Modification of Microelectrode Arrays with High Surface Area Dendritic Platinum 3D Structures: Enhanced Sensitivity for Oxygen Detection in Ionic Liquids


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Electrochemical Experiments and Electrodeposition Parameters
2.3. Oxygen Gas Experiments
2.4. Electrode Imaging
3. Results
3.1. Deposition and Characterization of 3D Nanostructured Microarrays
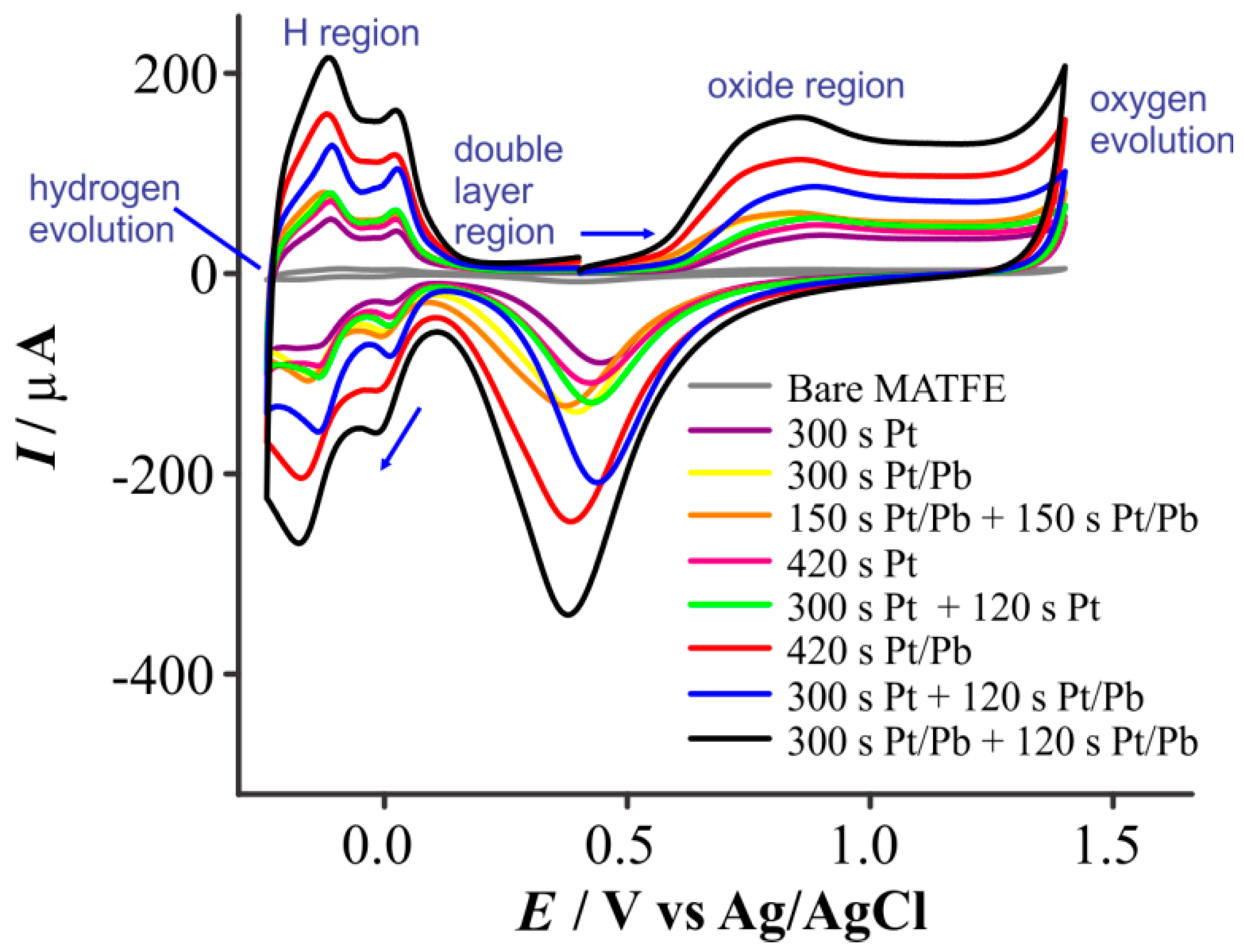
3.2. Electroactive Surface Area Calculation
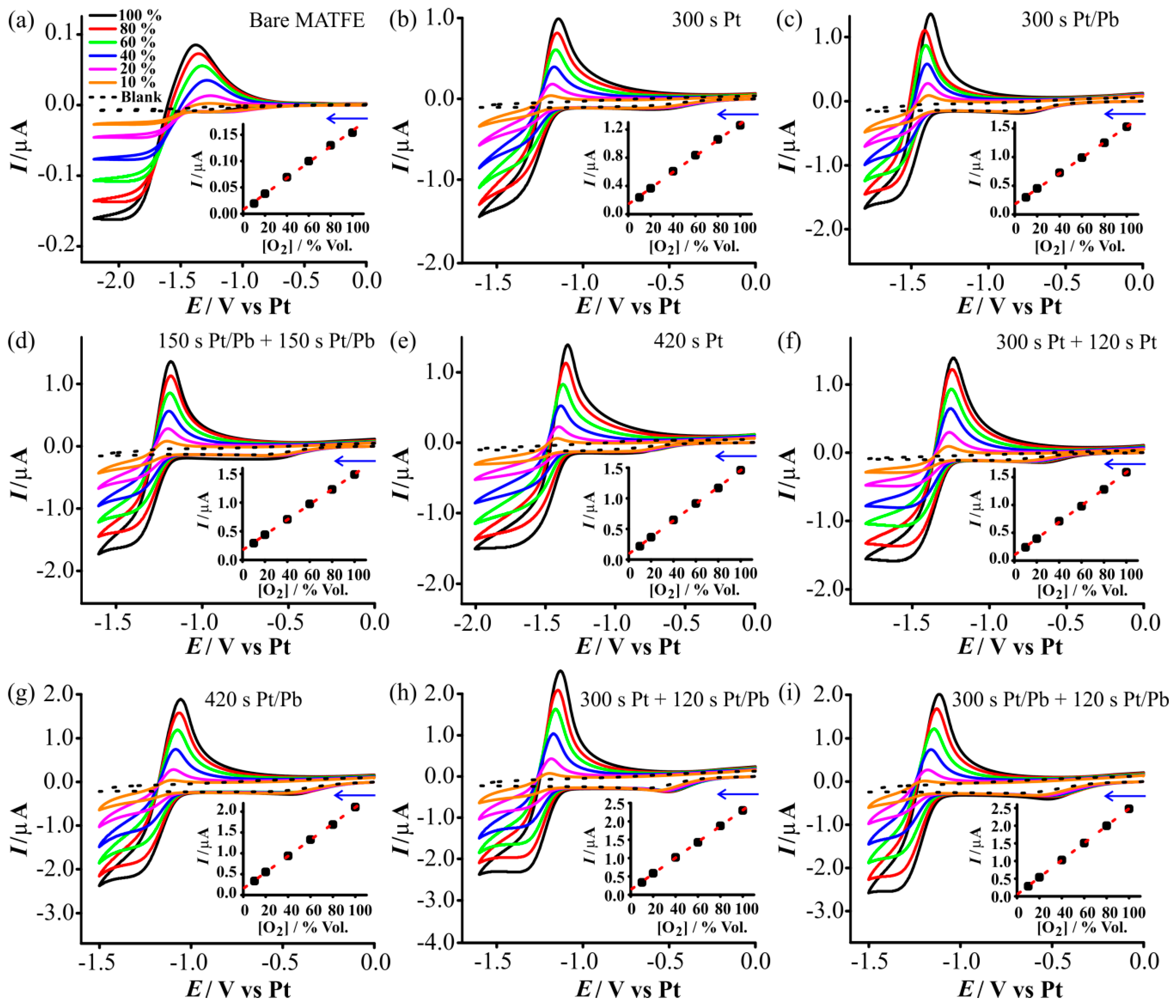
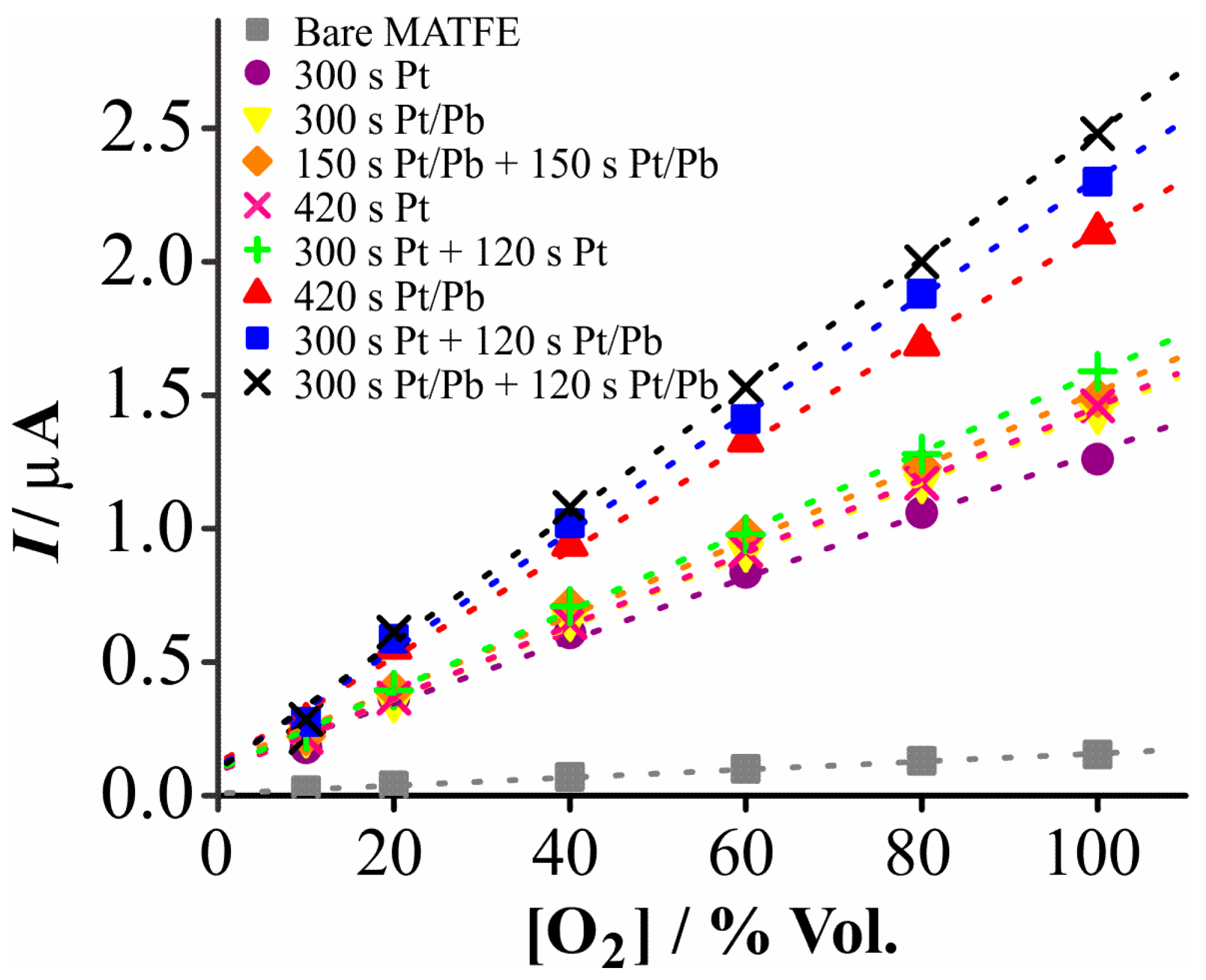
3.3. Electrochemical Reduction of Oxygen on the Prepared Electrodes
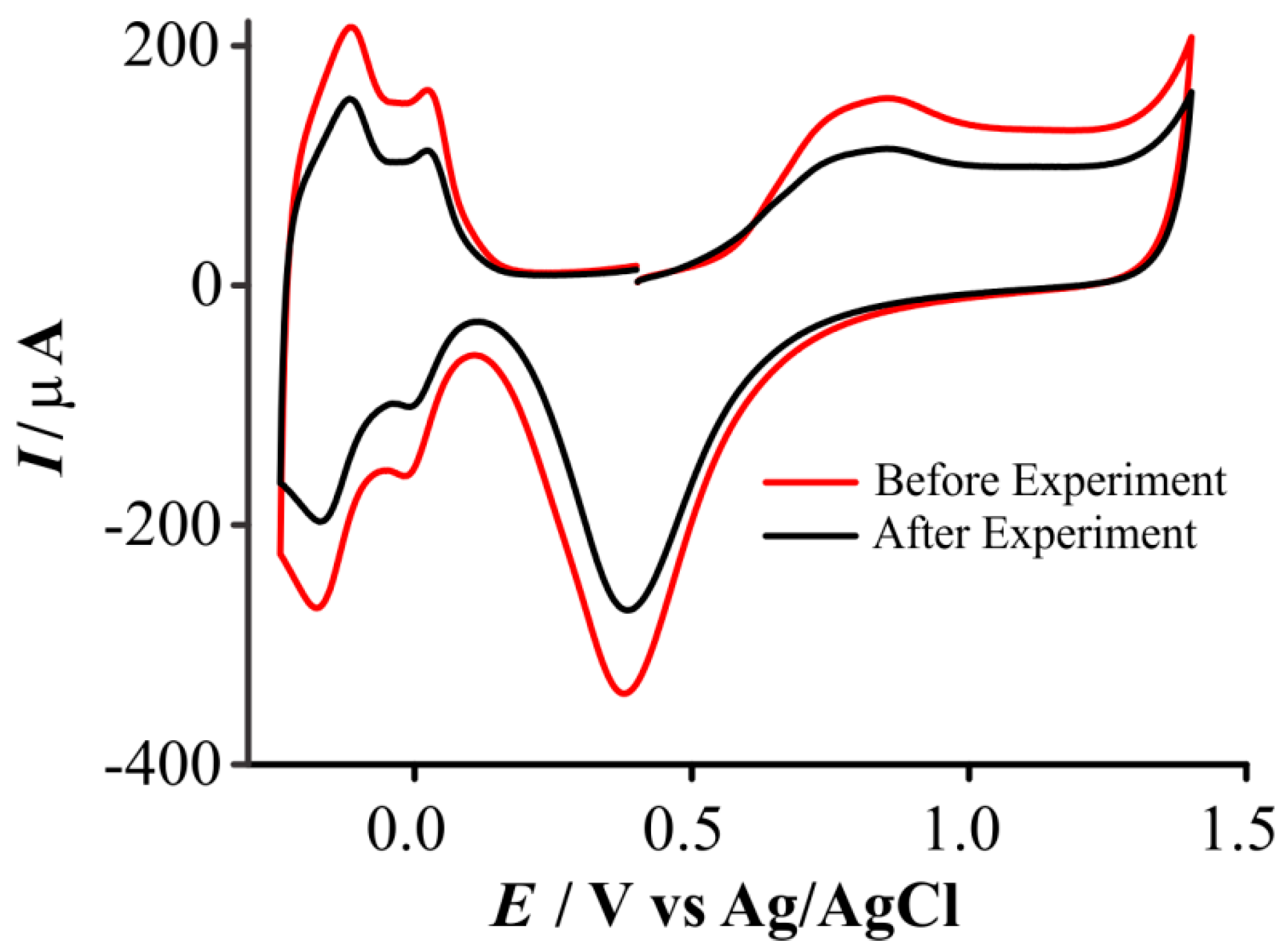
3.4. Loss of Electroactive Surface Area after Experiments in Ionic Liquids
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bakker, E. Electrochemical Sensors. Anal. Chem. 2004, 76, 3285–3298. [Google Scholar] [CrossRef] [PubMed]
- Stetter, J.R.; Penrose, W.R.; Yao, S. Sensors, Chemical Sensors, Electrochemical Sensors, and ECS. J. Electrochem. Soc. 2003, 150, S11–S16. [Google Scholar] [CrossRef]
- Kimmel, D.W.; LeBlanc, G.; Meschievitz, M.E.; Cliffel, D.E. Electrochemical Sensors and Biosensors. Anal. Chem. 2012, 84, 685–707. [Google Scholar] [CrossRef] [PubMed]
- Privett, B.J.; Shin, J.H.; Schoenfisch, M.H. Electrochemical Sensors. Anal. Chem. 2008, 80, 4499–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Granger, M.C.; Chen, Q.; Strojek, J.W.; Lister, T.E.; Swain, G.M. Boron-Doped Diamond Thin-Film Electrodes. Anal. Chem. News Featur. 1997, 69, 591–597. [Google Scholar] [CrossRef]
- Van Gerwen, P.; Laureyn, W.; Laureys, W.; Huyberechts, G.; Op De Beeck, M.; Baert, K.; Suls, J.; Sansen, W.; Jacobs, P.; Hermans, L.; et al. Nanoscaled interdigitated electrode arrays for biochemical sensors. Sens. Actuators B 1998, 49, 73–80. [Google Scholar] [CrossRef]
- Kadara, R.O.; Jenkinson, N.; Banks, C.E. Characterisation of commercially available electrochemical sensing platforms. Sens. Actuators B 2009, 138, 556–562. [Google Scholar] [CrossRef]
- Dominguez Renedo, O.; Alonso-Lomillo, M.A.; Arcos Martinez, M.J. Recent developments in the field of screen-printed electrodes and their related applications. Talanta 2007, 73, 202–219. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, F.G.; Compton, R.G. Regular Arrays of Microdisk Electrodes: Numerical Simulation as an Optimizing Tool to Maximize the Current Response and Minimize the Electrode Area Used. Electroanalysis 2006, 23, 2369–2374. [Google Scholar] [CrossRef]
- Tomčík, P. Microelectrode Arrays with Overlapped Diffusion Layers as Electroanalytical Detectors: Theory and Basic Applications. Sensors 2013, 13, 13659–13684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, T.J.; Compton, R.G. The cyclic and linear sweep voltammetry of regular and random arrays of microdisc electrodes: Theory. J. Electroanal. Chem. 2005, 585, 63–82. [Google Scholar] [CrossRef]
- Davies, T.J.; Ward-Jones, S.; Banks, C.E.; del Campo, J.; Mas, R.; Munoz, F.X.; Compton, R.G. The cyclic and linear sweep voltammetry of regular arrays of microdisc electrodes: Fitting of experimental data. J. Electroanal. Chem. 2005, 585, 51–62. [Google Scholar] [CrossRef]
- Huang, X.-J.; Aldous, L.; O’Mahony, A.M.; del Campo, F.J.; Compton, R.G. Toward Membrane-Free Amperometric Gas Sensors: A Microelectrode Array Approach. Anal. Chem. 2010, 82, 5238–5245. [Google Scholar] [CrossRef] [PubMed]
- Hussain, G.; Silvester, D.S. Detection of sub-ppm Concentrations of Ammonia in an Ionic Liquid: Enhanced Current Density Using “Filled” Recessed Microarrays. Anal. Chem. 2016, 88, 12453–12460. [Google Scholar] [CrossRef] [PubMed]
- Chiappe, C.; Pieraccini, D. Ionic liquids: Solvent properties and organic reactivity. J. Phys. Org. Chem. 2005, 18, 275–297. [Google Scholar] [CrossRef]
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-J.; O’Mahony, A.M.; Compton, R.G. Microelectrode Arrays for Electrochemistry: Approaches to Fabrication. Small 2009, 5, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Hussain, G.; Aldous, L.; Silvester, D.S. Preparation of Platinum-based ‘Cauliflower Microarrays’ for Enhanced Gas Sensing. Anal. Chim. Acta 2018. under revision. [Google Scholar]
- Gunawan, C.A.; Ge, M.; Zhao, C. Robust and versatile ionic liquid microarrays achieved by microcontact printing. Nat. Commun. 2014, 5, 3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, M.; Hussain, G.; Hibbert, D.B.; Silvester, D.S.; Zhao, C. Ionic Liquid-based Microchannels for Highly Sensitive and Fast Amperometric Detection of Toxic Gases. Electroanalysis 2018. under revision. [Google Scholar]
- Ott, A.; Jones, L.A.; Bhargava, S.K. Direct electrodeposition of porous platinum honeycomb structures. Electrochem. Commun. 2011, 13, 1248–1251. [Google Scholar] [CrossRef]
- Attard, G.D.; Bartlett, P.N.; Coleman, N.R.B.; Elliott, J.M.; Owen, J.R.; Wang, J.H. Mesoporous Platinum Films from Lyotropic Liquid Crystalline Phases. Science 1997, 278, 838–840. [Google Scholar] [CrossRef]
- Lertanantawong, B.; Hoshyargar, F.; O’Mullane, A.P. Directing Nanostructure Formation of Gold through the In Situ Underpotential Deposition of a Secondary Metal for the Detection of Nitrite Ions. ChemElectroChem 2018, 5, 911–916. [Google Scholar] [CrossRef]
- Plowman, B.J.; Mahajan, M.; O’Mullane, A.P.; Bhargava, S.K. Electrochemical detection of dopamine and cytochrome c at a nanostructured gold electrode. Electrochim. Acta 2010, 55, 8953–8959. [Google Scholar] [CrossRef]
- Sabri, Y.M.; Ippolito, S.J.; O’Mullane, A.P.; Tardio, J.; Bansal, V.; Bhargava, S.K. Creating gold nanoprisms directly on quartz crystal microbalance electrodes for mercury vapor sensing. Nanotechnology 2001, 22, 305501. [Google Scholar] [CrossRef] [PubMed]
- Plowman, B.J.; Abdelhamid, M.E.; Ippolito, S.J.; Bansal, V.; Bhargava, S.K.; O’Mullane, A.P. Electrocatalytic and SERS activity of Pt rich Pt-Pb nanostructures formed via the utilisation of in-situ underpotential deposition of lead. J. Solid State Electrochem. 2014, 18, 3345–3357. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Chen, L.; Li, J.; Li, J.; Wang, X. Shape-controlled fabrication of platinum electrocatalyst by pulse electrodeposition. Electrochem. Commun. 2008, 10, 476–479. [Google Scholar] [CrossRef]
- Demille, T.B.; Hughes, R.A.; Preston, A.S.; Adelung, R.; Mishra, Y.K.; Neretina, S. Light-Mediated Growth of Noble Metal Nanostructures (Au, Ag, Cu, Pt, Pd, Ru, Ir, Rh) From Micro- and Nanoscale ZnO Tetrapodal Backbones. Front. Chem. 2018, 6, 1–8. [Google Scholar] [CrossRef]
- Mishra, Y.K.; Adelung, R. ZnO tetrapod materials for functional applications. Mater. Today 2018, 21, 631–651. [Google Scholar] [CrossRef]
- Waibel, H.F.; Kleinert, M.; Kibler, L.A.; Kolb, D.M. Initial stages of Pt deposition on Au(111) and Au(100). Electrochim. Acta 2002, 47, 1461–1467. [Google Scholar] [CrossRef]
- Gotti, G.; Fajerwerg, K.; Evrard, D.; Gros, P. Electrodeposited gold nanoparticles on glassy carbon: Correlation between nanoparticles characteristics and oxygen reduction kinetics in neutral media. Electrochim. Acta 2014, 128, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Nasirpouri, F. Electrodeposition of 2D and 3D Meso and Nanostructures; Springer Series in Surface Sciences; Springer: Berlin/Heidelberg, Germany, 2017; Volume 62, pp. 123–185. [Google Scholar]
- Sheridan, E.; Hjelm, J.; Forster, R.J. Electrodeposition of gold nanoparticles on fluorine-doped tin oxide: Control of particle density and size distribution. J. Electroanal. Chem. 2007, 608, 1–7. [Google Scholar] [CrossRef]
- Ren, F.; Pearton, S.J. Semiconductor-Based Sensors; World Scientific: Singapore, 2016. [Google Scholar]
- Burk, J.J.; Buratto, S.K. Electrodeposition of Pt Nanoparticle Catalysts from H2Pt(OH)6 and Their Application in PEM Fuel Cells. J. Phys. Chem. C 2013, 117, 18957–18966. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; John Wiley: New York, NY, USA, 2001. [Google Scholar]
- Schroder, U.; Wadhawan, J.D.; Compton, R.G.; Marken, F.; Suarez, P.A.Z.; Consorti, C.S.; de Souza, R.F.; Dupont, J. Water-induced accelerated ion diffusion: Voltammetric studies in 1-methyl-3-[2,6-(S)-dimethylocten-2-yl]imidazolium tetrafluoroborate, 1-butyl-3-methylimidazolium tetrafluoroborate and hexafluorophosphate ionic liquids. New J. Chem. 2000, 24, 1009–1015. [Google Scholar] [CrossRef]
- Silvester, D.S.; Wain, A.J.; Aldous, L.; Hardacre, C.; Compton, R.G. Electrochemical reduction of nitrobenzene and 4-nitrophenol in the room temperature ionic liquid [C4dmim][N(Tf)2]. J. Electroanal. Chem. 2006, 596, 131–140. [Google Scholar] [CrossRef]
- Lee, J.; Murugappan, K.; Arrigan, D.W.M.; Silvester, D.S. Oxygen reduction voltammetry on platinum macrodisk and screen-printed electrodes in ionic liquids: Reaction of the electrogenerated superoxide species with compounds used in the paste of Pt screen-printed electrodes? Electrochim. Acta 2013, 101, 158–168. [Google Scholar] [CrossRef]
- Climent, V.; Feliu, J.M. Thirty years of platinum single crystal electrochemistry. J. Solid State Electrochem. 2011, 15, 1297–1315. [Google Scholar] [CrossRef]
- Jacobse, L.; Raaijman, S.J.; Koper, M.T.M. The reactivity of platinum microelectrodes. Phys. Chem. Chem. Phys. 2016, 18, 28451–28457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambunathan, K.; Shah, B.C.; Hudson, J.L.; Hillier, A.C. Scanning electrochemical microscopy of hydrogen electro-oxidation. Rate constant measurements and carbon monoxide poisoning on platinum. J. Electroanal. Chem. 2001, 500, 279–289. [Google Scholar] [CrossRef]
- Buzzeo, M.C.; Klymenko, O.V.; Wadhawan, J.D.; Hardacre, C.; Compton, R.G. Voltammetry of Oxygen in the Room-Temperature Ionic Liquids 1-Ethyl-3-methylimidazolium Bis((trifluoromethyl)sulfonyl)imide and Hexyltriethylammonium Bis((trifluoromethyl)sulfonyl)imide: One-Electron Reduction To Form Superoxide. Steady-State and Transient Behavior in the Same Cyclic Voltammogram Resulting from Widely Different Diffusion Coefficients of Oxygen and Superoxide. J. Phys. Chem. A 2003, 107, 8872–8878. [Google Scholar]
- Khan, A.; Gunawan, C.A.; Zhao, C. Oxygen Reduction Reaction in Ionic Liquids: Fundamentals and Applications in Energy and Sensors. ACS Sustain. Chem. Eng. 2017, 5, 3698–3715. [Google Scholar] [CrossRef]
- Evans, R.G.; Klymenko, O.V.; Saddoughi, S.A.; Hardacre, C.; Compton, R.G. Electroreduction of Oxygen in a Series of Room Temperature Ionic Liquids Composed of Group 15-Centered Cations and Anions. J. Phys. Chem. B 2004, 108, 7878–7886. [Google Scholar] [CrossRef]
- Wandt, J.; Lee, J.; Arrigan, D.W.M.; Silvester, D.S. A lithium iron phosphate reference electrode for ionic liquid electrolytes. Electrochem. Commun. 2018, 93, 148–151. [Google Scholar] [CrossRef]
- Noel, J.-M.; Yu, Y.; Mirkin, M.V. Dissolution of Pt at Moderately Negative Potentials during Oxygen Reduction in Water and Organic Media. Langmuir 2013, 29, 1346–1350. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Deposition Conditions and Plating Bath Composition | Deposition Time |
|---|---|---|
| 300 s Pt | Single step, 20 mM H2PtCl6 | 300 s |
| 300 s Pt/Pb | Single step, 20 mM H2PtCl6 + 2mM Pb(OAc)2 | 300 s |
| 150 s Pt/Pb + 150 s Pt/Pb | Two steps, both in 20 mM H2PtCl6 + 2mM Pb(OAc)2 | 150 s + 150 s = 300 s |
| 420 s Pt | Single step, 20 mM H2PtCl6 | 420 s |
| 300 s Pt + 120 s Pt | Two steps, both in 20 mM H2PtCl6 | 300 s + 120 s = 420 s |
| 420 s Pt/Pb | Single step, 20 mM H2PtCl6 + 2mM Pb(OAc)2 | 420 s |
| 300 s Pt + 120 s Pt/Pb | Two steps. First step: 20 mM H2PtCl6, second step: 20 mM H2PtCl6 + 2mM Pb(OAc)2 | 300 s + 120 s = 420 s |
| 300 s Pt/Pb + 120 s Pt/Pb | Two steps, both in 20 mM H2PtCl6 + 2mM Pb(OAc)2 | 300 s + 120 s = 420 s |
| Deposition Method | Diameter of Deposit/µm | QH = I × t/(µC) | ESA/(mm2) | Ip (100% O2)/(µA) | Sensitivity/(nA/%vol.) | LOD2/(% vol.) | E1/2 vs. Fc/Fc+/V |
|---|---|---|---|---|---|---|---|
| Bare MATFE | n/a | 2.13 | 1.01 | 0.15 | 1.5 | 5.8 | −1.45 |
| 300 s Pt | 22 ± 0.8 1 | 21.7 | 10.3 | 1.26 | 14.1 | 4.5 | −1.35 |
| 300 s Pt/Pb | 27 ± 0.6 | 31.6 | 15.0 | 1.43 | 13.7 | 2.4 | −1.37 |
| 150 s Pt/Pb + 150 s Pb/Pb | 25 ± 0.7 | 32.8 | 15.6 | 1.49 | 13.7 | 2.1 | −1.35 |
| 420 s Pt | 28 ± 0.8 | 28.9 | 13.8 | 1.46 | 13.9 | 2.3 | −1.35 |
| 300 s Pt + 120 s Pt | 30 ± 0.8 | 32.3 | 15.4 | 1.59 | 15.1 | 2.4 | −1.34 |
| 420 s Pt/Pb | 36 ± 0.9 | 68.1 | 32.4 | 2.11 | 19.8 | 2.4 | −1.34 |
| 300 s Pt + 120 s Pt/Pb | 37 ± 2.1 | 53.7 | 25.6 | 2.30 | 22.1 | 2.1 | −1.31 |
| 300 s Pt/Pb + 120 s Pt/Pb | 52 ± 4.2 | 93.3 | 44.4 | 2.48 | 24.3 | 1.5 | −1.34 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, G.; O’Mullane, A.P.; Silvester, D.S. Modification of Microelectrode Arrays with High Surface Area Dendritic Platinum 3D Structures: Enhanced Sensitivity for Oxygen Detection in Ionic Liquids. Nanomaterials 2018, 8, 735. https://doi.org/10.3390/nano8090735
Hussain G, O’Mullane AP, Silvester DS. Modification of Microelectrode Arrays with High Surface Area Dendritic Platinum 3D Structures: Enhanced Sensitivity for Oxygen Detection in Ionic Liquids. Nanomaterials. 2018; 8(9):735. https://doi.org/10.3390/nano8090735
Chicago/Turabian StyleHussain, Ghulam, Anthony P. O’Mullane, and Debbie S. Silvester. 2018. "Modification of Microelectrode Arrays with High Surface Area Dendritic Platinum 3D Structures: Enhanced Sensitivity for Oxygen Detection in Ionic Liquids" Nanomaterials 8, no. 9: 735. https://doi.org/10.3390/nano8090735