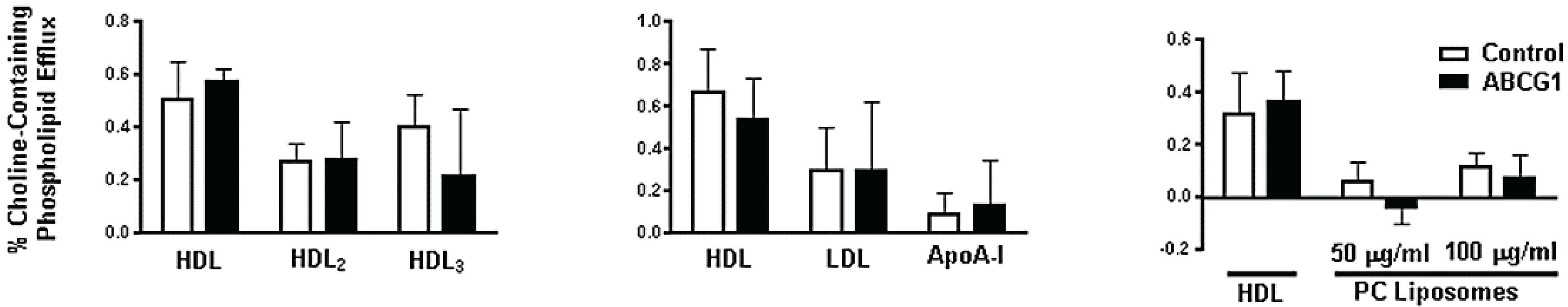
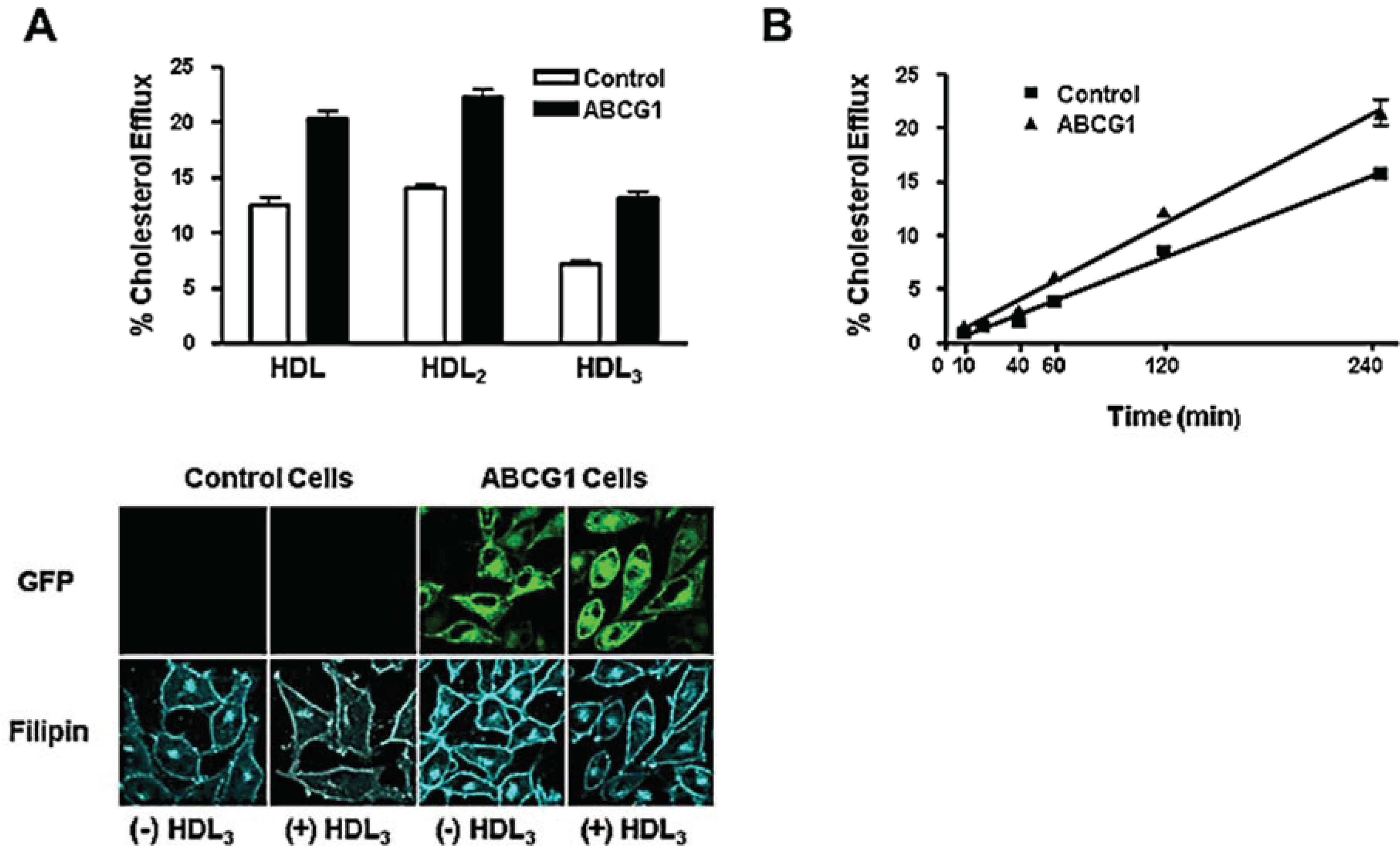
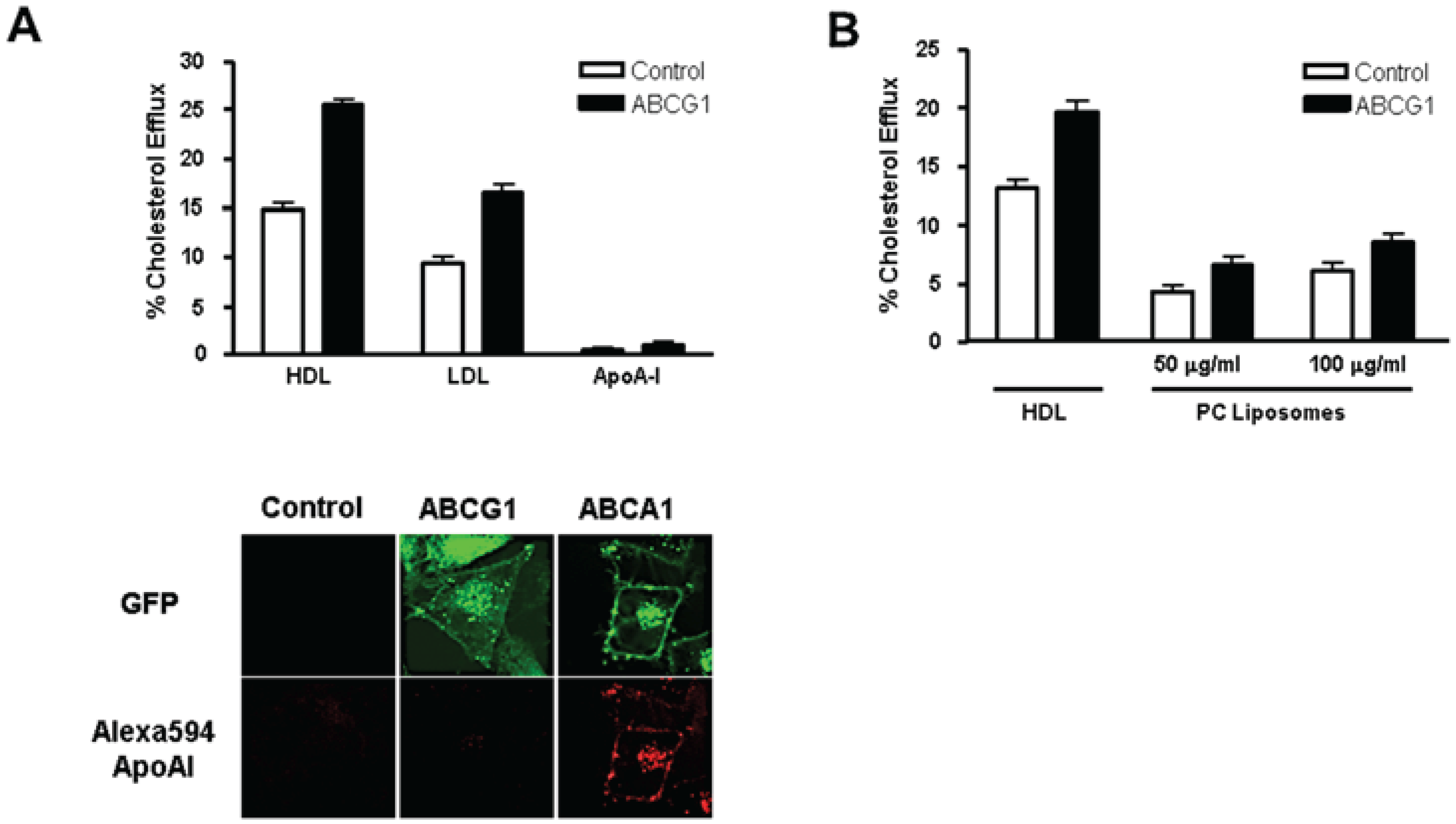
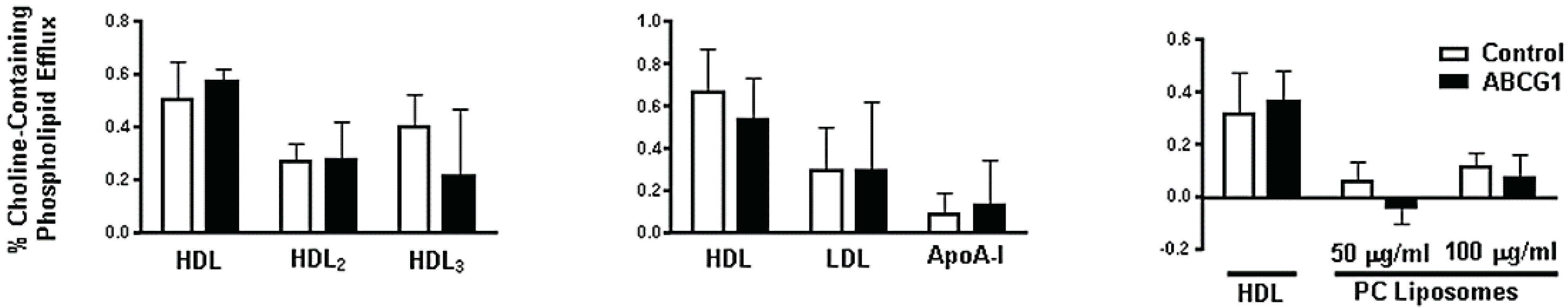
3.3. ABCG1-GFP Expression Increases Cellular Cholesterol Efflux to Extracellular Acceptors with a Lipid Surface
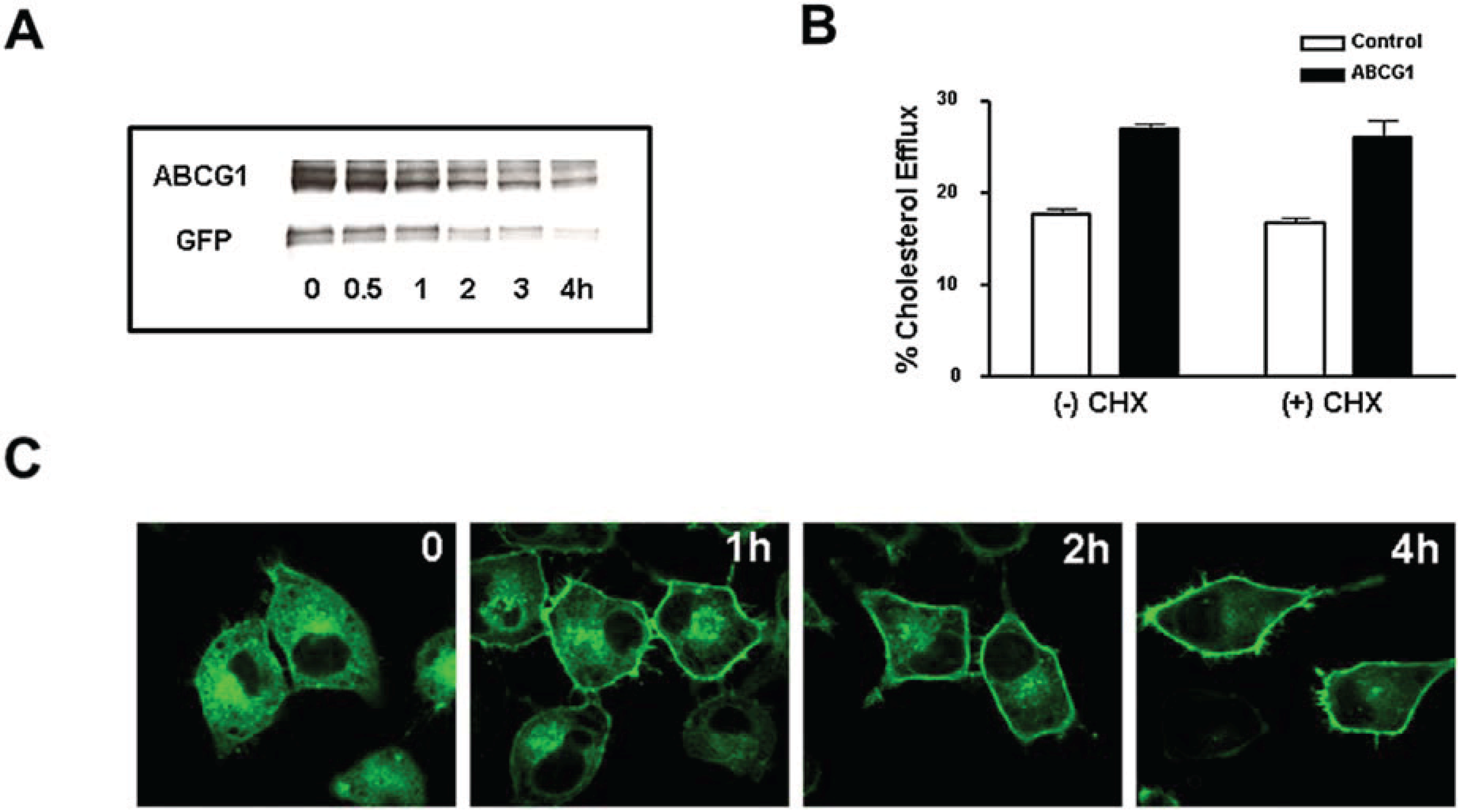
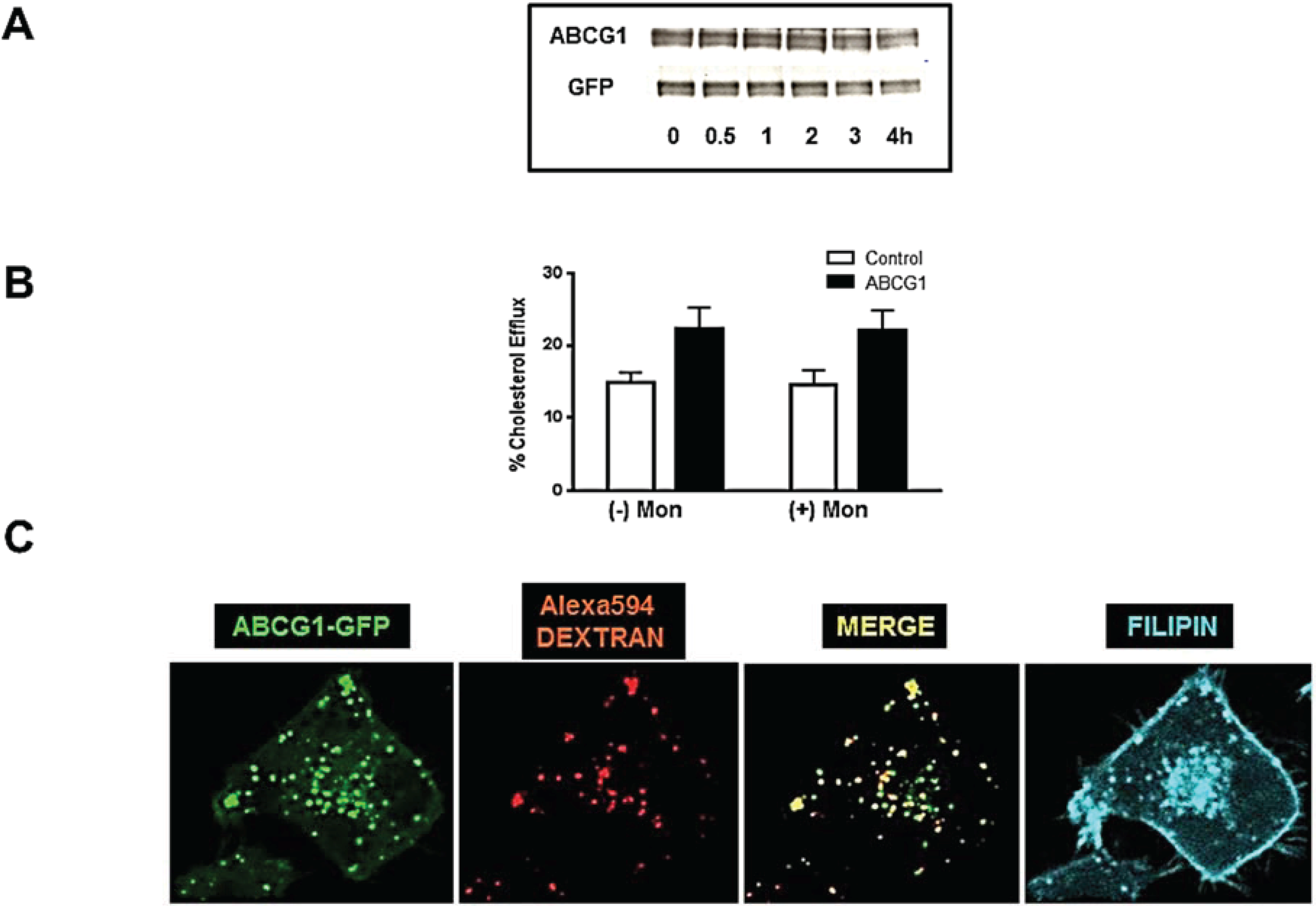
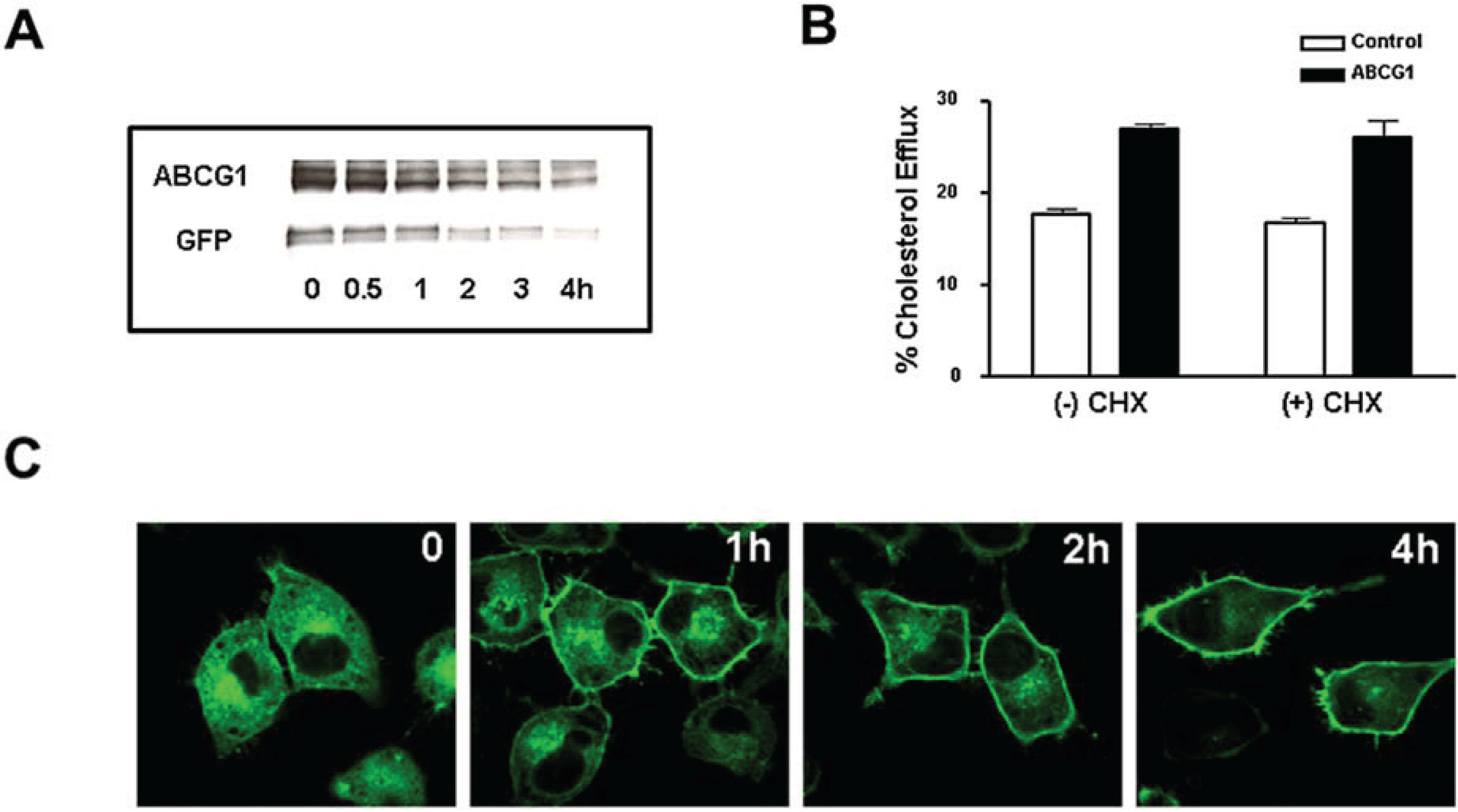
3.4. Cycloheximide-Treated ABCG1-Expressing Cells Maintain Sufficient Plasma Membrane and Endosomal ABCG1 to Sustain ABCG1-Mediated Cellular Sterol Efflux
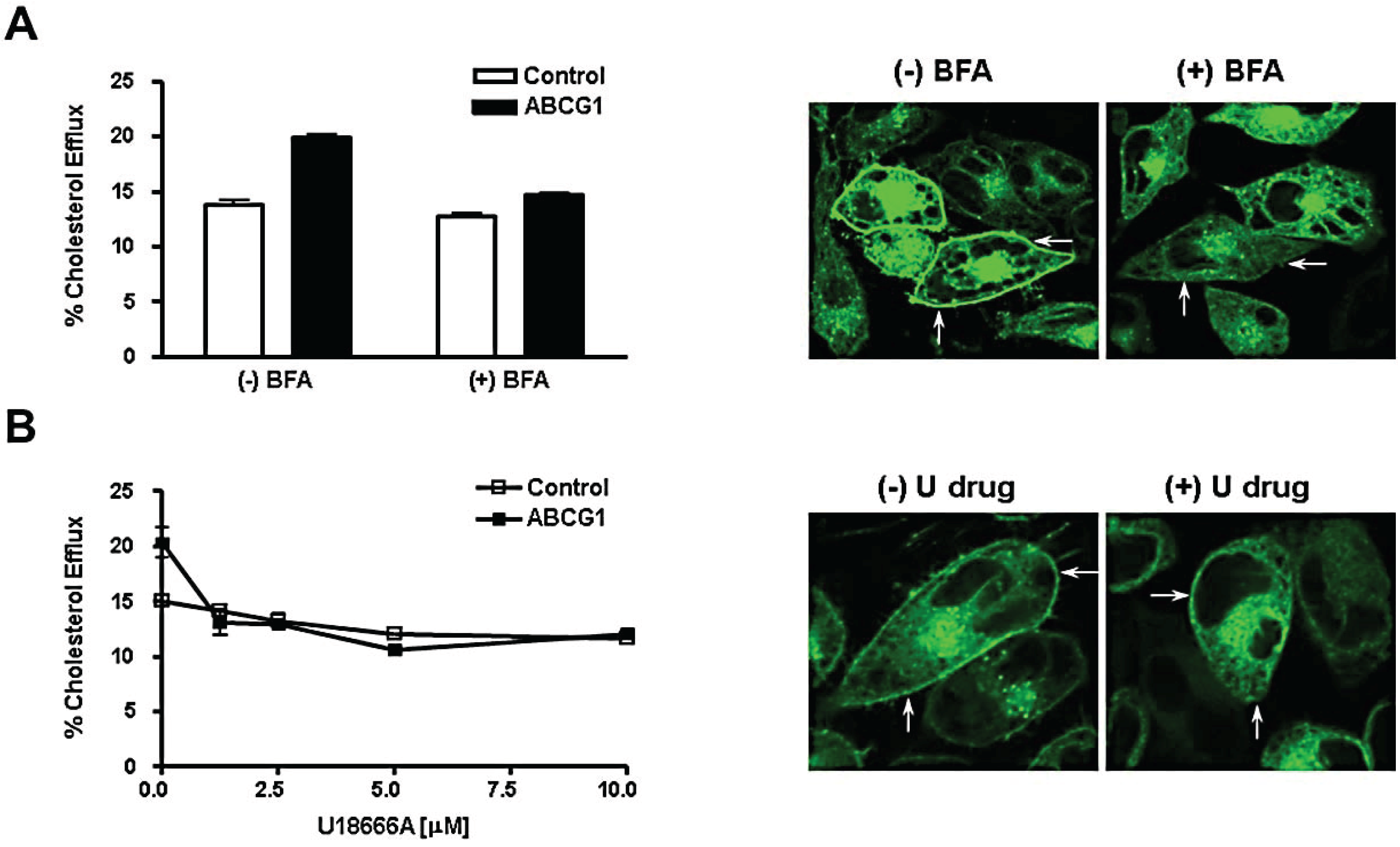
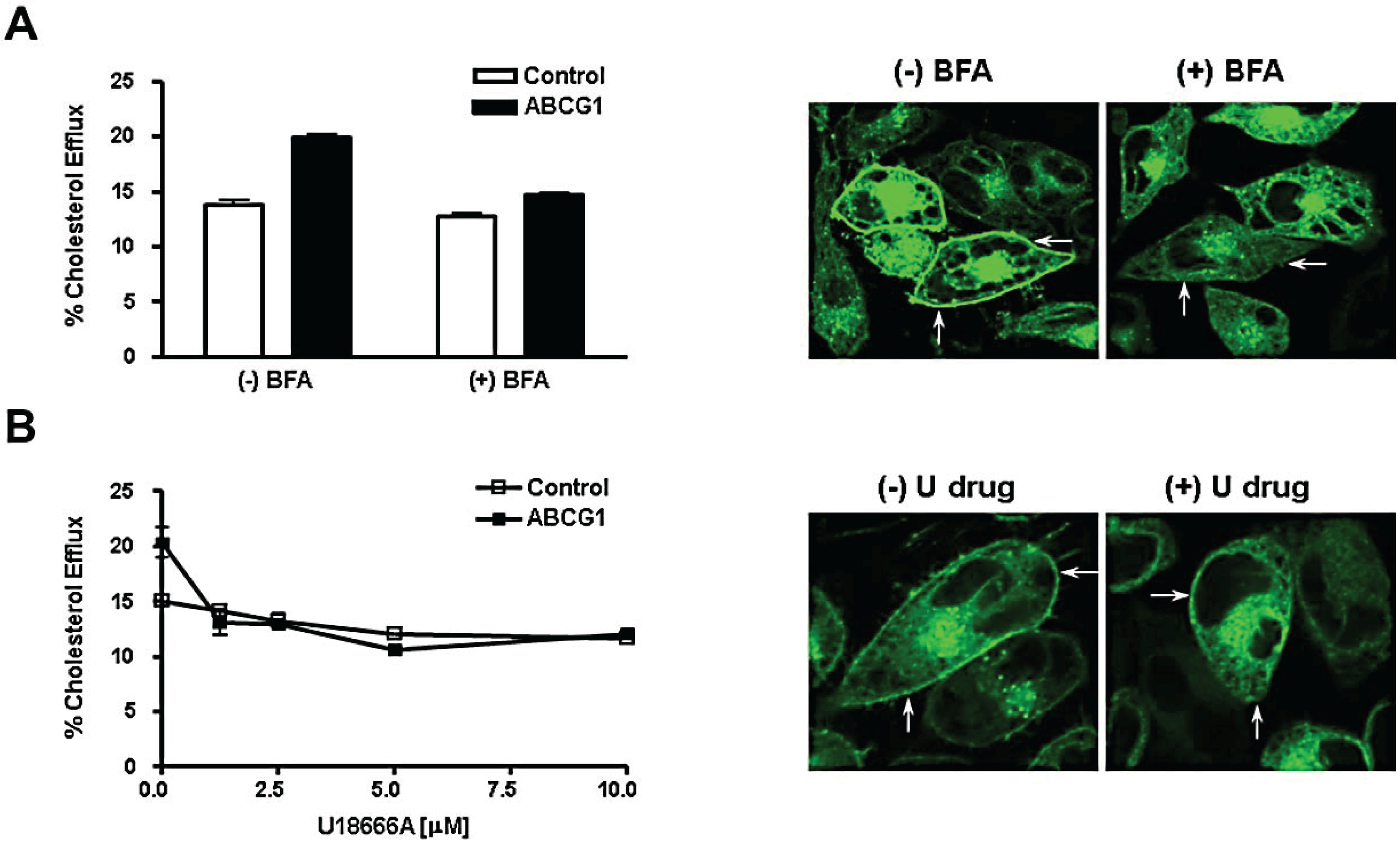
3.5. Brefeldin A and U18666A Block both Delivery of ABCG1-GFP to the Cell Surface and ABCG1-Mediated Cellular Cholesterol Efflux
3.6. ABCG1-GFP Trapped in Late Endocytic Compartments Can Still Mediate Enhanced Cellular Cholesterol Efflux
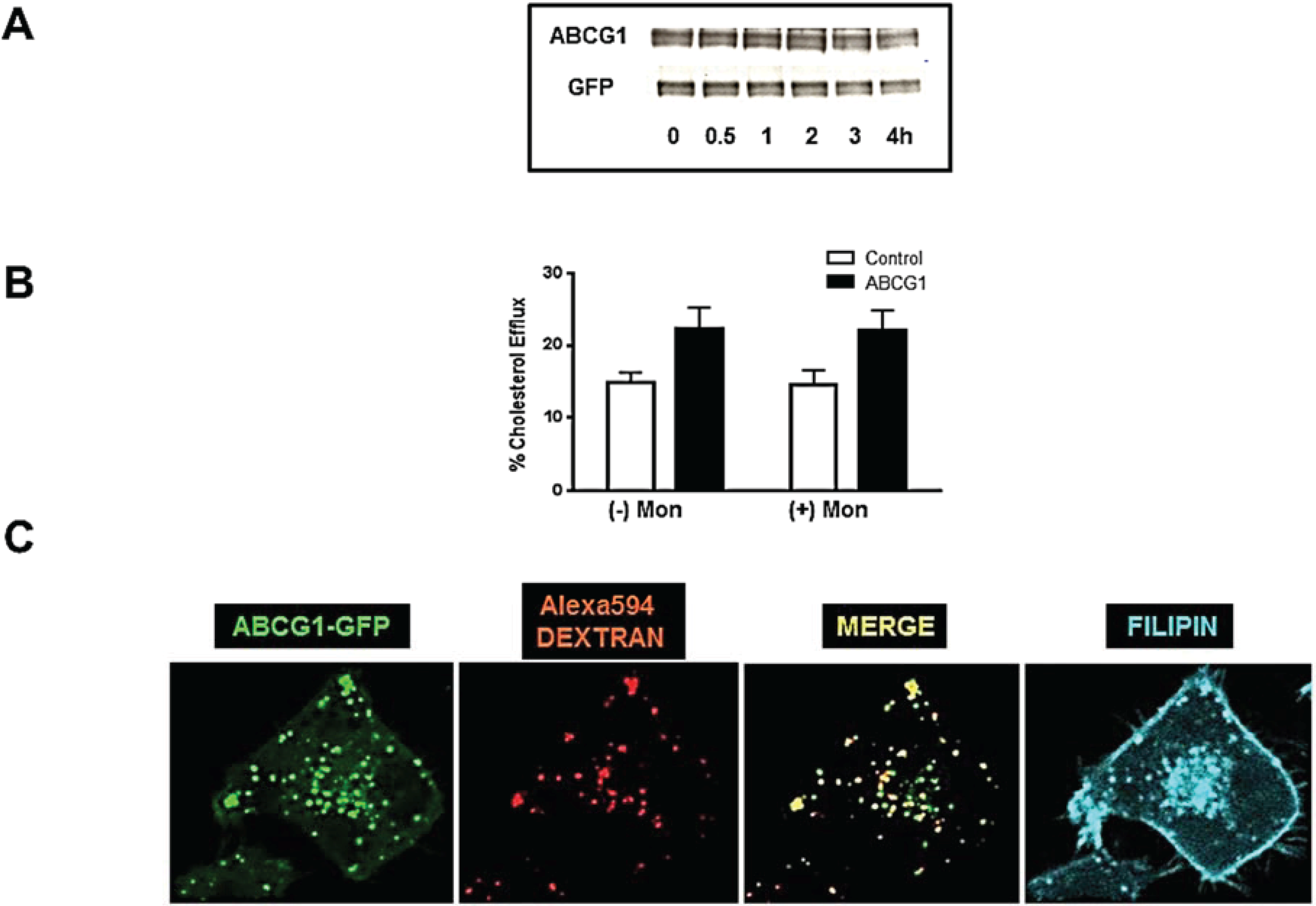
3.7. ABCG1-GFP Increases Endocytic Uptake of Fluid Phase Markers into Late Endocytic Compartments
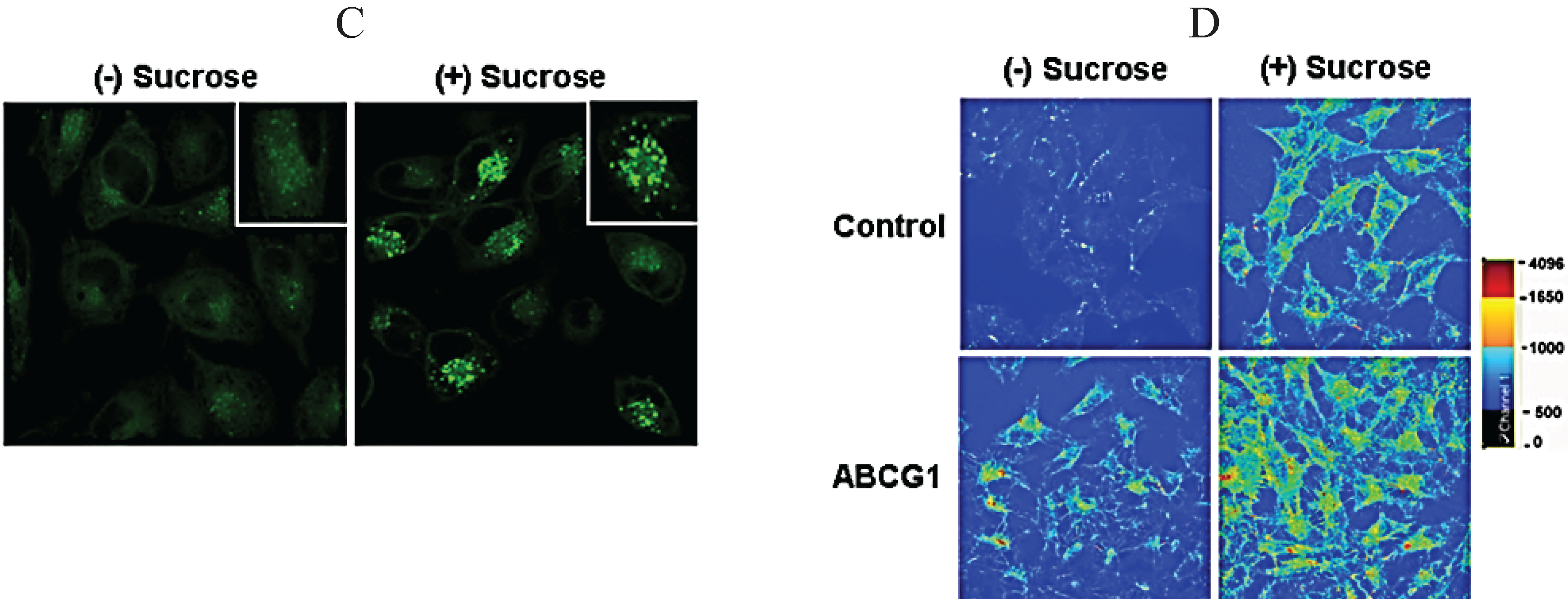
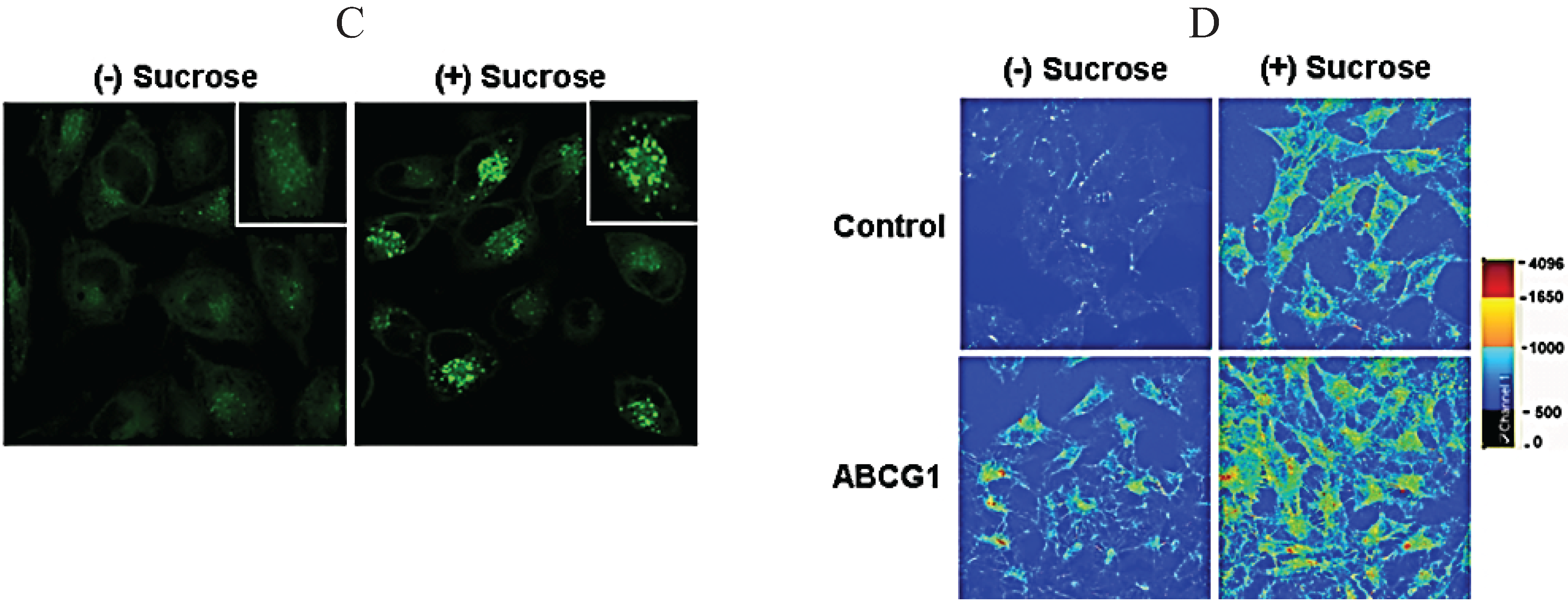
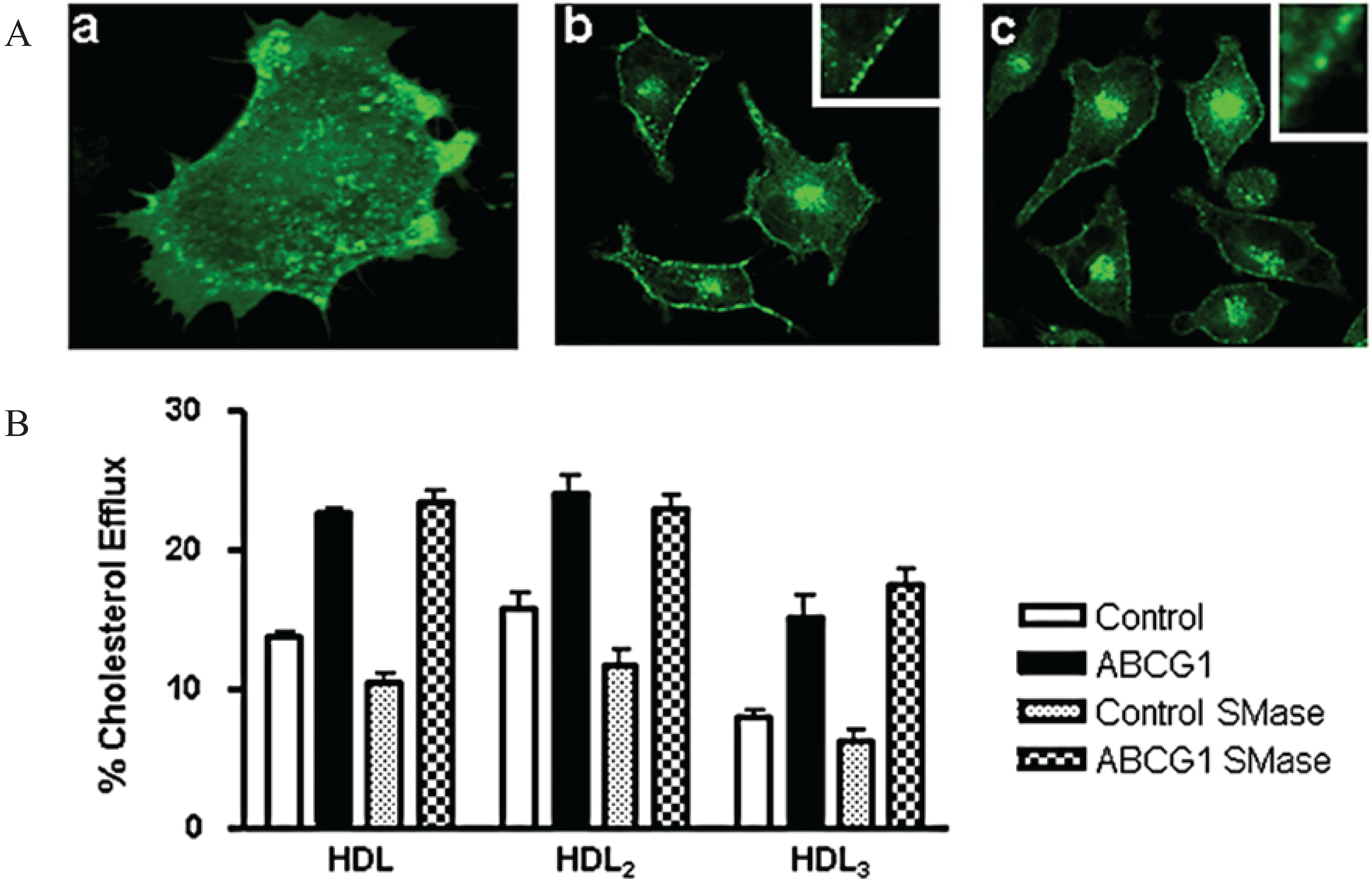
3.8. ABCG1-Mediated Cellular Cholesterol Efflux to HDL is Maintained When Cellular Sphingomyelin is Depleted
4. Conclusions
Supplementary Files
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Supplementary File 5Supplementary File 6Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neufeld, E.B.; Remaley, A.T.; Demosky, S.J.; Stonik, J.A.; Cooney, A.M.; Comly, M.; Dwyer, N.K.; Zhang, M.; Blanchette-Mackie, J.; Santamarina-Fojo, S.; et al. Cellular localization and trafficking of the human abca1 transporter. J. Biol. Chem. 2001, 276, 27584–27590. [Google Scholar] [CrossRef]
- Neufeld, E.B.; Stonik, J.A.; Demosky, S.J., Jr.; Knapper, C.L.; Combs, C.A.; Cooney, A.; Comly, M.; Dwyer, N.; Blanchette-Mackie, J.; Remaley, A.T.; et al. The abca1 transporter modulates late endocytic trafficking: Insights from the correction of the genetic defect in tangier disease. J. Biol. Chem. 2004, 279, 15571–15578. [Google Scholar] [CrossRef]
- Engelbrecht, S.; Kaltenborn, E.; Griese, M.; Kern, S. The surfactant lipid transporter ABCA3 is N-terminally cleaved inside LAMP3-positive vesicles. FEBS Lett. 2010, 584, 4306–4312. [Google Scholar] [CrossRef] [PubMed]
- Van der Kant, R.; Zondervan, I.; Janssen, L.; Neefjes, J. Cholesterol-binding molecules mln64 and orp1l mark distinct late endosomes with transporters abca3 and NPC1. J. Lipid Res. 2013, 54, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Wastney, M.; Patel, S.; Suresh, S.; Cooney, A.M.; Dwyer, N.K.; Roff, C.F.; Ohno, K.; Morris, J.A.; Carstea, E.D.; et al. The niemann-pick c1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem. 1999, 274, 9627–9635. [Google Scholar] [CrossRef]
- Santamarina-Fojo, S.; Remaley, A.T.; Neufeld, E.B.; Brewer, H.B., Jr. Regulation and intracellular trafficking of the abca1 transporter. J. Lipid Res. 2001, 42, 1339–1345. [Google Scholar] [PubMed]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Venkateswaran, A.; Tarr, P.T.; Xenarios, I.; Kudoh, J.; Shimizu, N.; Edwards, P.A. Characterization of the human ABCG1 gene: Liver X receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J. Biol. Chem. 2001, 276, 39438–39447. [Google Scholar] [CrossRef] [PubMed]
- Lorkowski, S.; Rust, S.; Engel, T.; Jung, E.; Tegelkamp, K.; Galinski, E.A.; Assmann, G.; Cullen, P. Genomic sequence and structure of the human ABCG1 (ABC8) gene. Biochem. Biophys. Res. Commun. 2001, 280, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Buchler, C.; Orso, E.; Kaminski, W.E.; Porsch-Ozcurumez, M.; Liebisch, G.; Kapinsky, M.; Diederich, W.; Drobnik, W.; Dean, M.; et al. ABCG1 (ABC8), the human homolog of the drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc. Natl. Acad. Sci. USA 2000, 97, 817–822. [Google Scholar] [CrossRef]
- Neufeld, E.B.; Sabol, S.; Remaley, A.T.; Ito, T.; Demosky, S.J.; Stonik, J.; Santamarina-Fojo, S.; Brewer, H.B. Cellular localization and trafficking of human ABCG1. Circulation 2001, 104, 147. [Google Scholar]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. Atp-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.M.; Oram, J.F. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J. Biol. Chem. 2005, 280, 30150–30157. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Barrera, G.C.; Nakamura, K.; Baldan, A.; Tarr, P.; Fishbein, M.C.; Frank, J.; Francone, O.L.; Edwards, P.A. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005, 1, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ranalletta, M.; Matsuura, F.; Peng, F.; Tall, A.R. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler. Thromb. Vascular Biol. 2006, 26, 1310–1316. [Google Scholar] [CrossRef]
- Xie, Q.; Engel, T.; Schnoor, M.; Niehaus, J.; Hofnagel, O.; Buers, I.; Cullen, P.; Seedorf, U.; Assmann, G.; Lorkowski, S. Cell surface localization of ABCG1 does not require LXR activation. Arterioscler. Thromb. Vascular Biol. 2006, 26, e143–e144. [Google Scholar] [CrossRef]
- Tarling, E.J.; Edwards, P.A. Atp binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl. Acad. Sci. USA 2011, 108, 19719–19724. [Google Scholar] [CrossRef] [PubMed]
- Baldan, A.; Tarr, P.; Vales, C.S.; Frank, J.; Shimotake, T.K.; Hawgood, S.; Edwards, P.A. Deletion of the transmembrane transporter ABCG1 results in progressive pulmonary lipidosis. J. Biol. Chem. 2006, 281, 29401–29410. [Google Scholar] [CrossRef] [PubMed]
- Sturek, J.M.; Castle, J.D.; Trace, A.P.; Page, L.C.; Castle, A.M.; Evans-Molina, C.; Parks, J.S.; Mirmira, R.G.; Hedrick, C.C. An intracellular role for ABCG1-mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J. Clin. Investig. 2010, 120, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Brewer, H.B., Jr.; Ronan, R.; Meng, M.; Bishop, C. Isolation and characterization of apolipoproteins A-I, A-II, and A-IV. Methods Enzymol. 1986, 128, 223–246. [Google Scholar] [PubMed]
- Remaley, A.T.; Thomas, F.; Stonik, J.A.; Demosky, S.J.; Bark, S.E.; Neufeld, E.B.; Bocharov, A.V.; Vishnyakova, T.G.; Patterson, A.P.; Eggerman, T.L.; et al. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J. Lipid Res. 2003, 44, 828–836. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Ghosh, R.N.; Gelman, D.L.; Maxfield, F.R. Quantification of low density lipoprotein and transferrin endocytic sorting hep2 cells using confocal microscopy. J. Cell Sci. 1994, 107, 2177–2189. [Google Scholar] [PubMed]
- Chen, C.S.; Bach, G.; Pagano, R.E. Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc. Natl. Acad. Sci. USA 1998, 95, 6373–6378. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, S.; Oram, J.F.; Asztalos, B.F.; Vaughan, A.M.; Lund-Katz, S.; Adorni, M.P.; Phillips, M.C.; Rothblat, G.H. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. J. Lipid Res. 2009, 50, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Klausner, R.D.; Donaldson, J.G.; Lippincott-Schwartz, J. Brefeldin A: Insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992, 116, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Cenedella, R.J. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 2009, 44, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, H.H.; Morre, D.J.; Rowe, L.D. Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim. Biophys. Acta 1990, 1031, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Jahraus, A.; Storrie, B.; Griffiths, G.; Desjardins, M. Evidence for retrograde traffic between terminal lysosomes and the prelysosomal/late endosome compartment. J. Cell Sci. 1994, 107, 145–157. [Google Scholar] [PubMed]
- Bright, N.A.; Reaves, B.J.; Mullock, B.M.; Luzio, J.P. Dense core lysosomes can fuse with late endosomes and are re-formed from the resultant hybrid organelles. J. Cell Sci. 1997, 110, 2027–2040. [Google Scholar] [PubMed]
- Zha, X.; Pierini, L.M.; Leopold, P.L.; Skiba, P.J.; Tabas, I.; Maxfield, F.R. Sphingomyelinase treatment induces atp-independent endocytosis. J. Cell Biol. 1998, 140, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Takanezawa, Y.; Hirata, T.; Shimizu, Y.; Misasa, K.; Kioka, N.; Arai, H.; Ueda, K.; Matsuo, M. Efflux of sphingomyelin, cholesterol, and phosphatidylcholine by ABCG1. J. Lipid Res. 2006, 47, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A. Lipid trafficking sans vesicles: Where, why, how? Cell 2010, 143, 870–874. [Google Scholar] [CrossRef]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Jauhiainen, M.; Hildebrand, R.B.; Willems van Dijk, K.; van Berkel, T.J.; Ehnholm, C.; van Eck, M.; Olkkonen, V.M. Expression of human OSBP-related protein 1L in macrophages enhances atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler. Thromb. Vascular Biol. 2007, 27, 1618–1624. [Google Scholar] [CrossRef]
- Neufeld, E.B.; O’Brien, K.; Walts, A.D.; Stonik, J.A.; Malide, D.; Combs, C.A.; Remaley, A.T. The human ABCG1 transporter mobilizes plasma membrane and late endosomal non-sphingomyelin-associated-cholesterol for efflux and esterification. Unpublished data. 2014. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neufeld, E.B.; O'Brien, K.; Walts, A.D.; Stonik, J.A.; Demosky, S.J., Jr.; Malide, D.; Combs, C.A.; Remaley, A.T. Cellular Localization and Trafficking of the Human ABCG1 Transporter. Biology 2014, 3, 781-800. https://doi.org/10.3390/biology3040781
Neufeld EB, O'Brien K, Walts AD, Stonik JA, Demosky SJ Jr., Malide D, Combs CA, Remaley AT. Cellular Localization and Trafficking of the Human ABCG1 Transporter. Biology. 2014; 3(4):781-800. https://doi.org/10.3390/biology3040781
Chicago/Turabian StyleNeufeld, Edward B., Katherine O'Brien, Avram D. Walts, John A. Stonik, Steven J. Demosky, Jr., Daniela Malide, Christian A. Combs, and Alan T. Remaley. 2014. "Cellular Localization and Trafficking of the Human ABCG1 Transporter" Biology 3, no. 4: 781-800. https://doi.org/10.3390/biology3040781