Structure-Dependent Immune Modulatory Activity of Protegrin-1 Analogs

Abstract
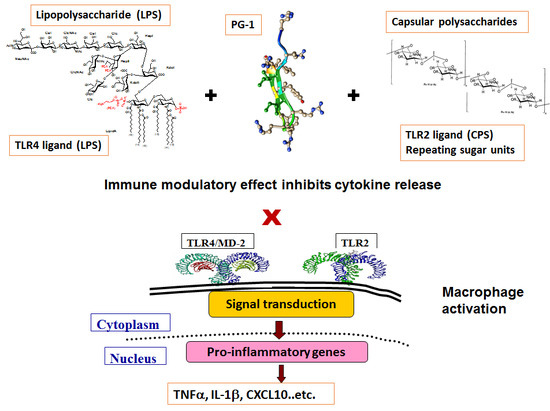
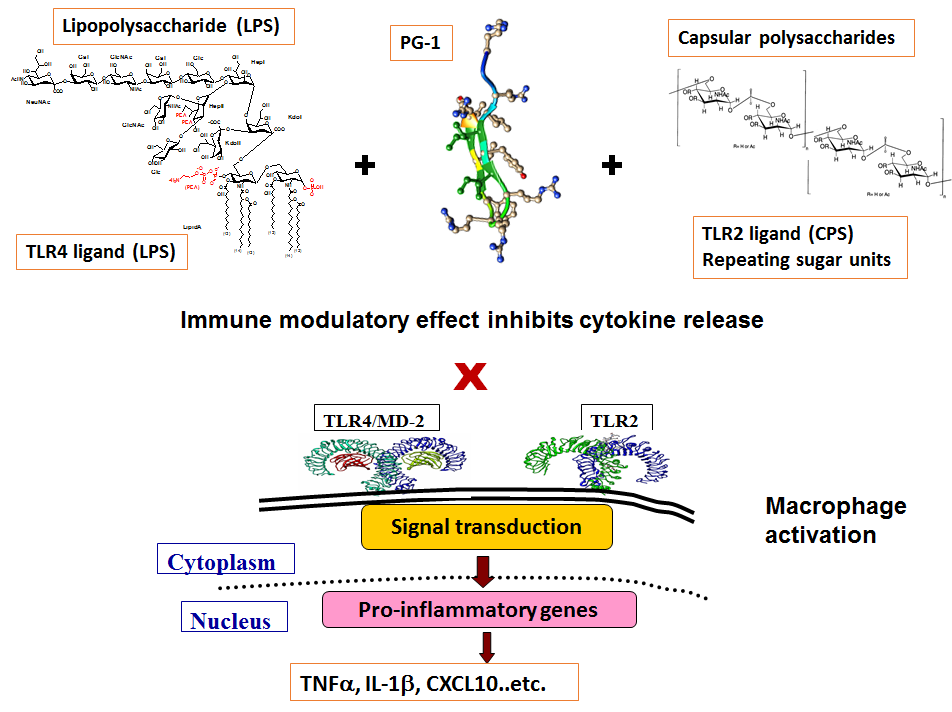
:
1. Introduction
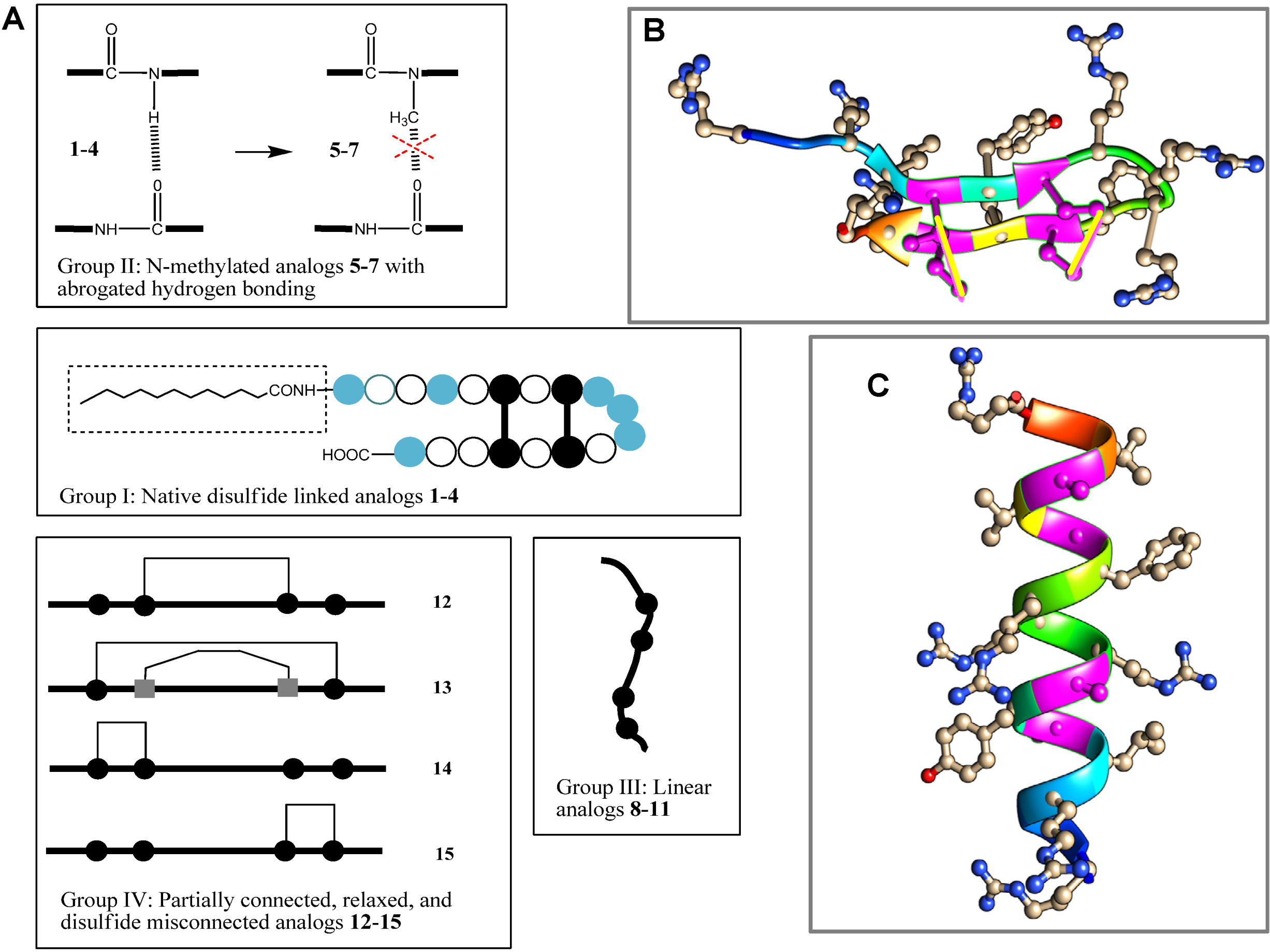
 = arginine or homoarginine (4);
= arginine or homoarginine (4);  = cysteine or
= cysteine or  homocysteine (13); absence of H-bonding in 5–7 is indicated (
homocysteine (13); absence of H-bonding in 5–7 is indicated (  )]; the dotted box indicates the acyl chain in analog 3. Three-dimensional structure model of PG-1 (B) and of its linearized non-cysteine containing analog 8 (C) with alanine replacements. The 3D structure of the PG-1 β-hairpin fold is predicted by I-TASSER based on the published crystal structures (PDB # 1PG1 and 1ZY6) and visualized using Chimera software. Cysteine residues that form disulfide bridges are magenta-colored. The 3D structure of a linearized analog 8 adopting a coil fold is achieved when four alanine residues are replaced by cysteines. The coil fold is predicted by I-TASSER and visualized by Chimera. Alanine residues are magenta-colored.
= arginine or homoarginine (4); = cysteine or homocysteine (13); absence of H-bonding in 5–7 is indicated ( )]; the dotted box indicates the acyl chain in analog 3. Three-dimensional structure model of PG-1 (B) and of its linearized non-cysteine containing analog 8 (C) with alanine replacements. The 3D structure of the PG-1 β-hairpin fold is predicted by I-TASSER based on the published crystal structures (PDB # 1PG1 and 1ZY6) and visualized using Chimera software. Cysteine residues that form disulfide bridges are magenta-colored. The 3D structure of a linearized analog 8 adopting a coil fold is achieved when four alanine residues are replaced by cysteines. The coil fold is predicted by I-TASSER and visualized by Chimera. Alanine residues are magenta-colored.
)]; the dotted box indicates the acyl chain in analog 3. Three-dimensional structure model of PG-1 (B) and of its linearized non-cysteine containing analog 8 (C) with alanine replacements. The 3D structure of the PG-1 β-hairpin fold is predicted by I-TASSER based on the published crystal structures (PDB # 1PG1 and 1ZY6) and visualized using Chimera software. Cysteine residues that form disulfide bridges are magenta-colored. The 3D structure of a linearized analog 8 adopting a coil fold is achieved when four alanine residues are replaced by cysteines. The coil fold is predicted by I-TASSER and visualized by Chimera. Alanine residues are magenta-colored.
= arginine or homoarginine (4); = cysteine or homocysteine (13); absence of H-bonding in 5–7 is indicated ( )]; the dotted box indicates the acyl chain in analog 3. Three-dimensional structure model of PG-1 (B) and of its linearized non-cysteine containing analog 8 (C) with alanine replacements. The 3D structure of the PG-1 β-hairpin fold is predicted by I-TASSER based on the published crystal structures (PDB # 1PG1 and 1ZY6) and visualized using Chimera software. Cysteine residues that form disulfide bridges are magenta-colored. The 3D structure of a linearized analog 8 adopting a coil fold is achieved when four alanine residues are replaced by cysteines. The coil fold is predicted by I-TASSER and visualized by Chimera. Alanine residues are magenta-colored.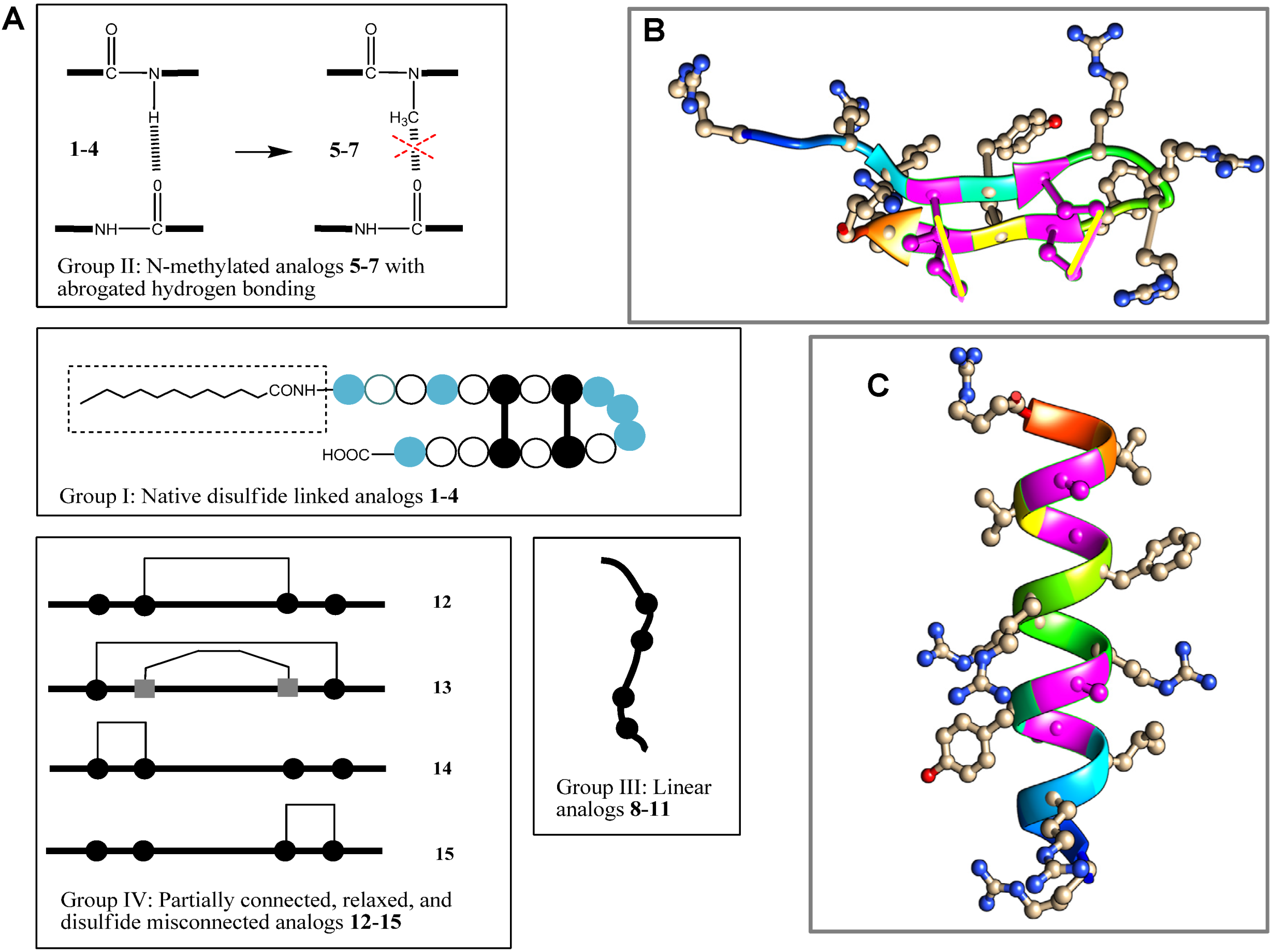
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp-ound | Sequence | LOS activity inhibition (%) | CPS activity inhibition (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 2 3 4 5 | 6 | 7 | 8 | 9 10 11 | 12 | 13 | 14 | 15 | 16 17 18 | |||
| 1 (I) | NH2-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 99.9 | 99 |
| 2 (I) | NH2-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 98.3 | 94.7 |
| 3 (I) | C12-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 74.2 | 22.5 |
| 4 (I) | NH2-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 100 | 100 |
| 5 (II) | NH2-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 60 | 52 |
| 6 (II) | NH2-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 16 | 21 |
| 7 (II) | NH2-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 0 | 15 |
| 8 (III) | NH2-R G G R L | A | Y | A | R R R | F | A | V | A | V G R-CONH2 | 33 | 23 |
| 9 (III) | NH2-R G G R L | C(Me) | Y | C(Me) | R R R | F | C(Me) | V | C(Me) | V G R-CONH2 | 0 | 5.7 |
| 10 (III) | NH2-R G G R L | M | Y | M | R R R | F | M | V | M | V G R-CONH2 | 4.6 | 24 |
| 11 (III) | NH2-R G G R L | M(O) | Y | M(O) | R R R | F | M(O) | V | M(O) | V G R-CONH2 | 20.5 | 4.6 |
| 12 (IV) | NH2-R G G R L | C(Me) | Y | C | R R R | F | C | V | C(Me) | V G R-CONH2 | 84.2 | 84.7 |
| 13 (IV) | NH2-R G G R L | C | Y | C | R R R | F | C | V | C | V G R-CONH2 | 99.9 | 88.4 |
| 14 (IV) | NH2-R G G R L | C | Y | C | R R R | F | C(Me) | V | C(Me) | V G R-CONH2 | 99.9 | 99.9 |
| 15 (IV) | NH2-R G G R L | C(Me) | Y | C(Me) | R R R | F | C | V | C | V G R-CONH2 | 88.4 | 91.6 |
 : homocysteine;
: homocysteine;  : S-methyl-cysteine; C12: dodecanoyl; F: N-methyl-phenylalanine; M(O): methionine oxide; R: homoarginine; and Y: N-methyl-tyrosine.
: S-methyl-cysteine; C12: dodecanoyl; F: N-methyl-phenylalanine; M(O): methionine oxide; R: homoarginine; and Y: N-methyl-tyrosine.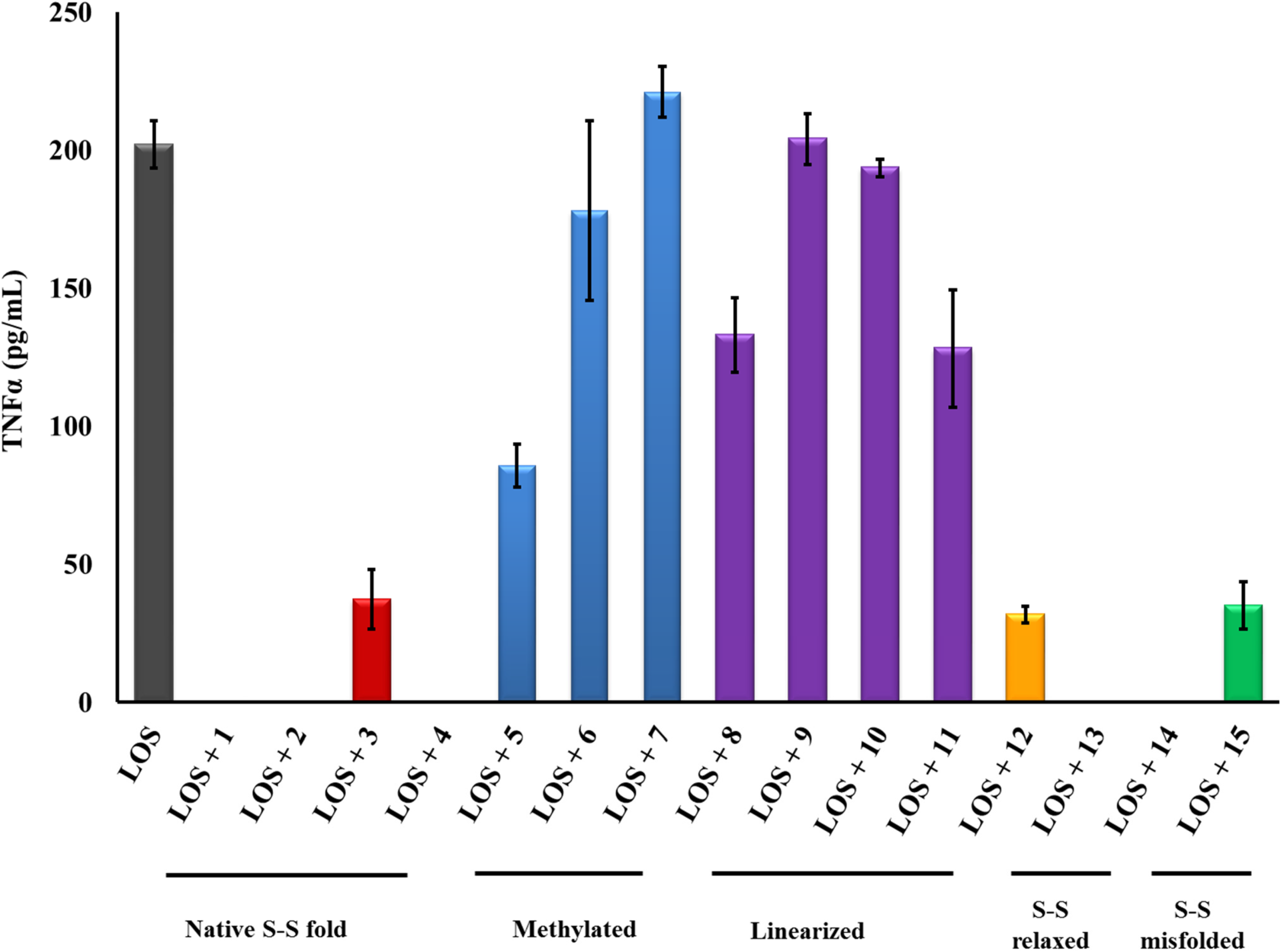
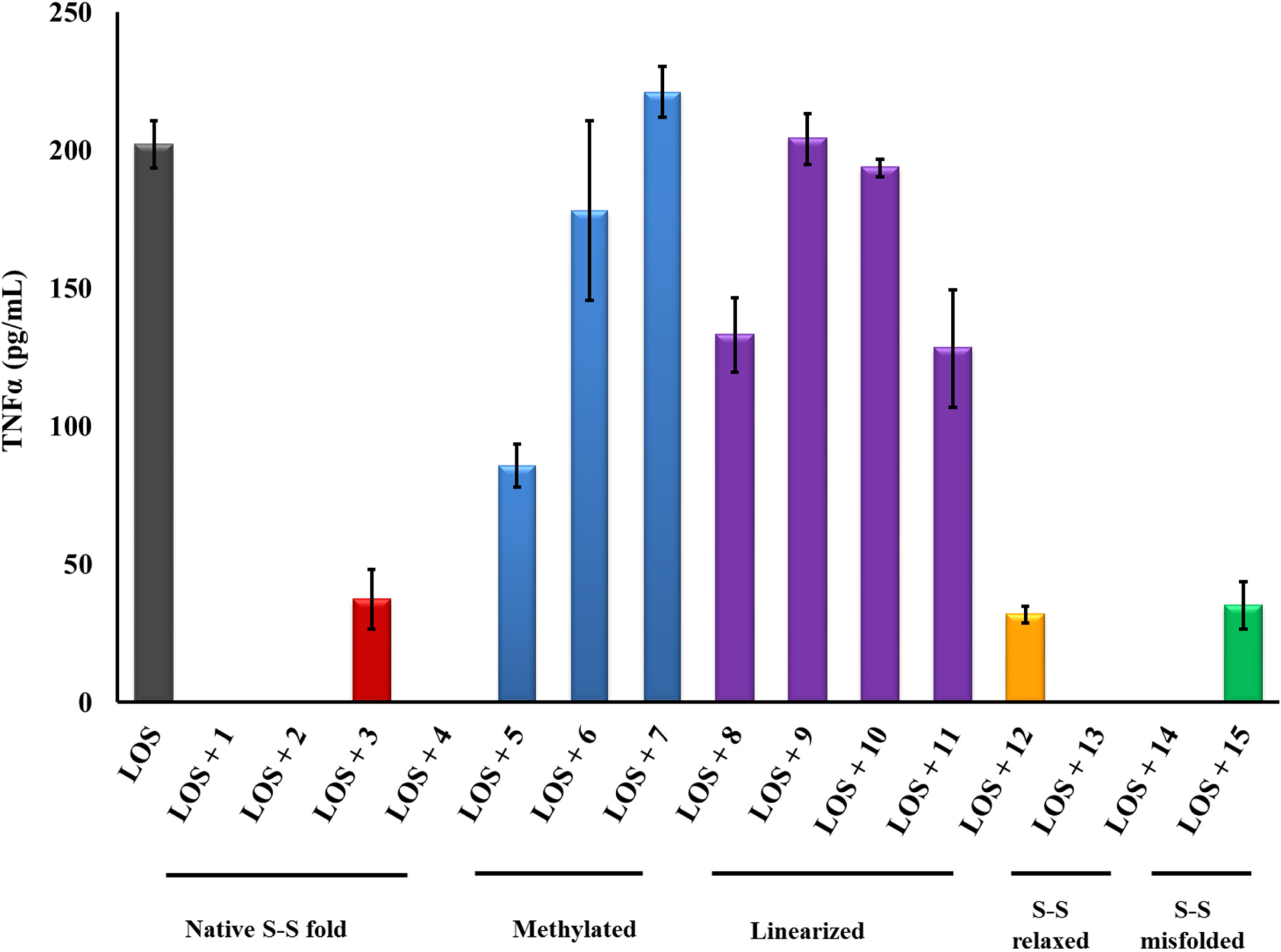
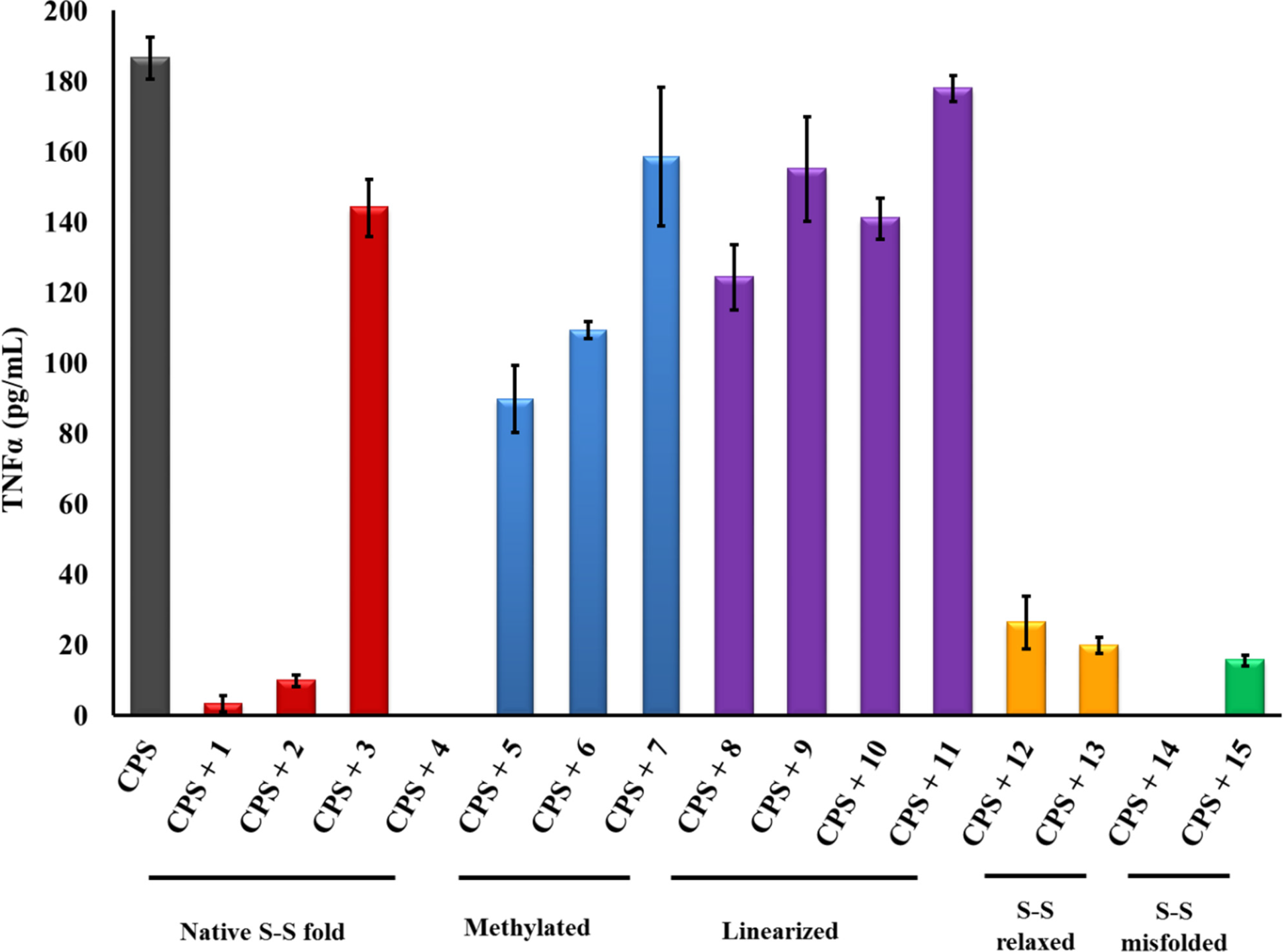
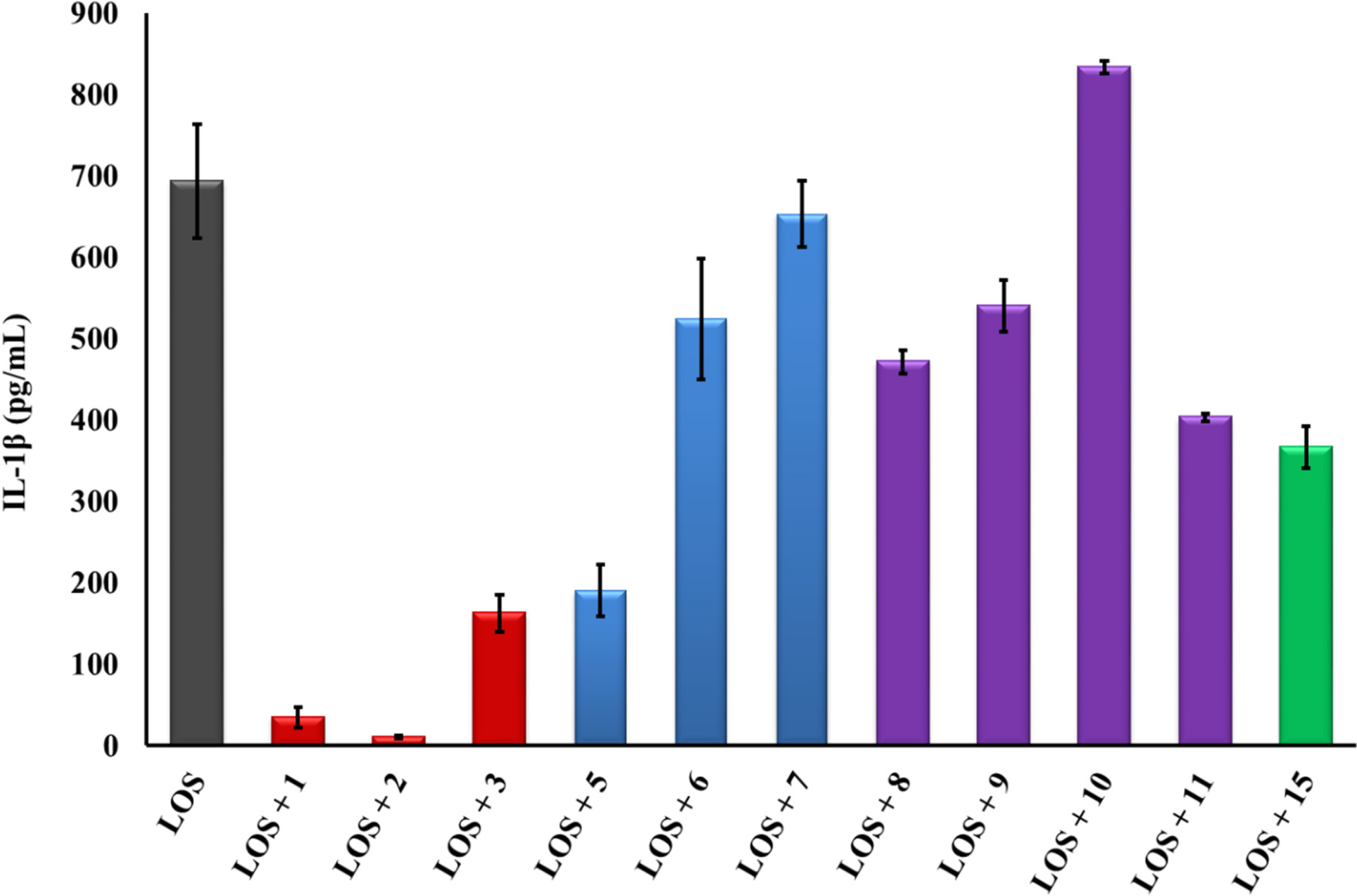
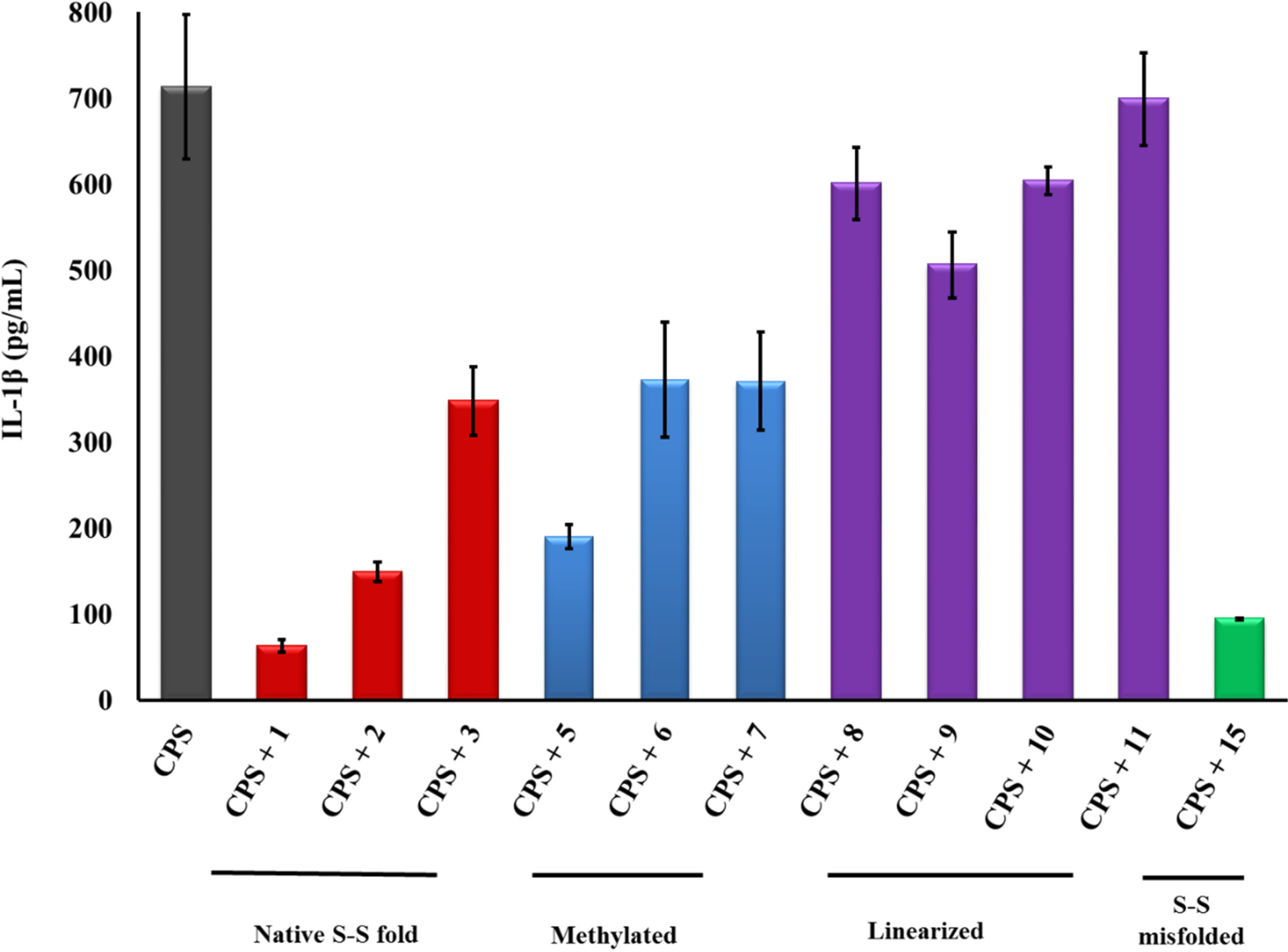
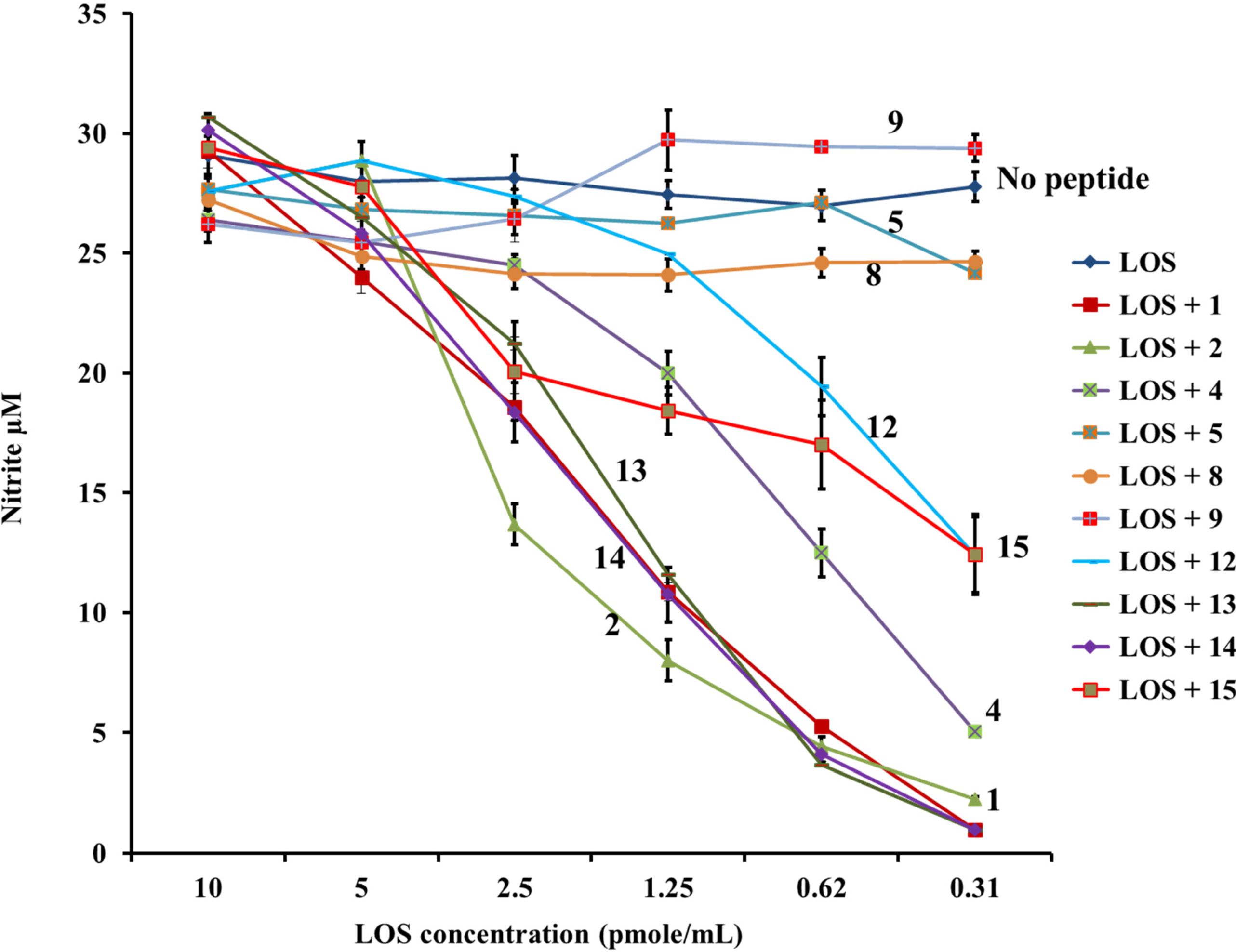
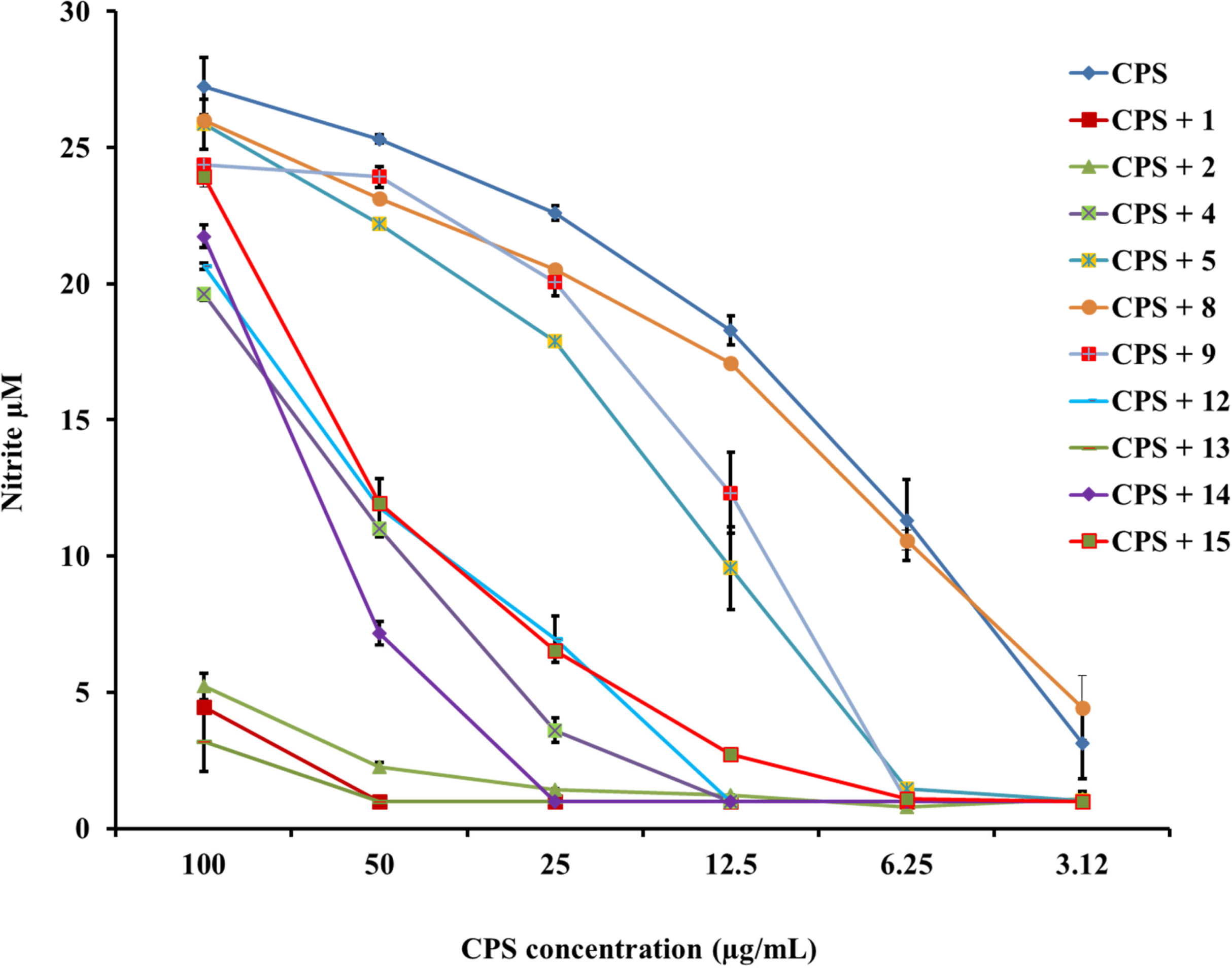
2.1. β-hairpin Analogs with Impaired H-Bonding
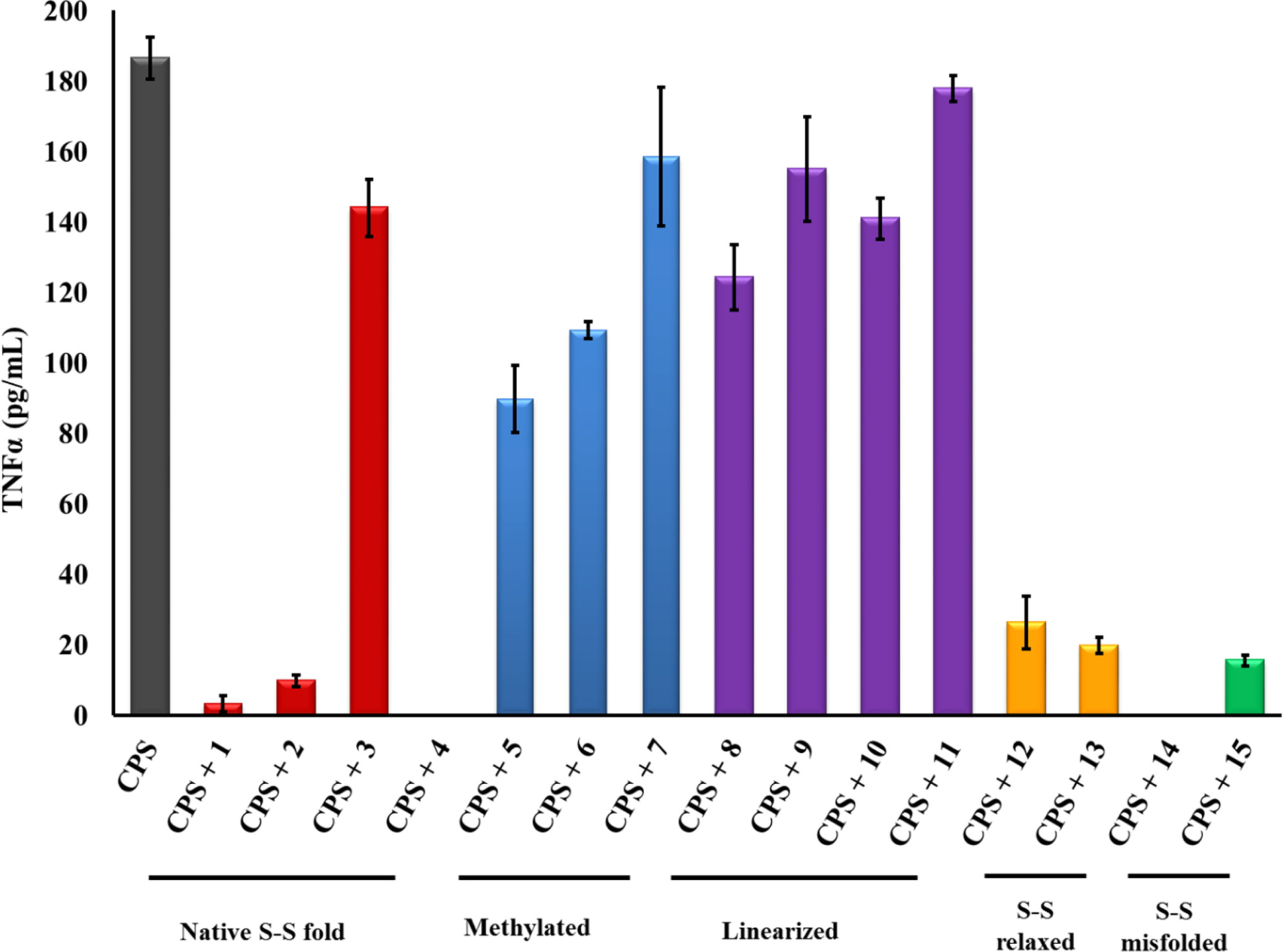
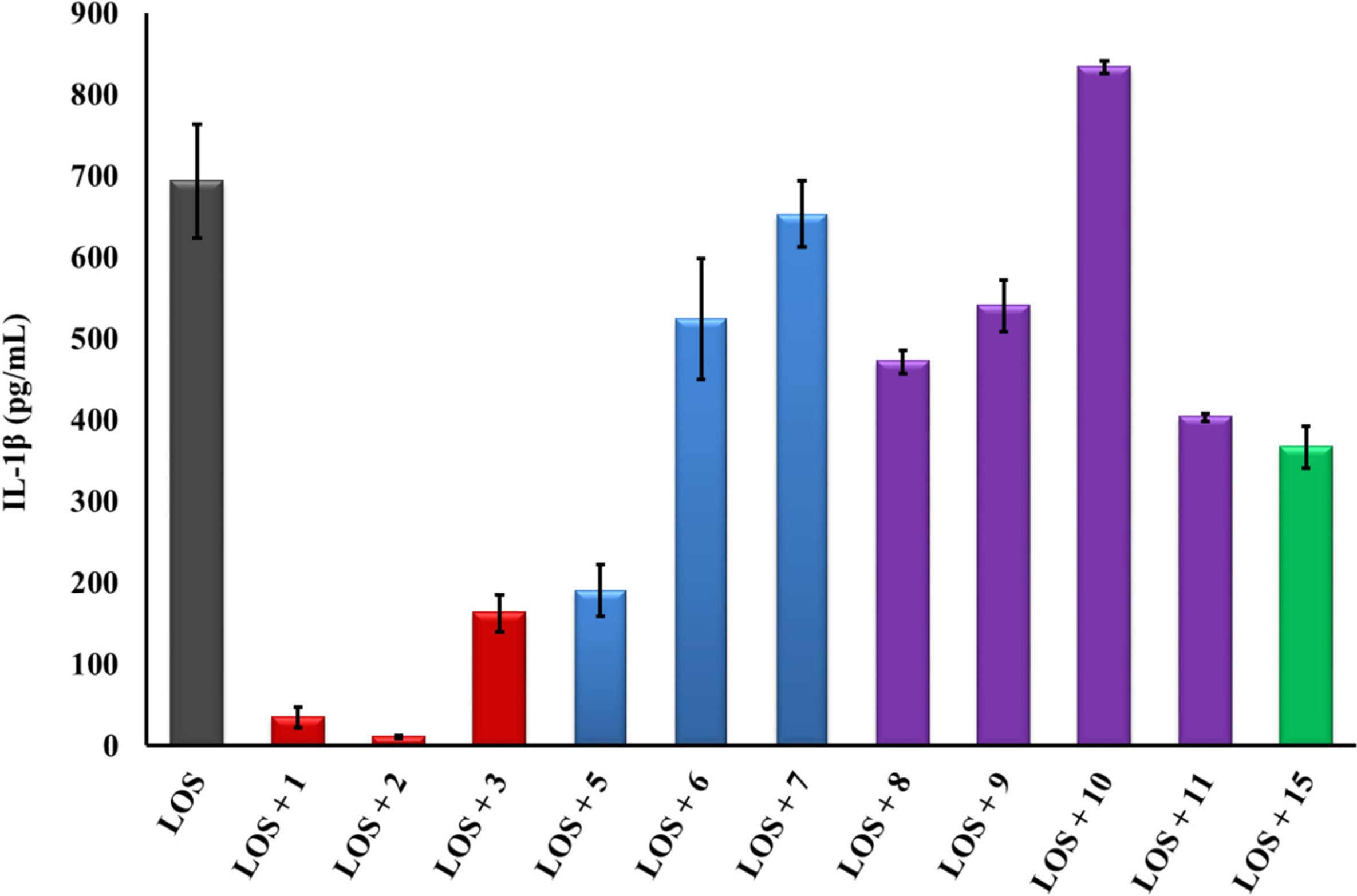
2.2. Linear PG-1 Analogs Lacking β-Hairpin
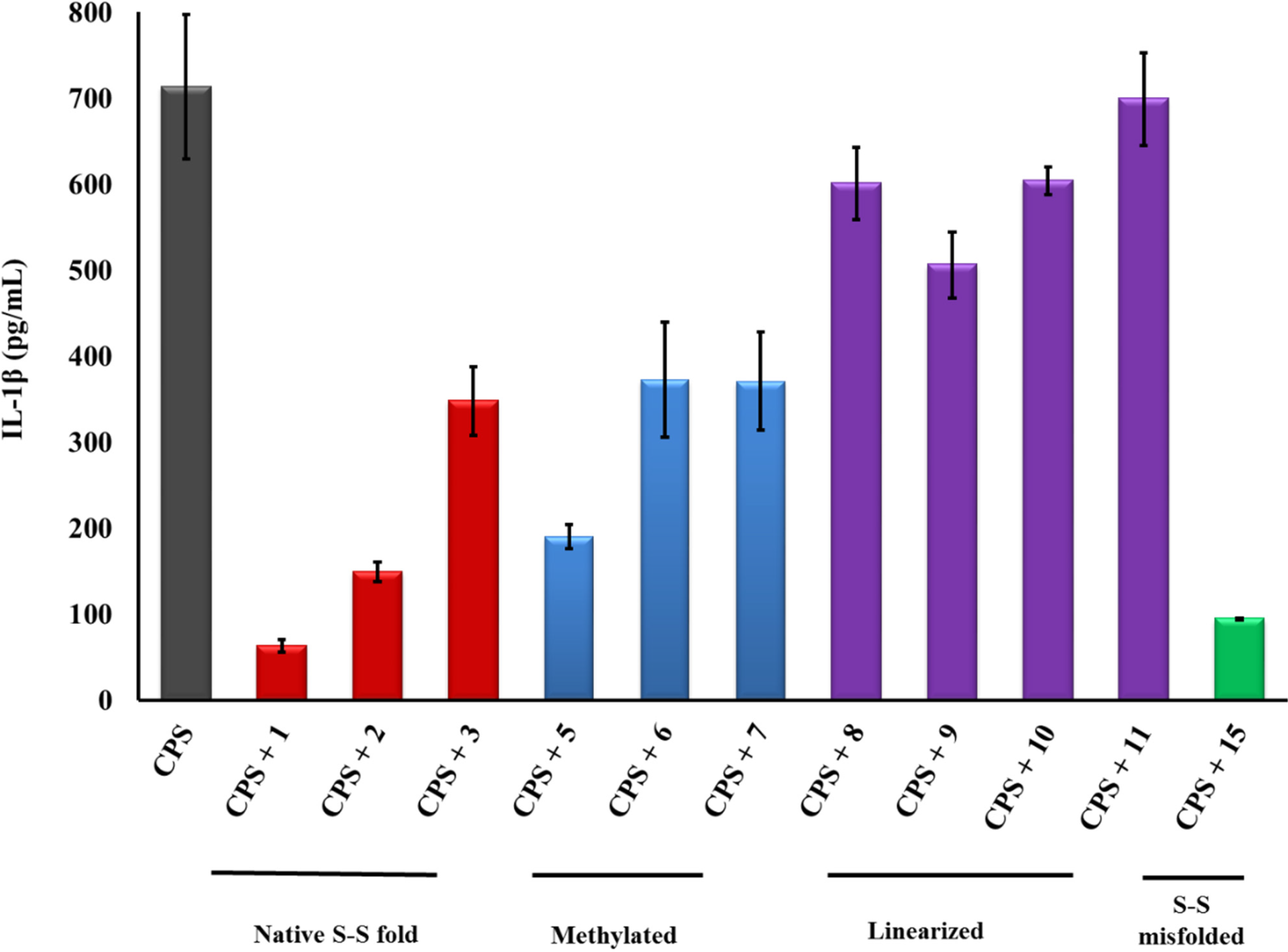
2.3. PG-1 Analogs with Altered Disulfide Connectivity

3. Experimental Section
3.1. Reagents
3.2. PG-1 Analog Synthesis
3.3. Cell Cultures
3.4. Cellular Activation
3.5. Cytokine Profiles
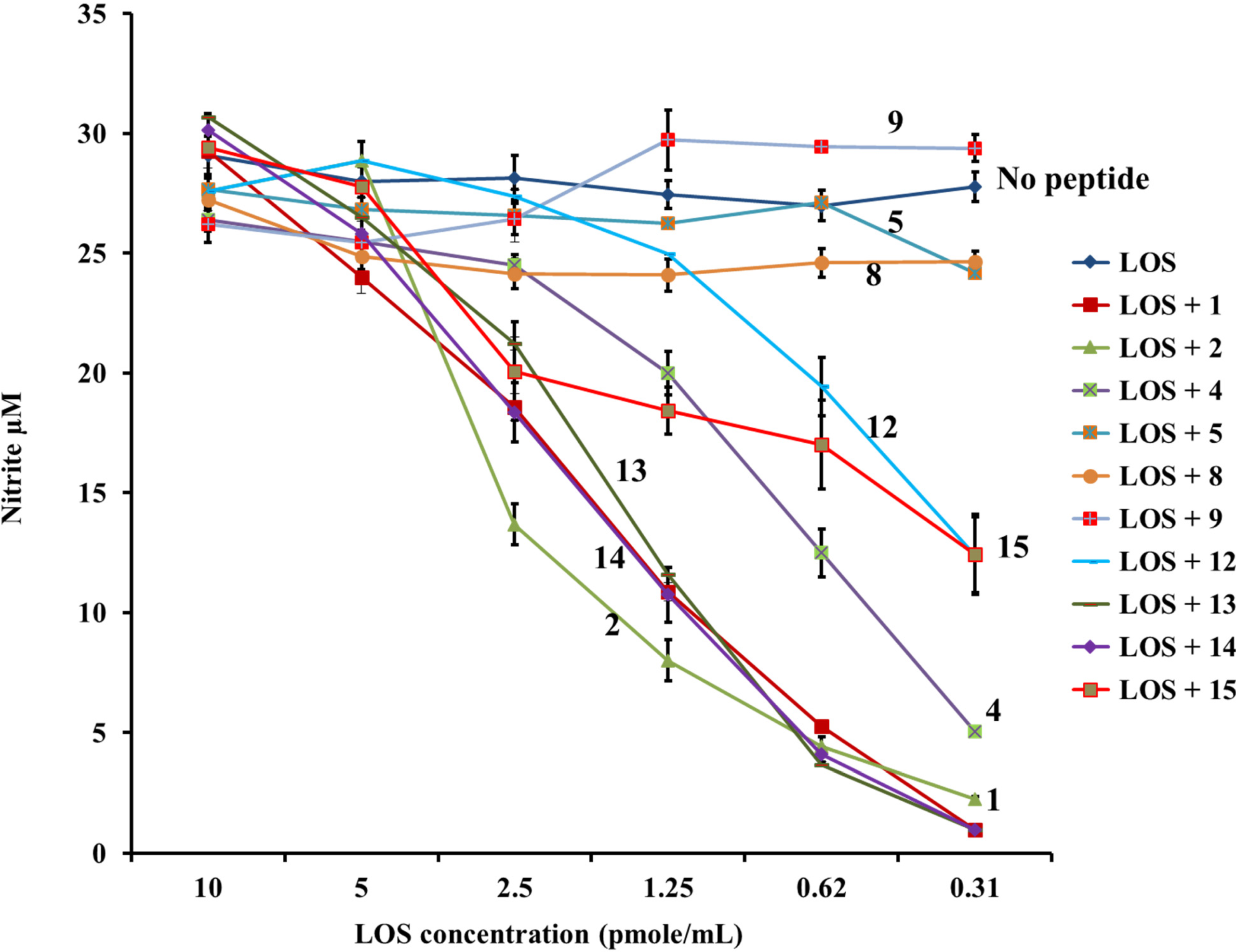
3.6. Nitric Oxide Induction by Murine Macrophages
3.7. Cellular Viability Assessment
3.8. Computational Modeling of PG-1’s 3-D Structure and Its Linearized Analogs
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beutler, B. Innate immune responses to microbial poisons: Discovery and function of the Toll-like receptors. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J. Innate immunity as a key element in host defense against methicillin resistant Staphylococcus aureus. Minerva Pediatr. 2009, 61, 503–514. [Google Scholar] [PubMed]
- Bartlett, J.A.; Fischer, A.J.; McCray, P.B., Jr. Innate immune functions of the airway epithelium. Contrib. Microbiol. 2008, 15, 147–163. [Google Scholar] [PubMed]
- Zasloff, M. Antibiotic peptides as mediators of innate immunity. Curr. Opin. Immunol. 1992, 4, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins and other antimicrobial peptides: A historical perspective and an update. Comb. Chem. High. Throughput Screen 2005, 8, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Chertov, O.; Yang, D.; Howard, O.M.; Oppenheim, J.J. Leukocyte granule proteins mobilize innate host defenses and adaptive immune responses. Immunol. Rev. 2000, 177, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Lu, W. Alpha-Defensins in human innate immunity. Immunol. Rev. 2012, 245, 84–112. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T. Cathelicidins: A family of endogenous antimicrobial peptides. Curr. Opin. Hematol. 2002, 9, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Miyasaki, K.T.; Lehrer, R.I. Beta-sheet antibiotic peptides as potential dental therapeutics. Int. J. Antimicrob Agents 1998, 9, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Lehrer, R.I. Antimicrobial peptides of vertebrates. Curr. Opin. Immunol. 1998, 10, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Lehrer, R. Cationic peptides: A new source of antibiotics. Trends Biotechnol. 1998, 16, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Adib-Conquy, M.; Cavaillon, J.M. Host inflammatory and anti-inflammatory response during sepsis. Pathol. Biol. 2012, 60, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Adib-Conquy, M.; Adrie, C.; Moine, P.; Asehnoune, K.; Fitting, C.; Pinsky, M.R.; Dhainaut, J.F.; Cavaillon, J.M. NF-KappaB expression in mononuclear cells of patients with sepsis resembles that observed in lipopolysaccharide tolerance. Am. J. Respir. Crit. Care Med. 2000, 162, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Hirsiger, S.; Simmen, H.P.; Werner, C.M.; Wanner, G.A.; Rittirsch, D. Danger signals activating the immune response after trauma. Mediat. Inflamm. 2012, 2012, e315941. [Google Scholar] [CrossRef]
- Mookherjee, N.; Brown, K.L.; Bowdish, D.M.; Doria, S.; Falsafi, R.; Hokamp, K.; Roche, F.M.; Mu, R.; Doho, G.H.; Pistolic, J.; et al. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J. Immunol. 2006, 176, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mookherjee, N.; Wee, K.; Bowdish, D.M.; Pistolic, J.; Li, Y.; Rehaume, L.; Hancock, R.E. Host defense peptide LL-37, in synergy with inflammatory mediator IL-1beta, augments immune responses by multiple pathways. J. Immunol. 2007, 179, 7684–7691. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Cirioni, O.; Ghiselli, R.; Mocchegiani, F.; Viticchi, C.; Orlando, F.; D'Amato, G.; del Prete, M.S.; Kamysz, W.; Lukasiak, J.; et al. Antiendotoxin activity of protegrin analog IB-367 alone or in combination with piperacillin in different animal models of septic shock. Peptides 2003, 24, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Cirioni, O.; Ghiselli, R.; Mocchegiani, F.; D'Amato, G.; del Prete, M.S.; Orlando, F.; Kamysz, W.; Lukasiak, J.; Saba, V.; et al. Administration of protegrin peptide IB-367 to prevent endotoxin induced mortality in bile duct ligated rats. Gut 2003, 52, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Weldon, S.; Buchanan, P.J.; Schock, B.; Ernst, R.K.; McAuley, D.F.; Tunney, M.M.; Irwin, C.R.; Elborn, J.S.; Taggart, C.C. Evaluation of the ability of LL-37 to neutralise LPS in vitro and ex vivo. PLoS One 2011, 6, e26525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zughaier, S.M.; Shafer, W.M.; Stephens, D.S. Antimicrobial peptides and endotoxin inhibit cytokine and nitric oxide release but amplify respiratory burst response in human and murine macrophages. Cell. Microbiol. 2005, 7, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Chow, L.N.; Mookherjee, N. Cationic host defence peptides: Multifaceted role in immune modulation and inflammation. J. Innate Immun. 2012, 4, 361–370. [Google Scholar] [PubMed]
- Semple, F.; MacPherson, H.; Webb, S.; Cox, S.L.; Mallin, L.J.; Tyrrell, C.; Grimes, G.R.; Semple, C.A.; Nix, M.A.; Millhauser, G.L.; et al. Human beta-defensin 3 affects the activity of pro-inflammatory pathways associated with MyD88 and TRIF. Eur. J. Immunol. 2011, 41, 3291–3300. [Google Scholar] [CrossRef] [PubMed]
- Semple, F.; Webb, S.; Li, H.N.; Patel, H.B.; Perretti, M.; Jackson, I.J.; Gray, M.; Davidson, D.J.; Dorin, J.R. Human beta-defensin 3 has immunosuppressive activity in vitro and in vivo. Eur. J. Immunol. 2010, 40, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, N.; Kaznessis, Y. Protegrin structure-activity relationships: Using homology models of synthetic sequences to determine structural characteristics important for activity. Peptides 2005, 26, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Fiddes, J.C. Protegrin antimicrobial peptides. Curr. Opin. Drug Discov. Dev. 2000, 3, e655. [Google Scholar]
- Yasin, B.; Lehrer, R.I.; Harwig, S.S.; Wagar, E.A. Protegrins: Structural requirements for inactivating elementary bodies of Chlamydia trachomatis. Infect. Immun. 1996, 64, 4863–4866. [Google Scholar] [PubMed]
- Harwig, S.S.; Waring, A.; Yang, H.J.; Cho, Y.; Tan, L.; Lehrer, R.I. Intramolecular disulfide bonds enhance the antimicrobial and lytic activities of protegrins at physiological sodium chloride concentrations. Eur. J. Biochem. 1996, 240, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, R.L.; Dieckmann, T.; Harwig, S.S.; Lehrer, R.I.; Eisenberg, D.; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996, 3, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, T.; He, Y.; Prieto, L. Membrane interactions and pore formation by the antimicrobial peptide protegrin. Biophys. J. 2013, 104, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Bolintineanu, D.S.; Vivcharuk, V.; Kaznessis, Y.N. Multiscale models of the antimicrobial Peptide protegrin-1 on gram-negative bacteria membranes. Int. J. Mol. Sci. 2012, 13, 11000–11011. [Google Scholar] [CrossRef] [PubMed]
- Gidalevitz, D.; Ishitsuka, Y.; Muresan, A.S.; Konovalov, O.; Waring, A.J.; Lehrer, R.I.; Lee, K.Y.C. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA 2003, 100, 6302–6307. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.L.; Ishitsuka, Y.; Cheng, Y.; Chien, K.; Waring, A.J.; Lehrer, R.I.; Lee, K.Y.C. Mechanism of supported membrane disruption by antimicrobial peptide protegrin-1. J. Phys. Chem. B 2006, 110, 21282–21286. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, Y.; Pham, D.S.; Waring, A.J.; Lehrer, R.I.; Lee, K.Y. Insertion selectivity of antimicrobial peptide protegrin-1 into lipid monolayers: Effect of head group electrostatics and tail group packing. Biochim. Biophys. Acta 2006, 1758, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Neville, F.; Ishitsuka, Y.; Hodges, C.S.; Konovalov, O.; Waring, A.J.; Lehrer, R.; Lee, K.Y.C.; Gidalevitz, D. Protegrin interaction with lipid monolayers: Grazing incidence X-ray diffraction and X-ray reflectivity study. Soft Matter 2008, 4, 1665–1674. [Google Scholar] [CrossRef]
- Hong, M.; Su, Y. Structure and dynamics of cationic membrane peptides and proteins: Insights from solid-state NMR. Protein Sci. 2011, 20, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Bolintineanu, D.; Hazrati, E.; Davis, H.T.; Lehrer, R.I.; Kaznessis, Y.N. Antimicrobial mechanism of pore-forming protegrin peptides: 100 pores to kill E. coli. Peptides 2010, 31, 1–8. [Google Scholar] [CrossRef]
- Mohanram, H.; Bhattacharjya, S. Cysteine deleted protegrin-1 (CDP-1): Anti-bacterial activity, outer-membrane disruption and selectivity. Biochim. Biophys. Acta 2014, 1840, 3006–3016. [Google Scholar] [CrossRef] [PubMed]
- Tamamura, H.; Murakami, T.; Horiuchi, S.; Sugihara, K.; Otaka, A.; Takada, W.; Ibuka, T.; Waki, M.; Yamamoto, N.; Fujii, N. Synthesis of protegrin-related peptides and their antibacterial and anti-human immunodeficiency virus activity. Chem. Pharm. Bull. 1995, 43, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Arce, F.T.; Mustata, M.; Ramachandran, S.; Capone, R.; Nussinov, R.; Lal, R. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 2011, 100, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiselli, R.; Giacometti, A.; Cirioni, O.; Mocchegiani, F.; Silvestri, C.; Orlando, F.; Kamysz, W.; Licci, A.; Nadolski, P.; Vittoria, A.D.; et al. Pretreatment with the protegrin IB-367 affects Gram-positive biofilm and enhances the therapeutic efficacy of linezolid in animal models of central venous catheter infection. J. Parenter Enteral Nutr. 2007, 31, 463–468. [Google Scholar] [CrossRef]
- Bolintineanu, D.S.; Langham, A.A.; Davis, H.T.; Kaznessis, Y.N. Molecular dynamics simulations of three protegrin-type antimicrobial peptides: Interplay between charges at the termini, beta-sheet structure and amphiphilic interactions. Mol. Simul. 2007, 33, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Zhu, X.; Chou, S.; Shan, A.; Li, W.; Jiang, J. Antimicrobial potency and selectivity of simplified symmetric-end peptides. Biomaterials 2014, 35, 8028–8039. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Demoulins, T.; Python, S.; Summerfield, A. Porcine cathelicidins efficiently complex and deliver nucleic acids to plasmacytoid dendritic cells and can thereby mediate bacteria-induced IFN-alpha responses. J. Immunol. 2014, 193, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.R.; Epand, R.F.; Weisblum, B.; Epand, R.M.; Gellman, S.H. Roles of salt and conformation in the biological and physicochemical behavior of protegrin-1 and designed analogues: Correlation of antimicrobial, hemolytic, and lipid bilayer-perturbing activities. Biochemistry 2006, 45, 15718–15730. [Google Scholar]
- Bogucka, K.; Krolicka, A.; Kamysz, W.; Ossowski, T.; Lukasiak, J.; Lojkowska, E. Activities of synthetic peptides against human pathogenic bacteria. Pol. J. Microbiol. 2004, 53, 41–44. [Google Scholar] [PubMed]
- Steinstraesser, L.; Klein, R.D.; Aminlari, A.; Fan, M.H.; Khilanani, V.; Remick, D.G.; Su, G.L.; Wang, S.C. Protegrin-1 enhances bacterial killing in thermally injured skin. Crit. Care Med. 2001, 29, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Mosca, D.A.; Hurst, M.A.; So, W.; Viajar, B.S.; Fujii, C.A.; Falla, T.J. IB-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob Agents Chemother. 2000, 44, 1803–1808. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Falla, T.J.; Liu, H.; Hurst, M.A.; Fujii, C.A.; Mosca, D.A.; Embree, J.R.; Loury, D.J.; Radel, P.A.; Chang, C.C.; et al. Development of protegrins for the treatment and prevention of oral mucositis: Structure-activity relationships of synthetic protegrin analogues. Biopolymers 2000, 55, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Rodziewicz-Motowidlo, S.; Mickiewicz, B.; Greber, K.; Sikorska, E.; Szultka, L.; Kamysz, E.; Kamysz, W. Antimicrobial and conformational studies of the active and inactive analogues of the protegrin-1 peptide. FEBS J. 2010, 277, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Waring, A.J.; Ruchala, P.; Hong, M. Structures of beta-hairpin antimicrobial protegrin peptides in lipopolysaccharide membranes: Mechanism of gram selectivity obtained from solid-state nuclear magnetic resonance. Biochemistry 2011, 50, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Waring, A.J.; Hong, M. Arginine dynamics in a membrane-bound cationic beta-hairpin peptide from solid-state NMR. ChemBioChem 2008, 9, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Zughaier, S.M. Neisseria meningitidis capsular polysaccharides induce inflammatory responses via TLR2 and TLR4-MD-2. J. Leukoc. Biol. 2011, 89, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Zughaier, S.M.; Svoboda, P.; Pohl, J.; Stephens, D.S.; Shafer, W.M. The human host defense peptide LL-37 interacts with Neisseria meningitidis capsular polysaccharides and inhibits inflammatory mediators release. PLoS One 2010, 5, e13627. [Google Scholar] [CrossRef] [PubMed]
- Fattorini, L.; Gennaro, R.; Zanetti, M.; Tan, D.; Brunori, L.; Giannoni, F.; Pardini, M.; Orefici, G. In vitro activity of protegrin-1 and beta-defensin-1, alone and in combination with isoniazid, against Mycobacterium tuberculosis. Peptides 2004, 25, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Ericksen, B.; Ling, M.; Toth, E.; Shi, J.; Li, X.; Galliher-Beckley, A.; Lan, L.; Zou, G.; Zhan, C.; et al. Structural and functional analysis of the pro-domain of human cathelicidin, LL-37. Biochemistry 2013, 52, 1547–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai-Larsen, Y.; Agerberth, B. The role of the multifunctional peptide LL-37 in host defense. Front. Biosci. 2008, 13, 3760–3767. [Google Scholar] [CrossRef] [PubMed]
- Zughaier, S.M.; Tzeng, Y.L.; Zimmer, S.M.; Datta, A.; Carlson, R.W.; Stephens, D.S. Neisseria meningitidis lipooligosaccharide structure-dependent activation of the macrophage CD14/Toll-like receptor 4 pathway. Infect. Immun. 2004, 72, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Prise, K.M.; Gaal, J.C.; Pearson, C.K. Increased protein ADPribosylation in HeLa cells exposed to the anti-cancer drug methotrexate. Biochim. Biophys. Acta 1986, 887, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Waring, A.J.; Hong, M. Intermolecular packing and alignment in an ordered beta-hairpin antimicrobial peptide aggregate from 2D solid-state NMR. J. Am. Chem. Soc. 2005, 127, 13919–13927. [Google Scholar] [CrossRef]
- Mak, P.; Pohl, J.; Dubin, A.; Reed, M.S.; Bowers, S.E.; Fallon, M.T.; Shafer, W.M. The increased bactericidal activity of a fatty acid-modified synthetic antimicrobial peptide of human cathepsin G correlates with its enhanced capacity to interact with model membranes. Int. J. Antimicrob. Agents 2003, 21, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Shafer, W.M.; Hubalek, F.; Huang, M.; Pohl, J. Bactericidal activity of a synthetic peptide (CG 117–136) of human lysosomal cathepsin G is dependent on arginine content. Infect. Immun. 1996, 64, 4842–4845. [Google Scholar]
- Shafer, W.M.; Katzif, S.; Bowers, S.; Fallon, M.; Hubalek, M.; Reed, M.S.; Veprek, P.; Pohl, J. Tailoring an antibacterial peptide of human lysosomal cathepsin G to enhance its broad-spectrum action against antibiotic-resistant bacterial pathogens. Curr. Pharm. Des. 2002, 8, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Andreev, K.; Bianchi, C.; Laursen, J.S.; Citterio, L.; Hein-Kristensen, L.; Gram, L.; Kuzmenko, I.; Olsen, C.A.; Gidalevitz, D. Guanidino groups greatly enhance the action of antimicrobial peptidomimetics against bacterial cytoplasmic membranes. Biochim. Biophys. Acta 2014, 1838, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Hristova, K.; Wimley, W.C. A look at arginine in membranes. J. Membr. Biol. 2011, 239, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Waring, A.J.; Hong, M. Effects of arginine density on the membrane-bound structure of a cationic antimicrobial peptide from solid-state NMR. Biochim. Biophys. Acta 2009, 1788, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Waring, A.J.; Hong, M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007, 129, 11438–11446. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.L.; Ambrose, K.D.; Zughaier, S.; Zhou, X.; Miller, Y.K.; Shafer, W.M.; Stephens, D.S. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 2005, 187, 5387–5396. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.T.; Wang, W.; Shamova, O.; Lehrer, R.I.; Schiller, N.L. Binding of protegrin-1 to Pseudomonas aeruginosa and Burkholderia cepacia. Respir. Res. 2002, 3, e18. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-Methylation of peptides: A new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Ma, B.; Nussinov, R. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 2007, 7, e21. [Google Scholar] [CrossRef]
- Buffy, J.J.; Waring, A.J.; Hong, M. Determination of peptide oligomerization in lipid bilayers using 19F spin diffusion NMR. J. Am. Chem. Soc. 2005, 127, 4477–4483. [Google Scholar] [PubMed]
- Mangoni, M.E.; Aumelas, A.; Charnet, P.; Roumestand, C.; Chiche, L.; Despaux, D.; Grassy, G.; Calas, B.; Chavanieu, A. Change in membrane permeability induced by protegrin 1: Implication of disulphide bridges for pore formation. FEBS Lett. 1996, 383, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.R.; Huck, B.R.; Weisblum, B.; Gellman, S.H. Design of non-cysteine-containing antimicrobial beta-hairpins: Structure-activity relationship studies with linear protegrin-1 analogues. Biochemistry 2002, 41, 12835–12842. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.; Domadia, P.N.; Torres, J.; Hallock, K.J.; Ramamoorthy, A.; Bhattacharjya, S. NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles: Mechanism of outer membrane permeabilization. J. Biol. Chem. 2010, 285, 3883–3895. [Google Scholar] [CrossRef] [PubMed]
- Kushibiki, T.; Kamiya, M.; Aizawa, T.; Kumaki, Y.; Kikukawa, T.; Mizuguchi, M.; Demura, M.; Kawabata, S.; Kawano, K. Interaction between tachyplesin I, an antimicrobial peptide derived from horseshoe crab, and lipopolysaccharide. Biochim. Biophys. Acta 2014, 1844, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Doran, K.S.; Banerjee, A.; Disson, O.; Lecuit, M. Concepts and mechanisms: Crossing host barriers. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Dignass, A.U. Intestinal barrier function. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P.; Estes, M.K. Microbes and microbial toxins: Paradigms for microbial-mucosal interactions. VIII. Pathological consequences of rotavirus infection and its enterotoxin. Am. J. Physiol. Gastrointest Liver Physiol. 2001, 281, G303–G310. [Google Scholar] [PubMed]
- Marchetti, G.; Tincati, C.; Silvestri, G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin. Microbiol. Rev. 2013, 26, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Szeto, C.C.; Kwan, B.C.; Chow, K.M.; Lai, K.B.; Chung, K.Y.; Leung, C.-B.; Li, P.K.T. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS One 2011, 6, e25333. [Google Scholar] [CrossRef] [PubMed]
- Volkmer-Engert, R.; Landgraf, C.; Schneider-Mergener, J. Charcoal surface-assisted catalysis of intramolecular disulfide bond formation in peptides. J. Pept. Res. 1998, 51, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.D.; Harwig, S.S.; Shafer, W.M.; Lehrer, R.I. Protegrin structure and activity against Neisseria gonorrhoeae. Infect. Immun. 1997, 65, 636–639. [Google Scholar] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, e40. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zughaier, S.M.; Svoboda, P.; Pohl, J. Structure-Dependent Immune Modulatory Activity of Protegrin-1 Analogs. Antibiotics 2014, 3, 694-713. https://doi.org/10.3390/antibiotics3040694
Zughaier SM, Svoboda P, Pohl J. Structure-Dependent Immune Modulatory Activity of Protegrin-1 Analogs. Antibiotics. 2014; 3(4):694-713. https://doi.org/10.3390/antibiotics3040694
Chicago/Turabian StyleZughaier, Susu M., Pavel Svoboda, and Jan Pohl. 2014. "Structure-Dependent Immune Modulatory Activity of Protegrin-1 Analogs" Antibiotics 3, no. 4: 694-713. https://doi.org/10.3390/antibiotics3040694