Whole Genome Sequencing and a New Bioinformatics Platform Allow for Rapid Gene Identification in D. melanogaster EMS Screens

Abstract
:1. Introduction
2. Methods
2.1. Sample Preparation
2.2. Illumina Whole-genome Sequencing
2.3. Data Analysis and Variant Detection
 with a max score of 99. The QUAL score is the Phred probability that a variant polymorphism exists at a given site given the sequencing data, where a score of 10 indicates a 1 in 10 chance of error and a score of 100 indicates a 1 in 100 chance of error.
with a max score of 99. The QUAL score is the Phred probability that a variant polymorphism exists at a given site given the sequencing data, where a score of 10 indicates a 1 in 10 chance of error and a score of 100 indicates a 1 in 100 chance of error.3. Results
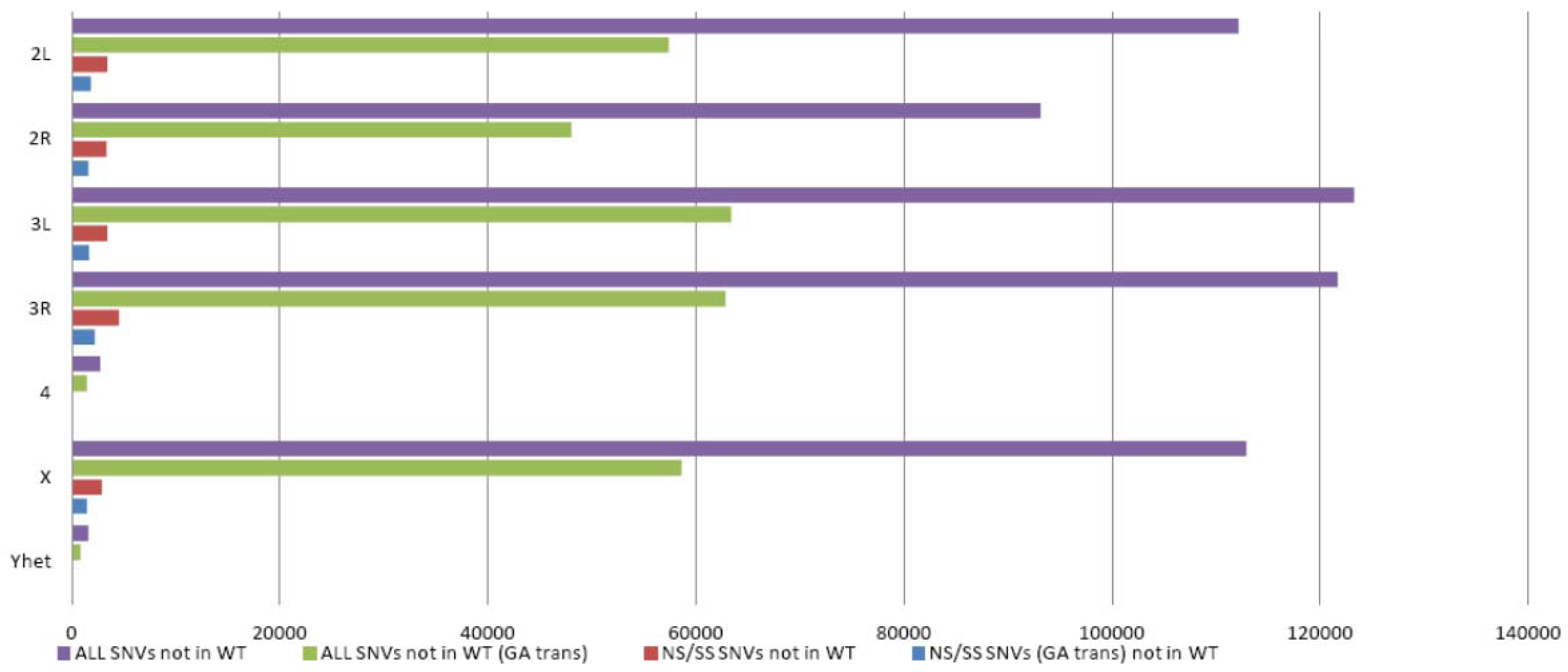
3.1. Whole Genome Sequencing Analysis of D. melanogaster

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | # of reads | # of reads aligned | % reads aligned | Avg depth | # of SNVs | # of high quality SNVs | # of NS/SS cSNV | # of NS/SS cSNV on 3L | Unique NS/SS cSNV on 3L |
|---|---|---|---|---|---|---|---|---|---|
| Background | 42,278,410 | 37,650,186 | 88.1 | 27.1 | 412,362 | 88,593 | 21,745 | 2,851 | 25 |
| Mutant 1 | 83,425,870 | 72,784,178 | 87.2 | 44.6 | 849,658 | 456,778 | 35,172 | 6,544 | 863 |
| Mutant 2 | 43,972,798 | 38,172,143 | 86.81 | 23.4 | 647,655 | 427,332 | 25,054 | 5,252 | 533 |
| Mutant 3 | 125,284,692 | 111,178,422 | 88.7 | 68.1 | 657,987 | 231,449 | 32,114 | 4,913 | 48 |
| Total in Mutants | 2,155,300 | 1,115,559 | 92,340 | 16,709 | 1,444 |
| Chromsome | Variants per Mbp |
|---|---|
| 2L | 11023 |
| 2R | 9848 |
| 3L | 11043 |
| 3R | 8554 |
| 4 | 3331 |
| X | 8108 |
| Y | 72 |

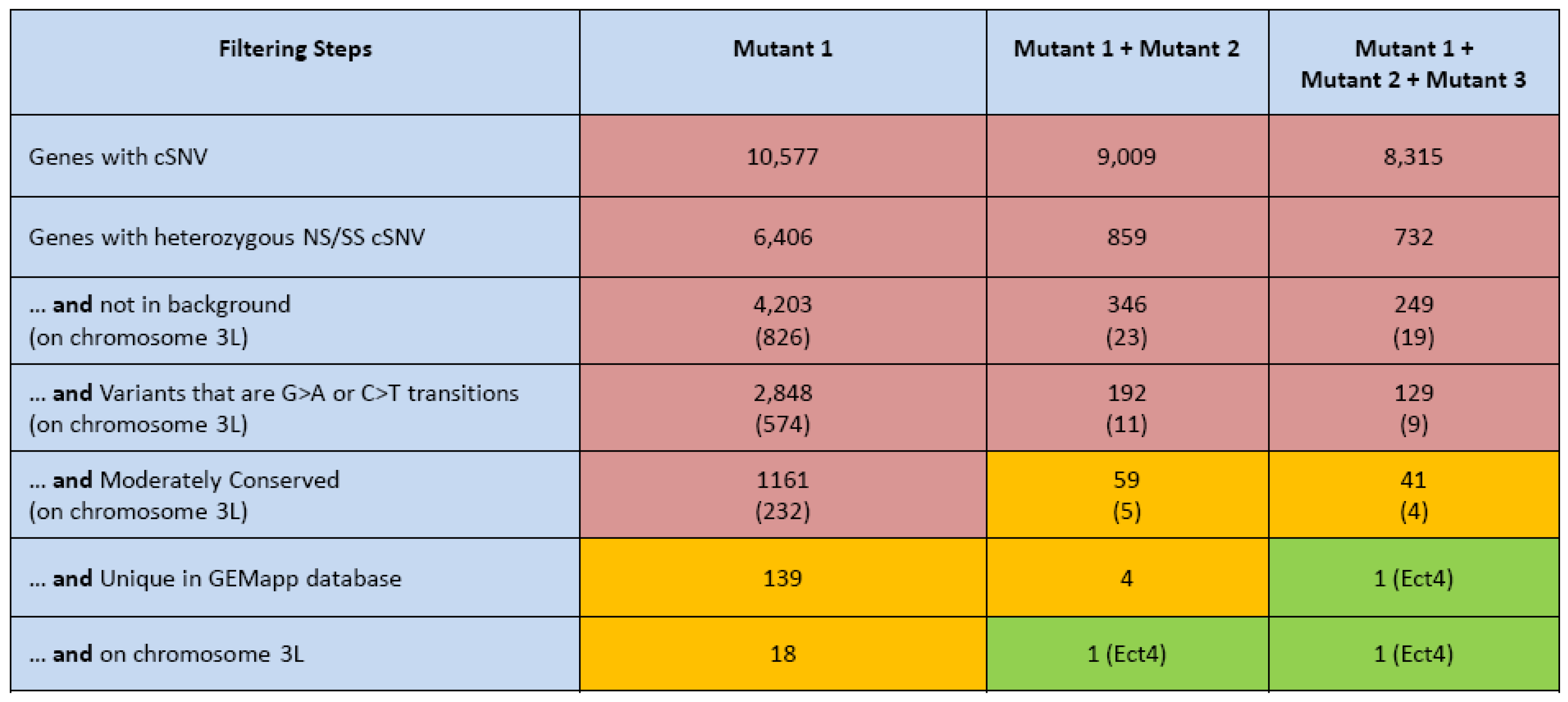
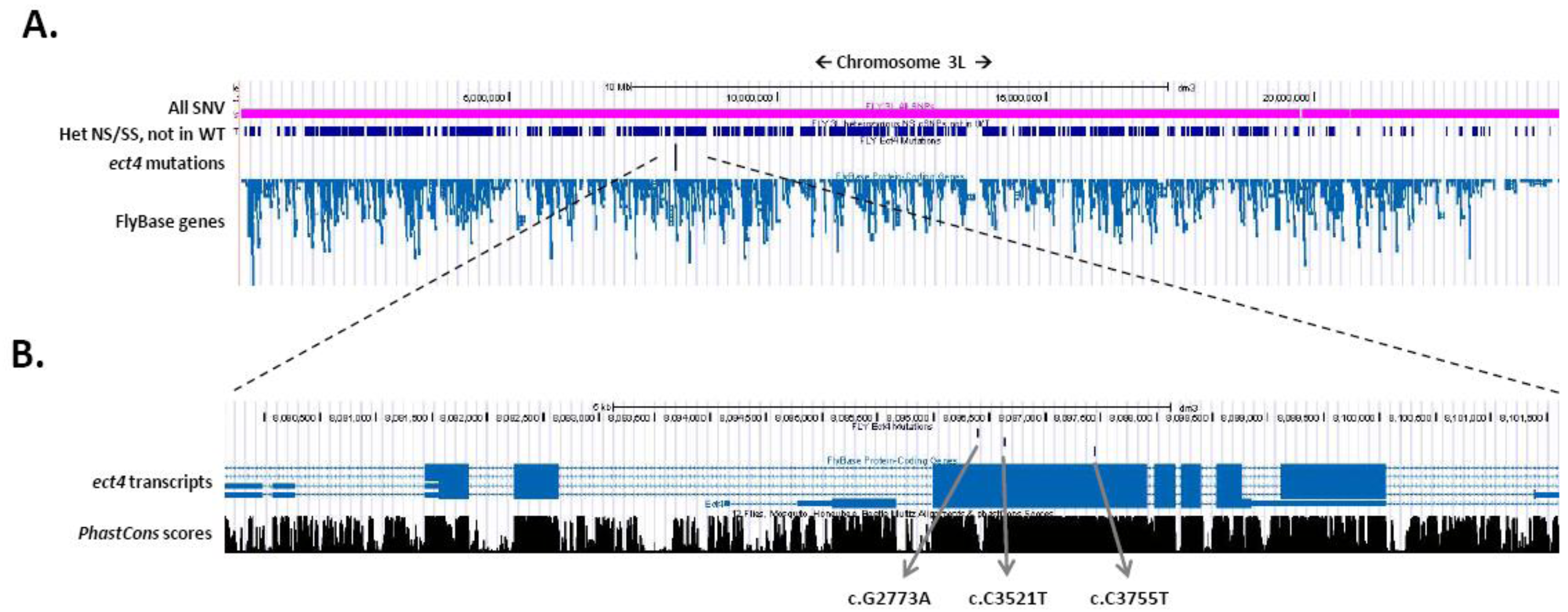
3.2. Gene Identification Applying WGS
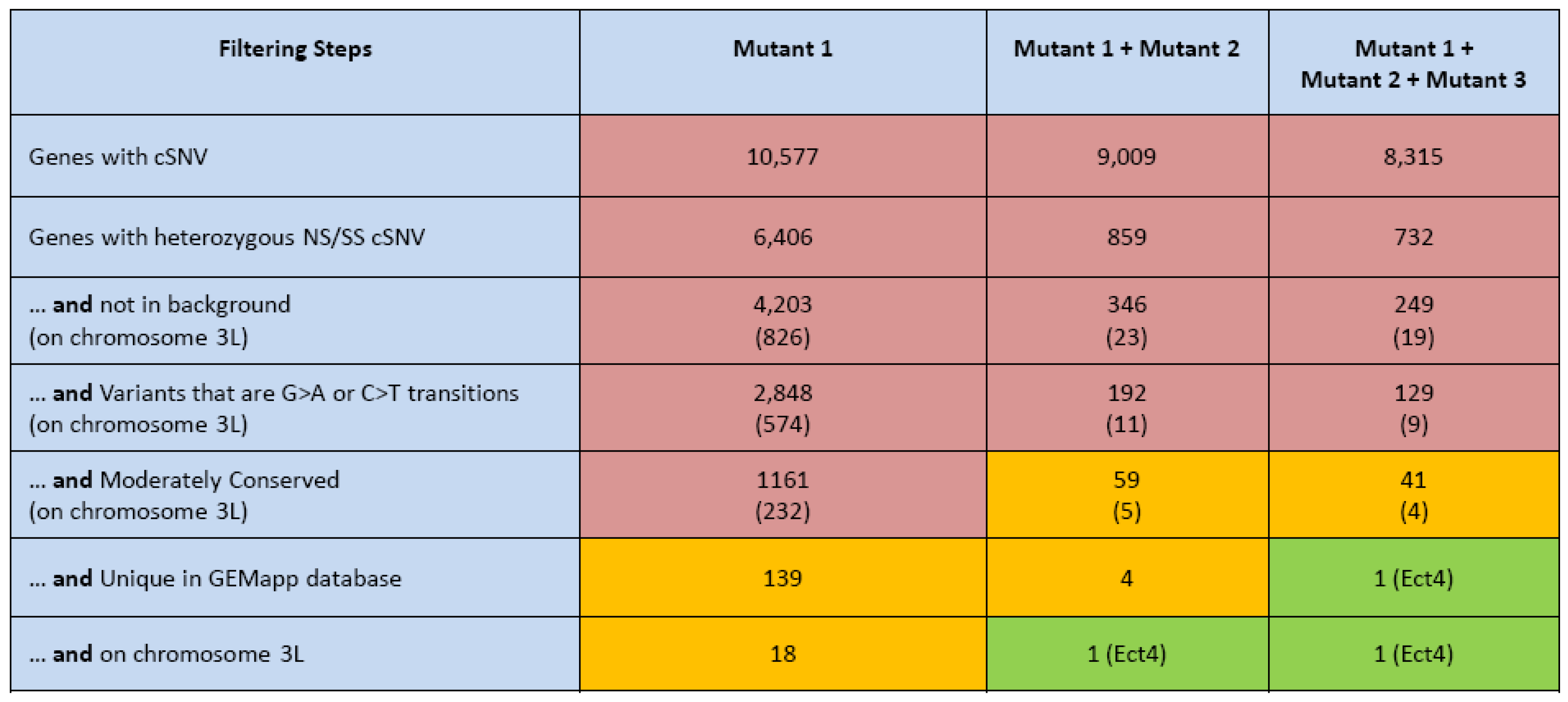
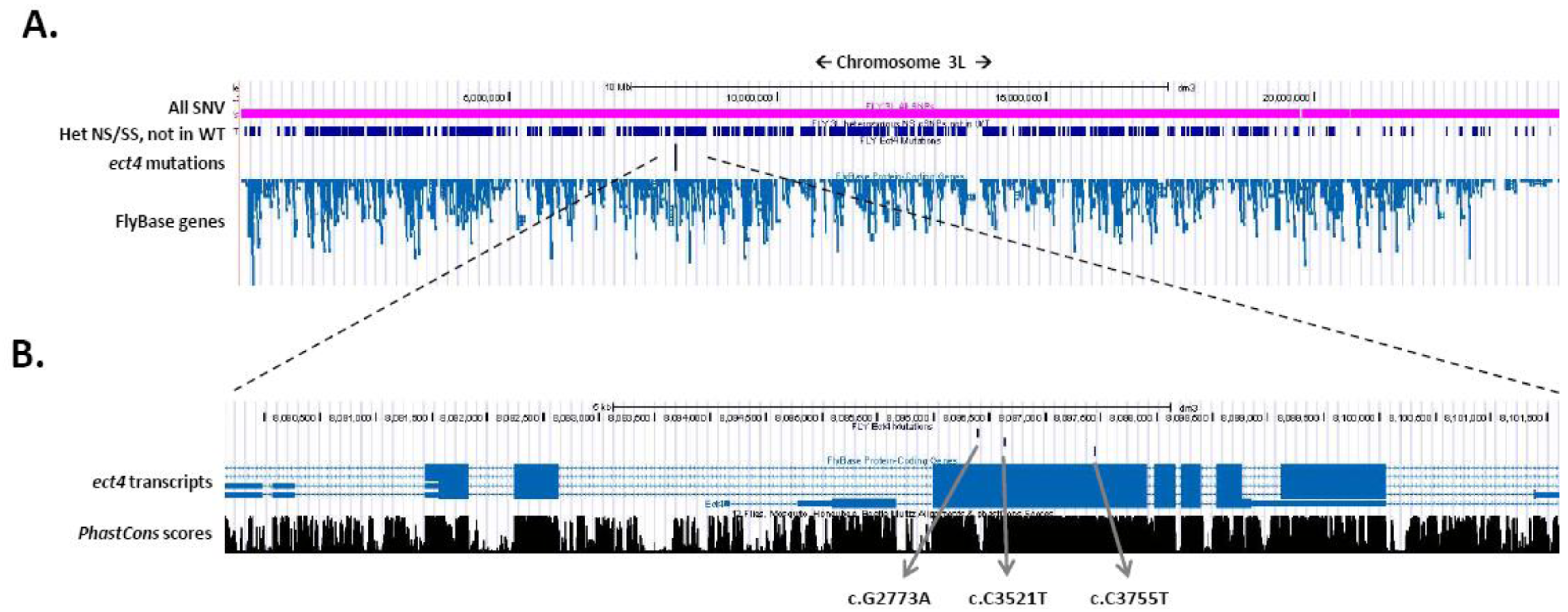
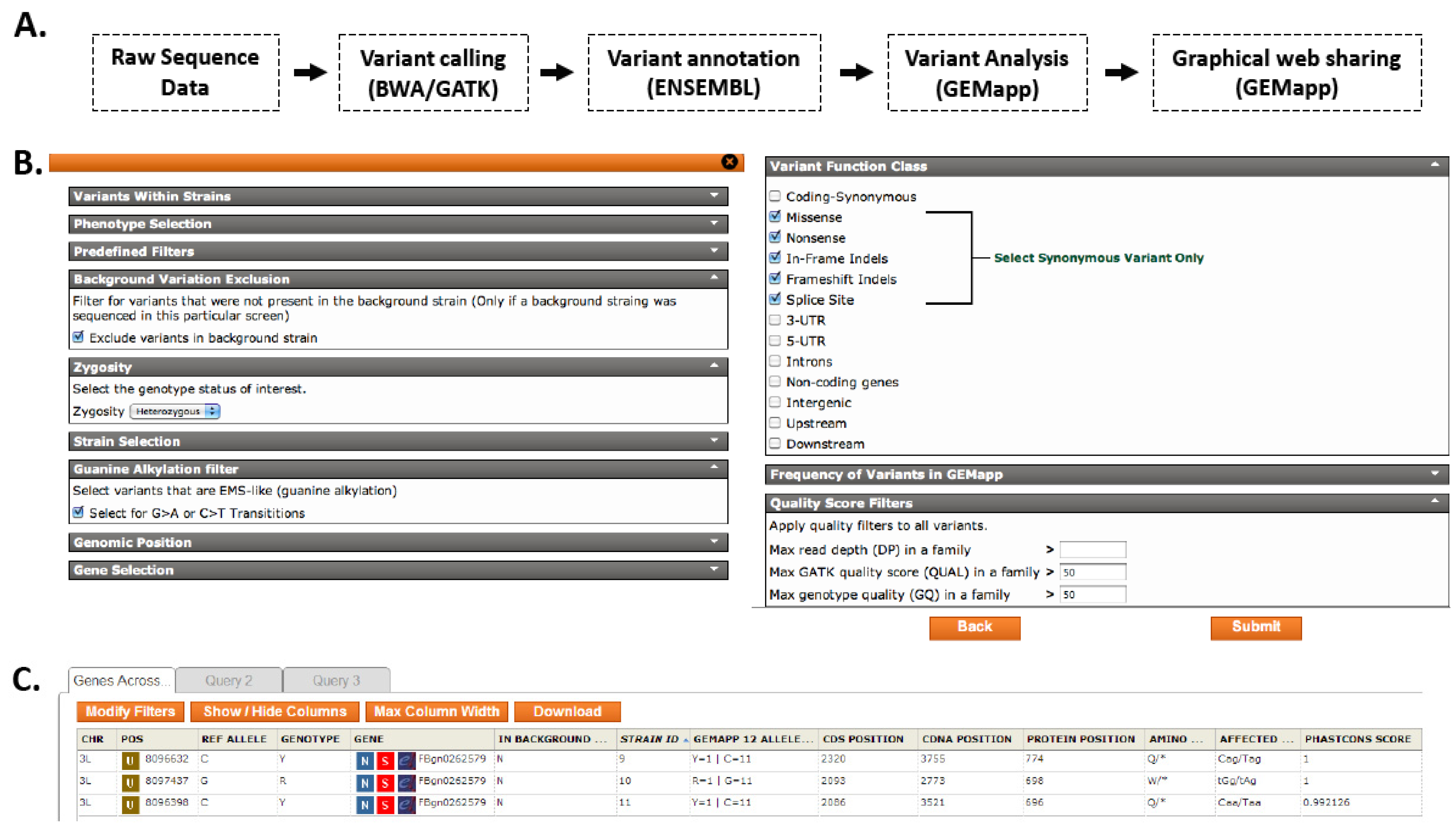
3.3. The Genomes Management Application (GEM.app)—A Novel Tool for Rapid Genome Analysis and Comparison

4. Discussion
- If a single mutant line exists, the parallel sequencing of the background strain and mapping to a chromosomal arm will reduce the number of genes carrying a strong coding variant to less than 20, possibly even identifying a strong mutation outright.
- It is most advantageous to add a second non-complementing strain to filter for genes that contain strong variants in both strains. In this case, further mapping of the gene is not even necessary in most cases (Figure 2).
- Adding more Drosophila genomes, related or unrelated to a project, has a great potential to further eliminate variants with a low frequency in the Drosophila population; or alternatively, nucleotide positions that are frequently hit by the EMS mutagen.
5. Conclusions
Acknowledgments
References
- Rubin, G.M.; Lewis, E.B. A brief history of Drosophila's contributions to genome research. Science 2000, 287, 2216–2218. [Google Scholar] [CrossRef]
- St. Johnston, D. The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 2002, 3, 176–188. [Google Scholar] [CrossRef]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar]
- Blumenstiel, J.P.; Noll, A.C.; Griffiths, J.A.; Perera, A.G.; Walton, K.N.; Gilliland, W.D.; Hawley, R.S.; Staehling-Hampton, K. Identification of EMS-induced mutations in Drosophila melanogaster by whole-genome sequencing. Genetics 2009, 182, 25–32. [Google Scholar] [CrossRef]
- Wang, H.; Chattopadhyay, A.; Li, Z.; Daines, B.; Li, Y.; Gao, C.; Gibbs, R.; Zhang, K.; Chen, R. Rapid identification of heterozygous mutations in Drosophila melanogaster using genomic capture sequencing. Genome Res. 2010, 20, 981–988. [Google Scholar] [CrossRef]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; MacDonald, J.M.; Ziegenfuss, J.S.; Milde, S.; Hou, Y.J.; Nathan, C.; Ding, A.; Brown, R.H., Jr.; Conforti, L.; Coleman, M.; Tessier-Lavigne, M.; Zuchner, S.; Freeman, M.R. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar]
- Smith, D.R.; Quinlan, A.R.; Peckham, H.E.; Makowsky, K.; Tao, W.; Woolf, B.; Shen, L.; Donahue, W.F.; Tusneem, N.; Stromberg, M.P.; Stewart, D.A.; et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008, 18, 1638–1642. [Google Scholar] [CrossRef]
- Drake, J.; Baltz, R. Biochemistry of Mutagenesis. Annu. Rev. Biochem. 1976, 45, 11–37. [Google Scholar] [CrossRef]
- Cooper, J.L.; Greene, E.A.; Till, B.J.; Codomo, C.A.; Wakimoto, B.T.; Henikoff, S. Retention of induced mutations in a Drosophila reverse-genetic resource. Genetics 2008, 180, 661–667. [Google Scholar] [CrossRef]
- Bentley, A.; MacLennan, B.; Calvo, J.; Dearolf, C.R. Targeted recovery of mutations in Drosophila. Genetics 2000, 156, 1169–1173. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; DePristo, M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; Weinstock, G.M.; Wilson, R.K.; Gibbs, R.A.; Kent, W.J.; Miller, W.; Haussler, D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genoms. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; Jensen, L.J.; von Mering, C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gonzalez, M.A.; Van Booven, D.; Hulme, W.; Ulloa, R.H.; Lebrigio, R.F.A.; Osterloh, J.; Logan, M.; Freeman, M.; Zuchner, S. Whole Genome Sequencing and a New Bioinformatics Platform Allow for Rapid Gene Identification in D. melanogaster EMS Screens. Biology 2012, 1, 766-777. https://doi.org/10.3390/biology1030766
Gonzalez MA, Van Booven D, Hulme W, Ulloa RH, Lebrigio RFA, Osterloh J, Logan M, Freeman M, Zuchner S. Whole Genome Sequencing and a New Bioinformatics Platform Allow for Rapid Gene Identification in D. melanogaster EMS Screens. Biology. 2012; 1(3):766-777. https://doi.org/10.3390/biology1030766
Chicago/Turabian StyleGonzalez, Michael A., Derek Van Booven, William Hulme, Rick H. Ulloa, Rafael F. Acosta Lebrigio, Jeannette Osterloh, Mary Logan, Marc Freeman, and Stephan Zuchner. 2012. "Whole Genome Sequencing and a New Bioinformatics Platform Allow for Rapid Gene Identification in D. melanogaster EMS Screens" Biology 1, no. 3: 766-777. https://doi.org/10.3390/biology1030766