Organozinc Precursor-Derived Crystalline ZnO Nanoparticles: Synthesis, Characterization and Their Spectroscopic Properties


Abstract
:
1. Introduction
2. Results and Discussion
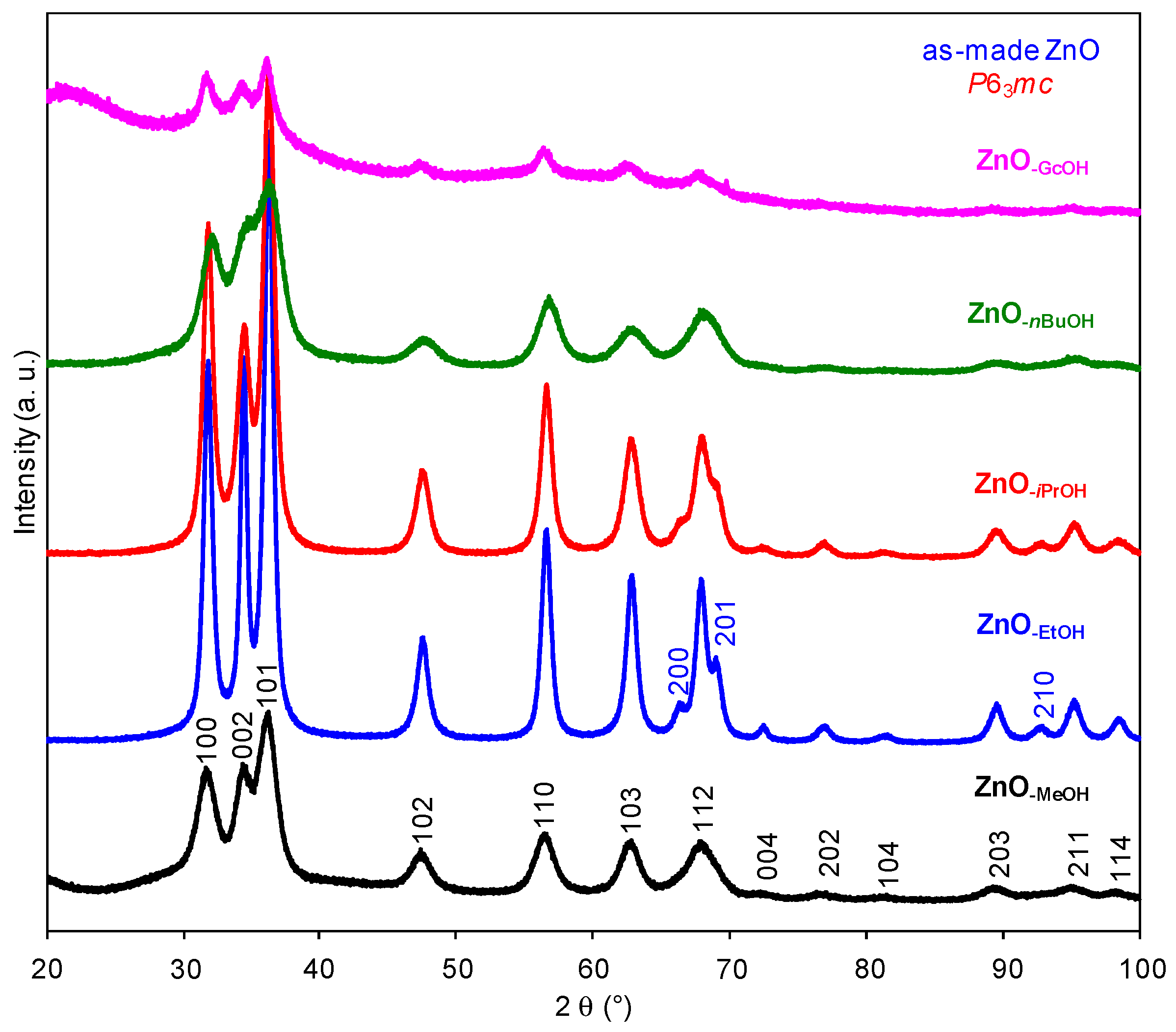
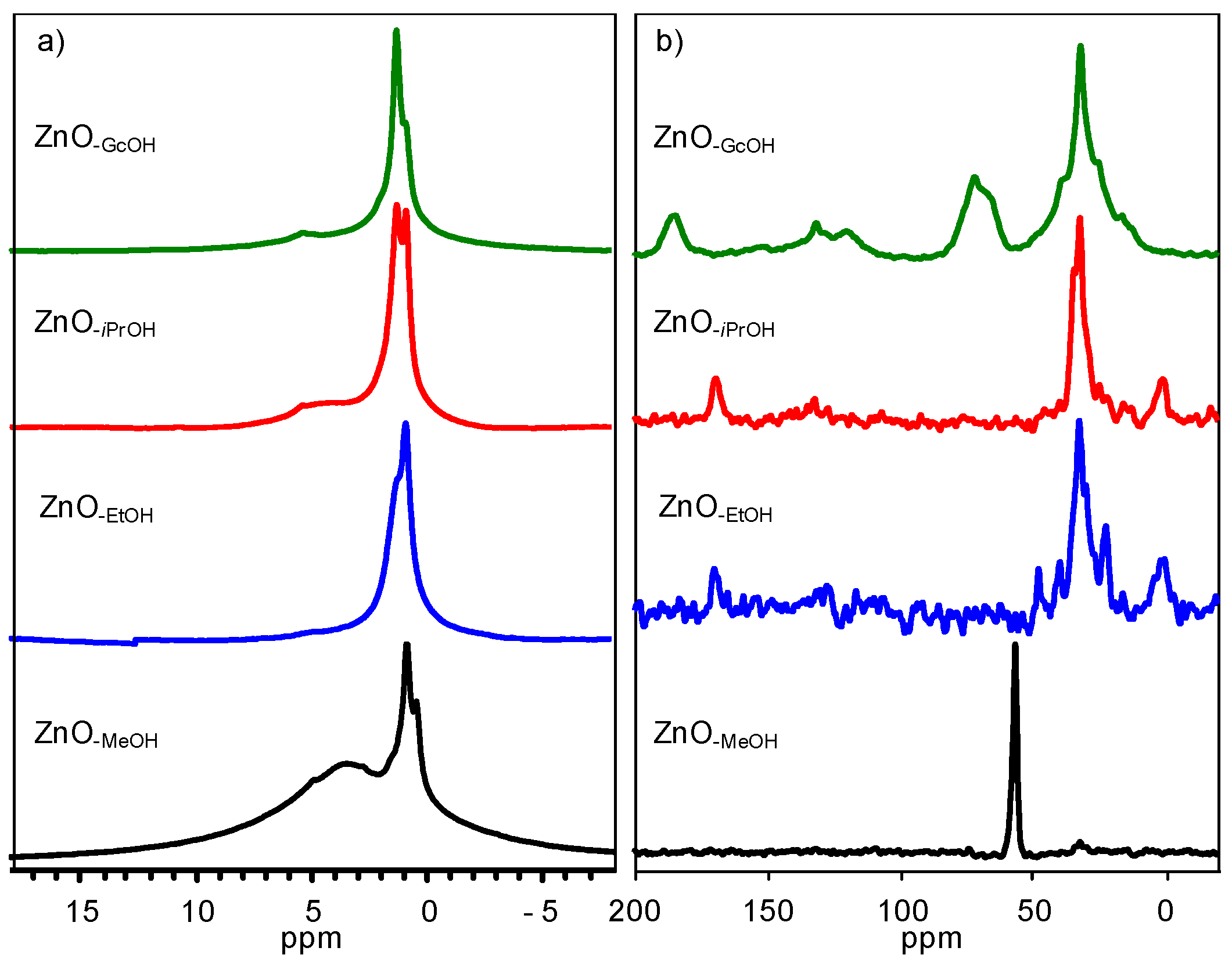
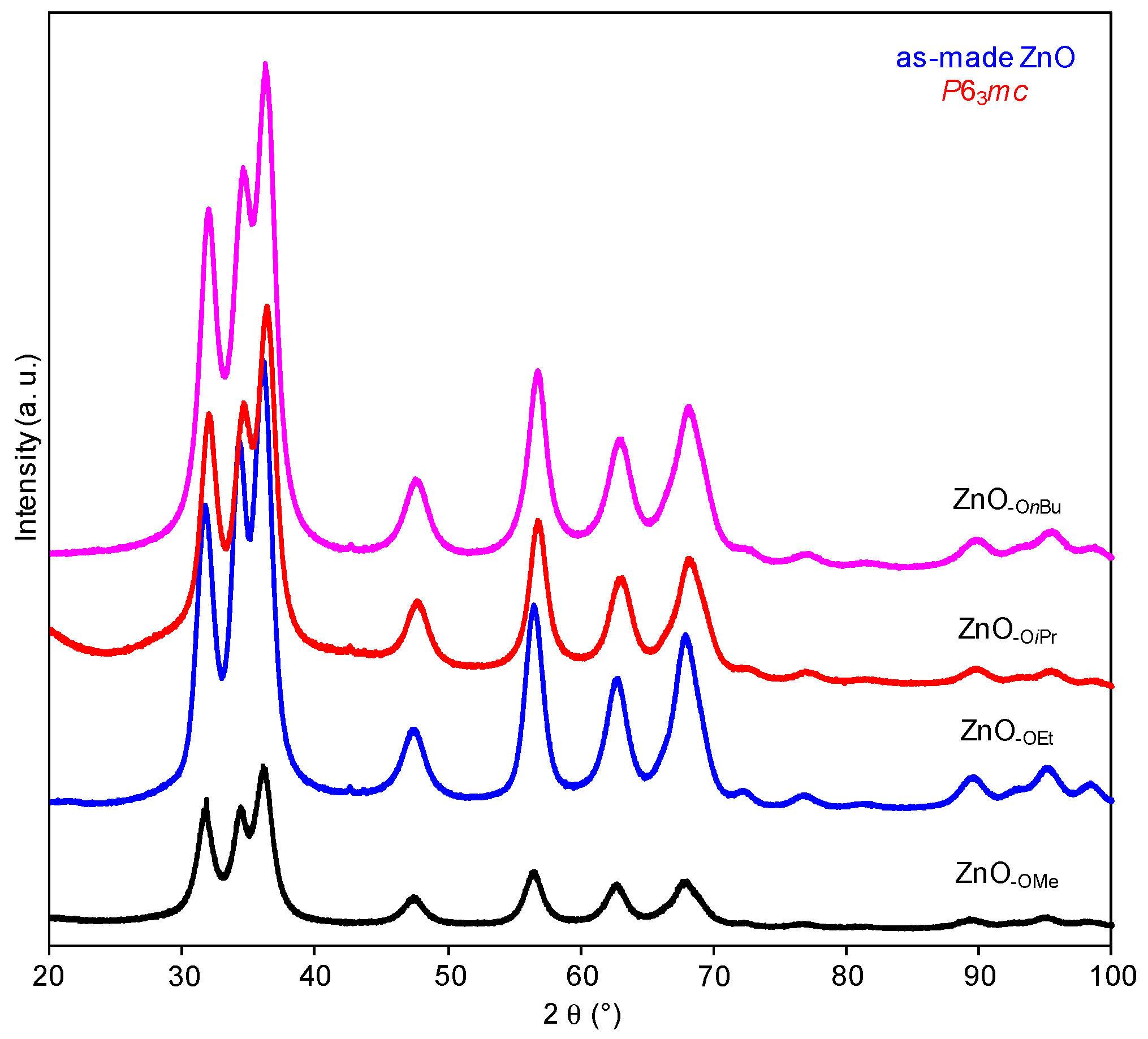
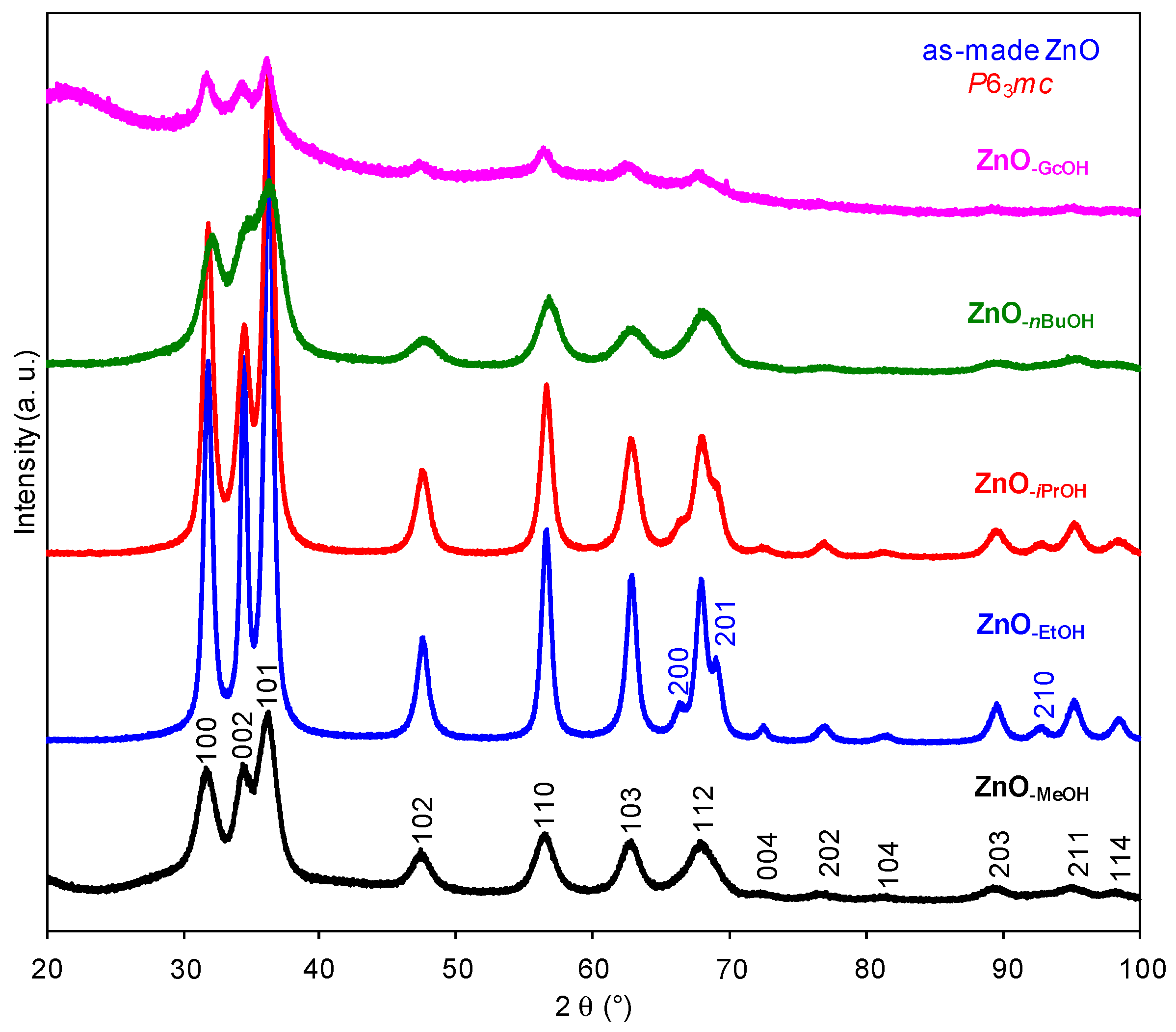
2.1. Structural Characterization of a Series of Nano-Structured Zinc Oxide Particles ZnO-ROH, ZnO-OR and ZnO-R (R = Me, Et, iPr, nBu, Gc)
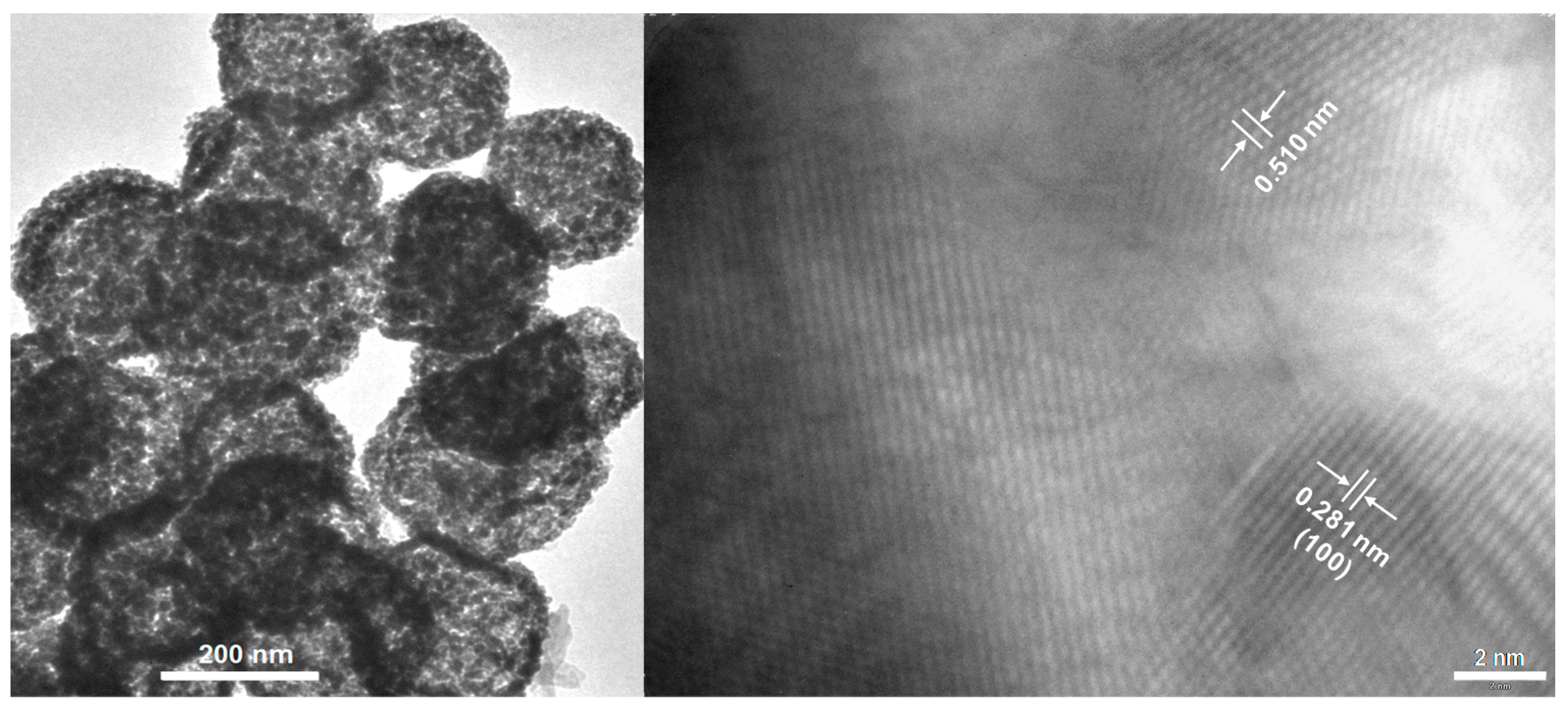
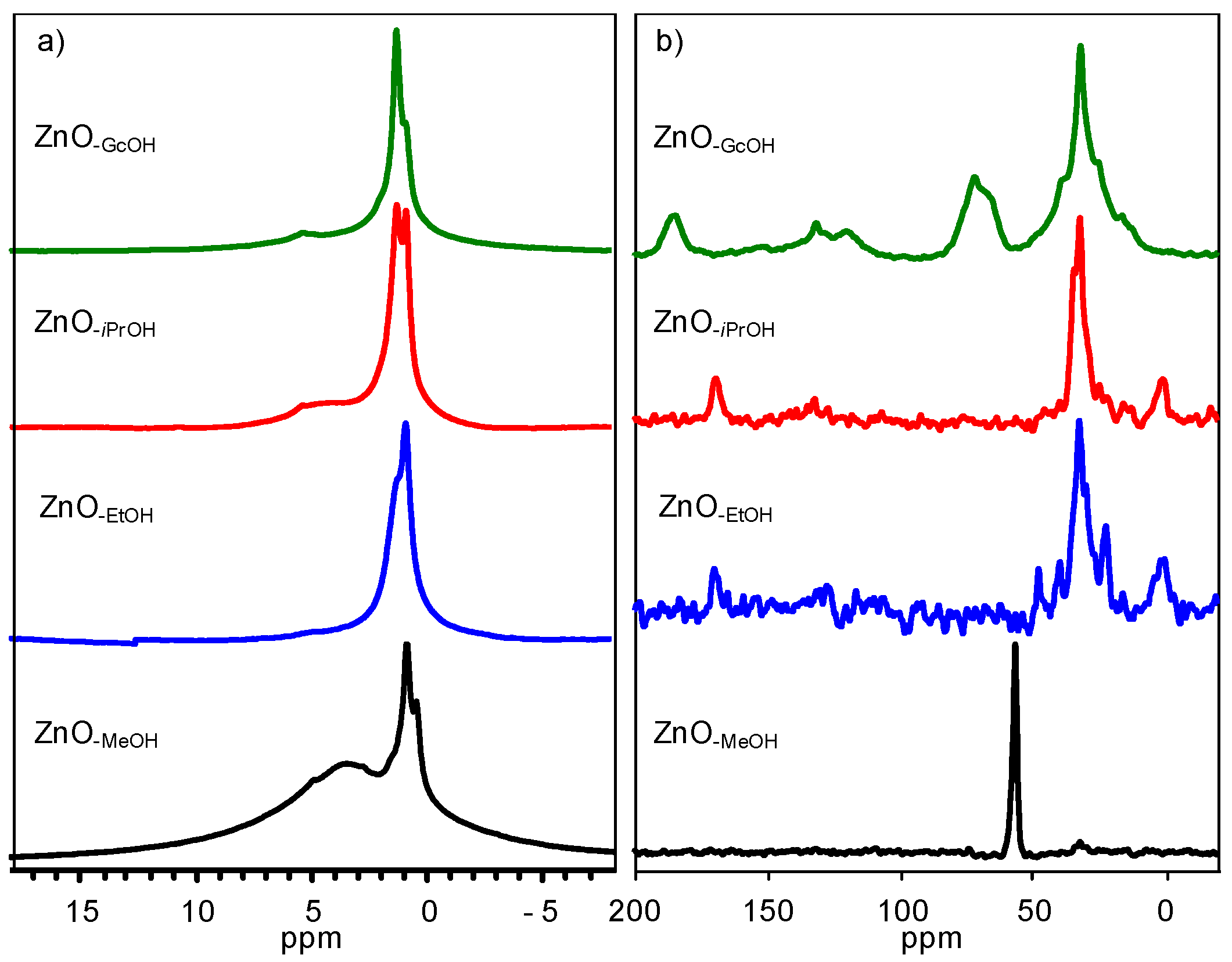
2.1.1. Crystalline ZnO-MeOH NPs
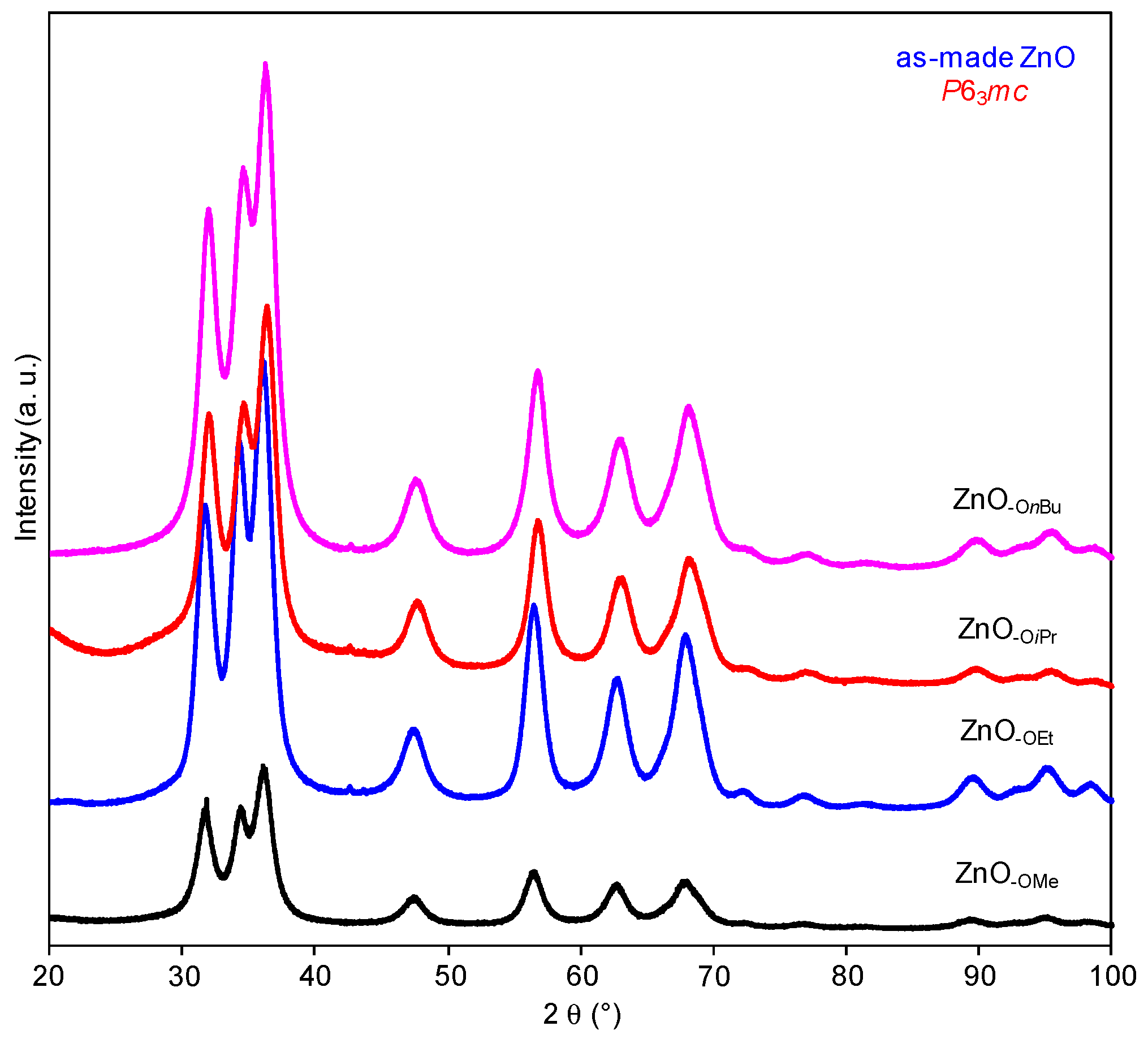
2.1.2. Alkyl Alcohol Effect
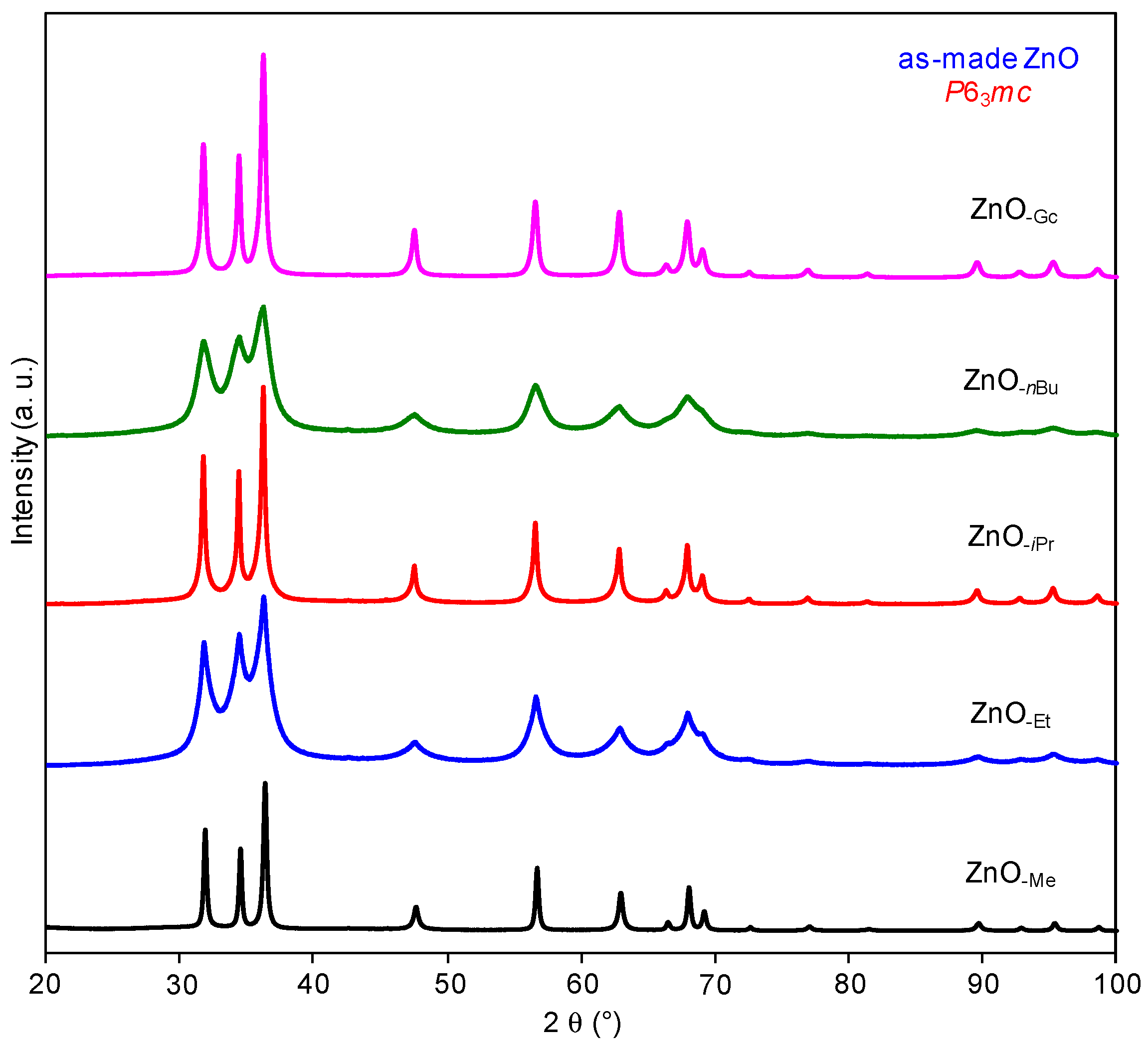
2.1.3. Zinc Precursor Effect
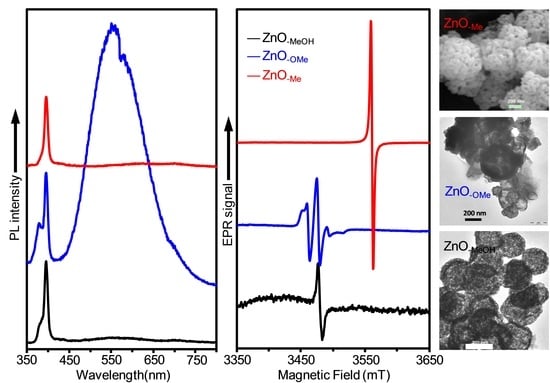
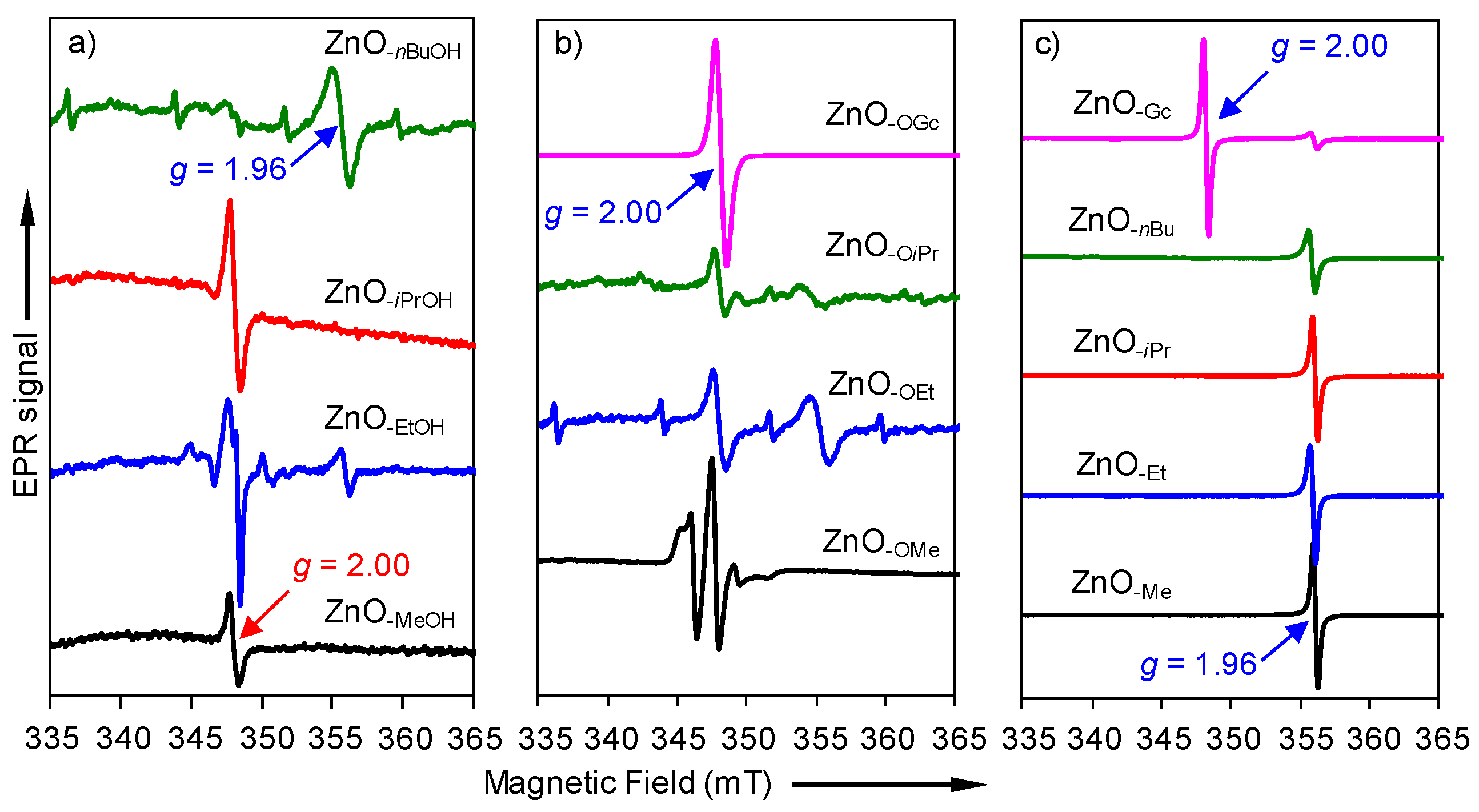
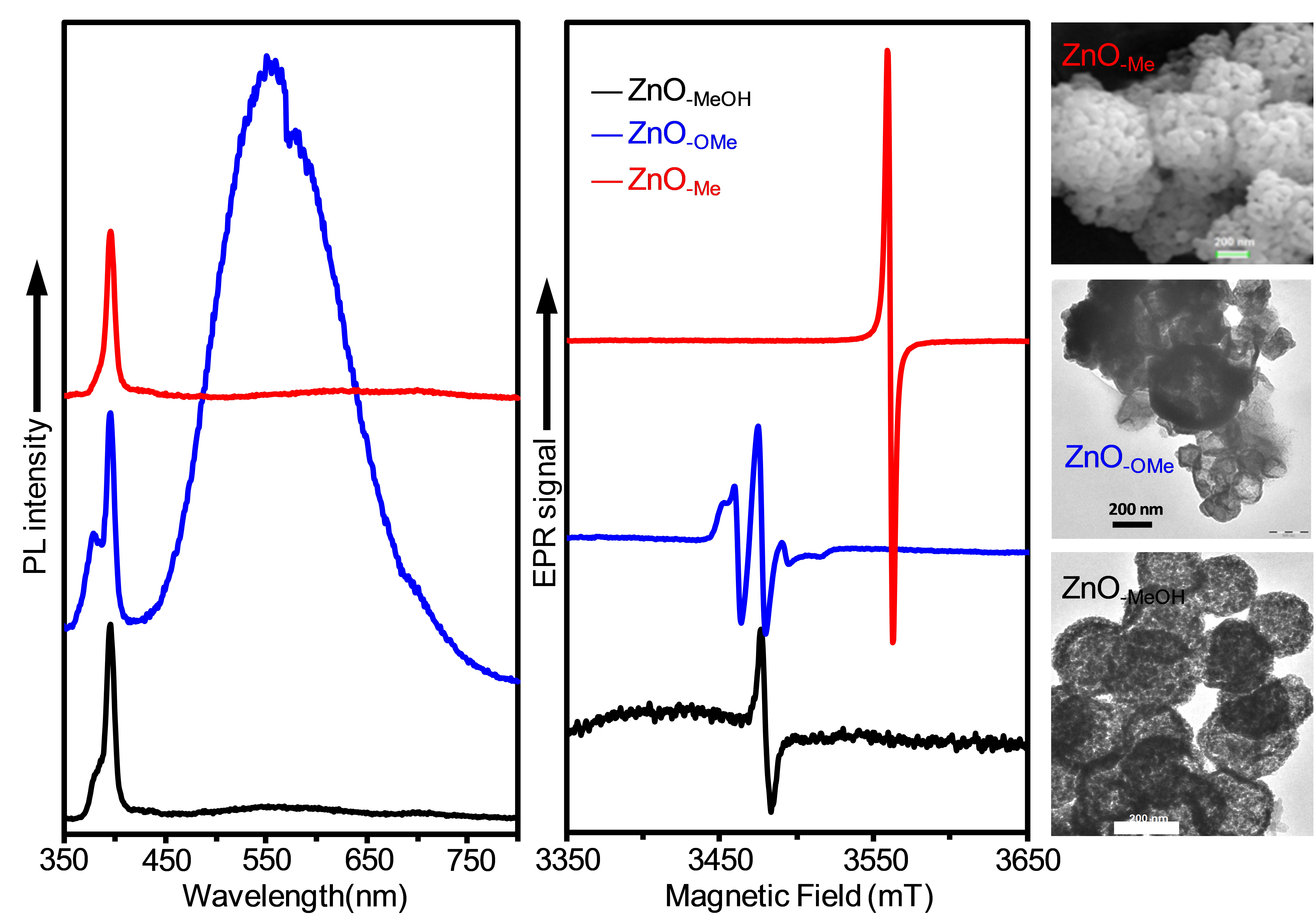
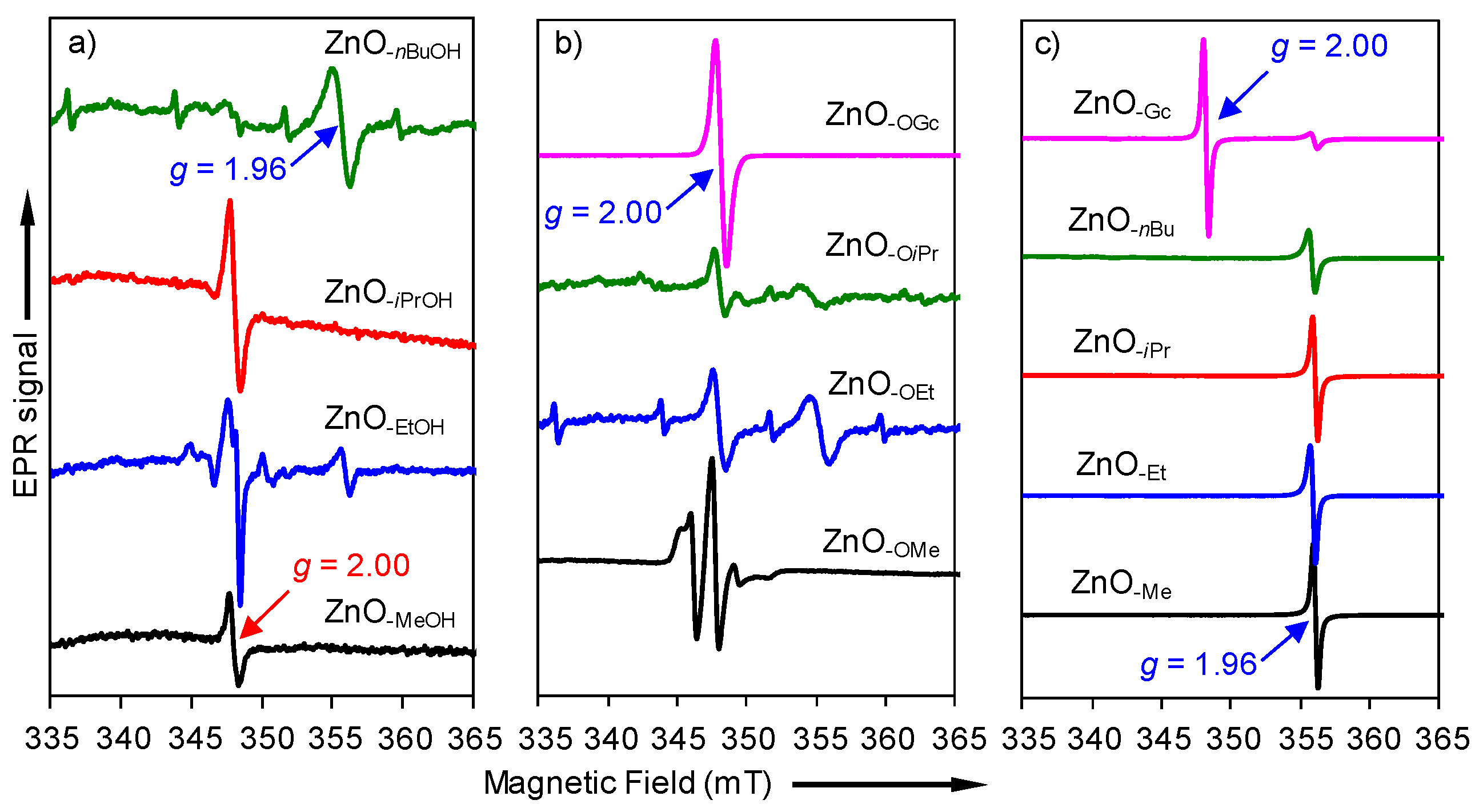
2.2. Electron Paramagnetic Resonance (EPR) Spectra of a Series of Nano-Structured Zinc Oxide Particles ZnO-ROH, ZnO-OR and ZnO-R (R = Me, Et, iPr, nBu, Gc)
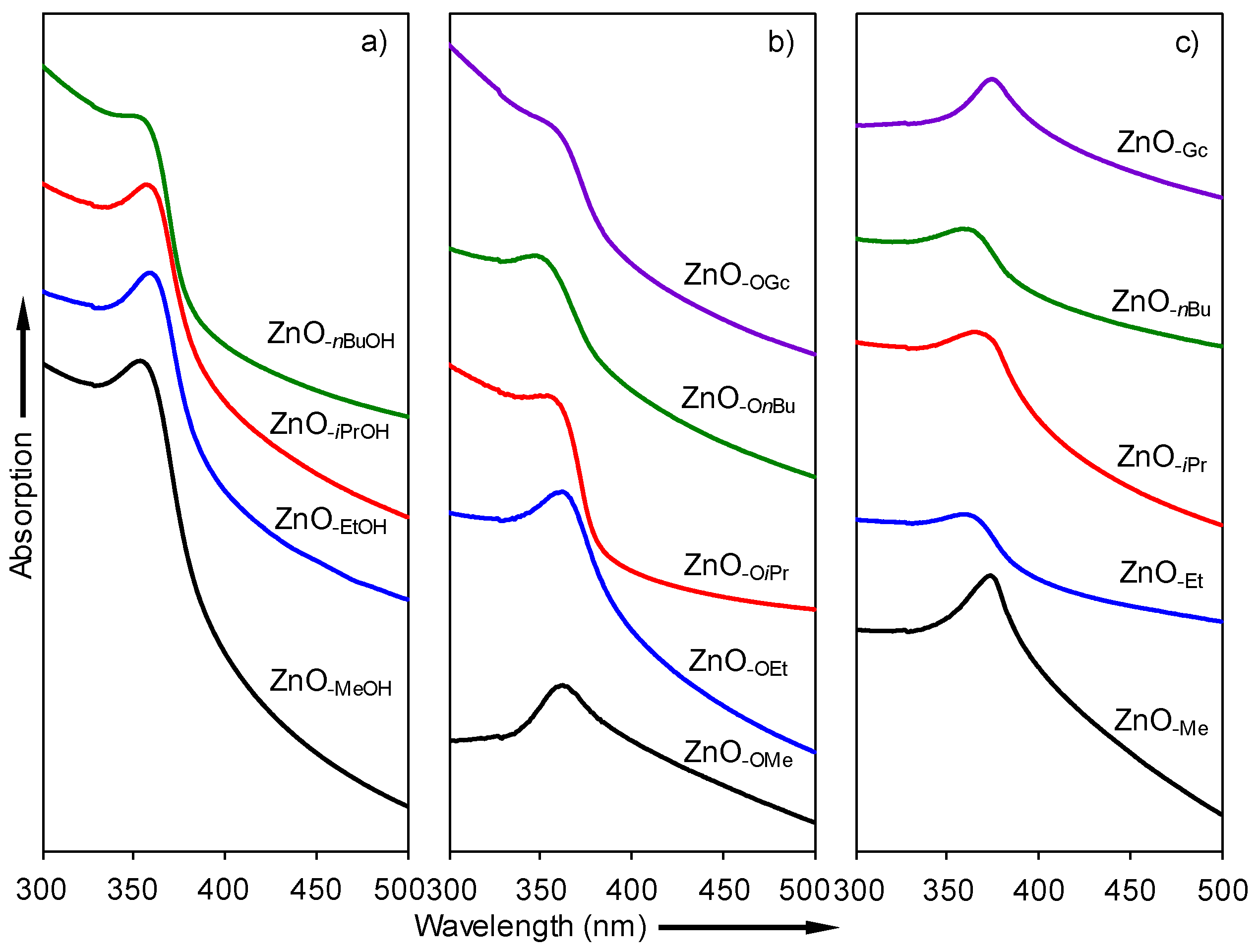
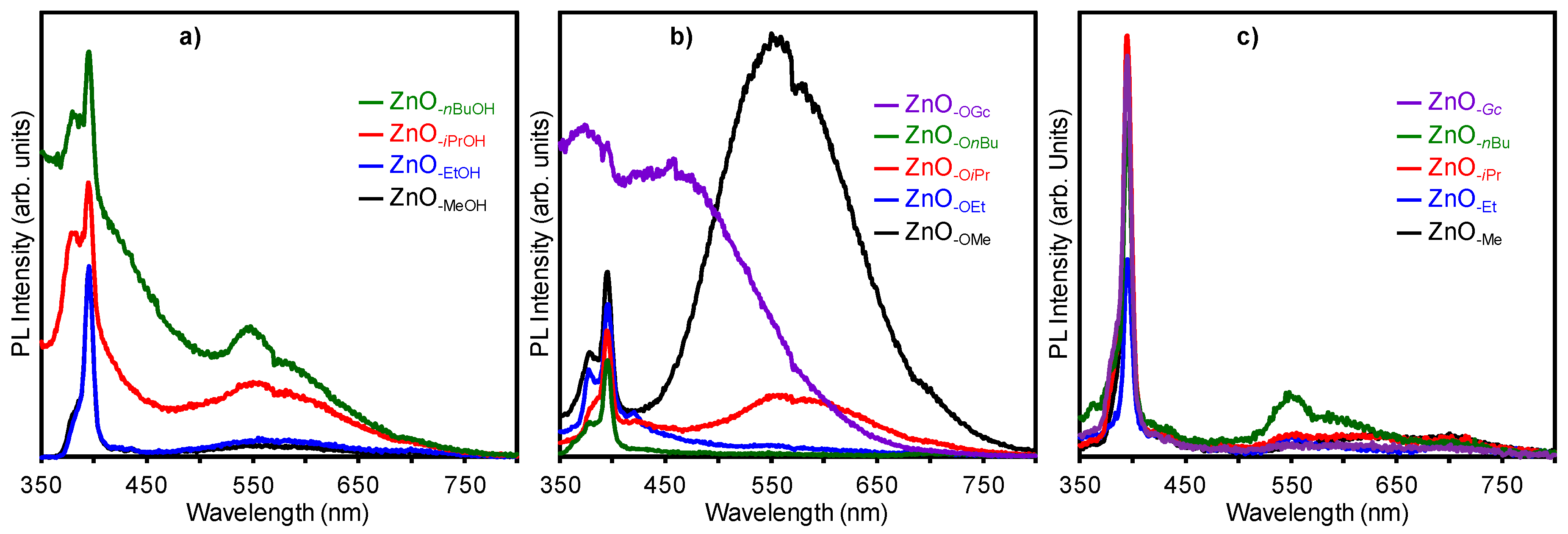
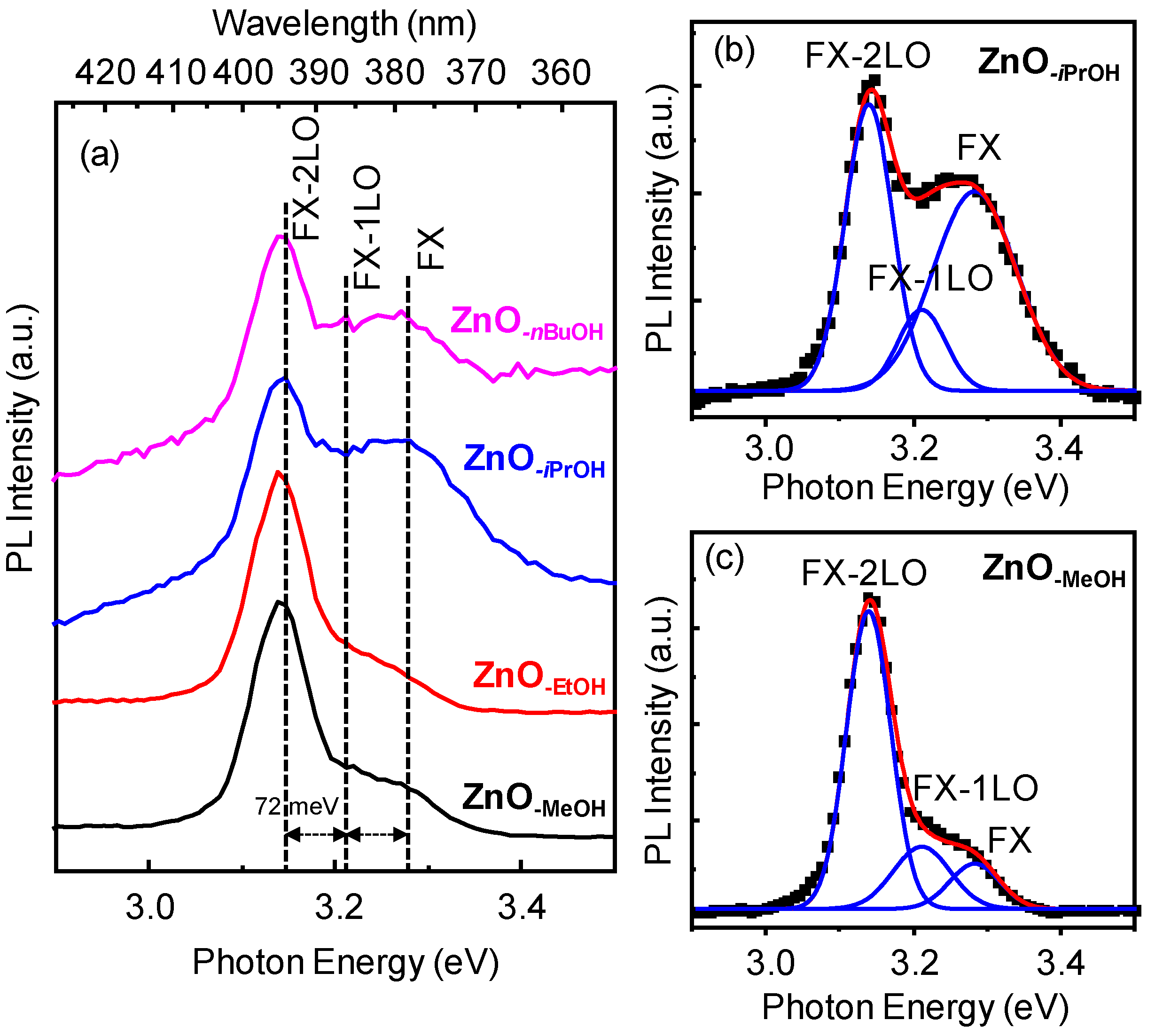
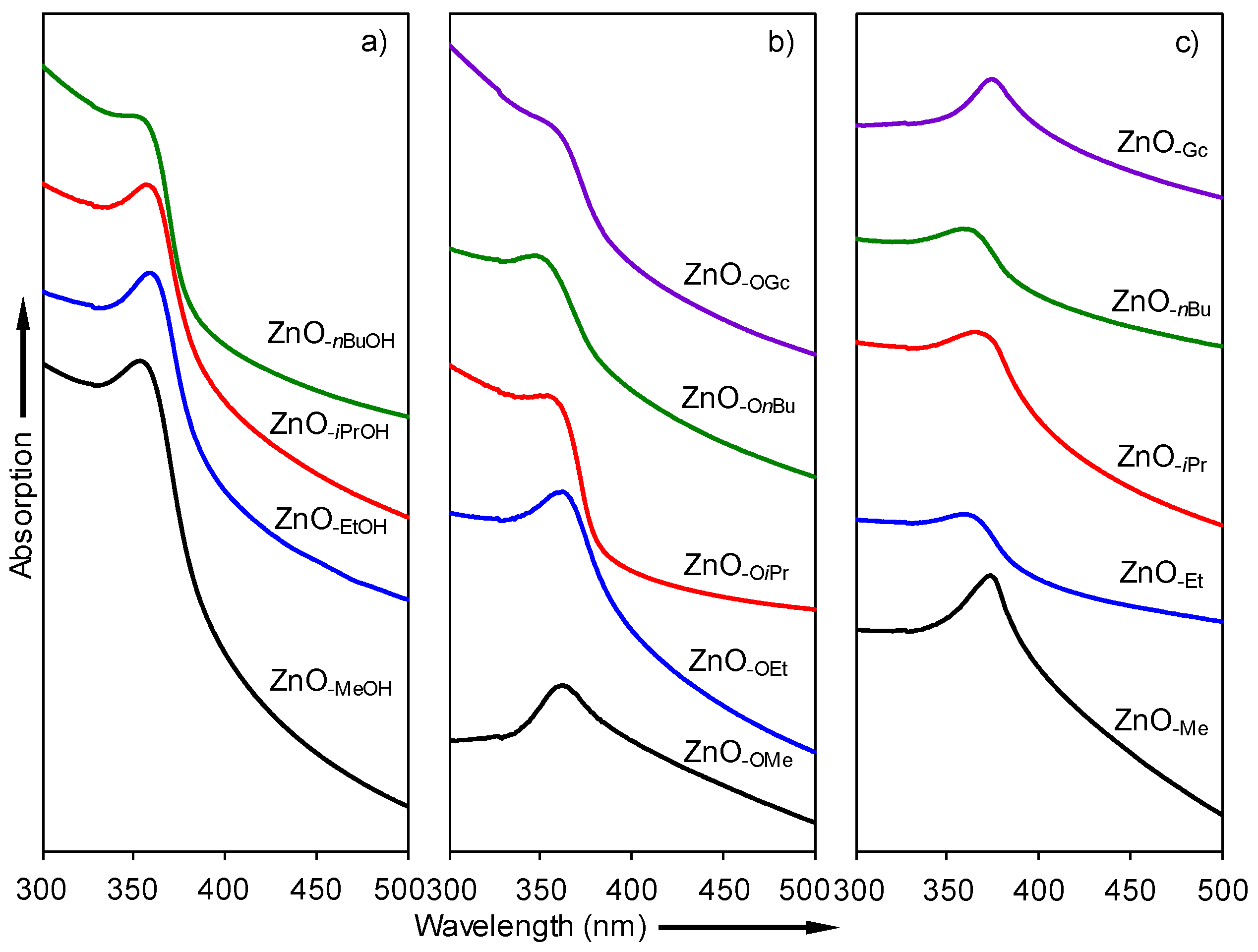
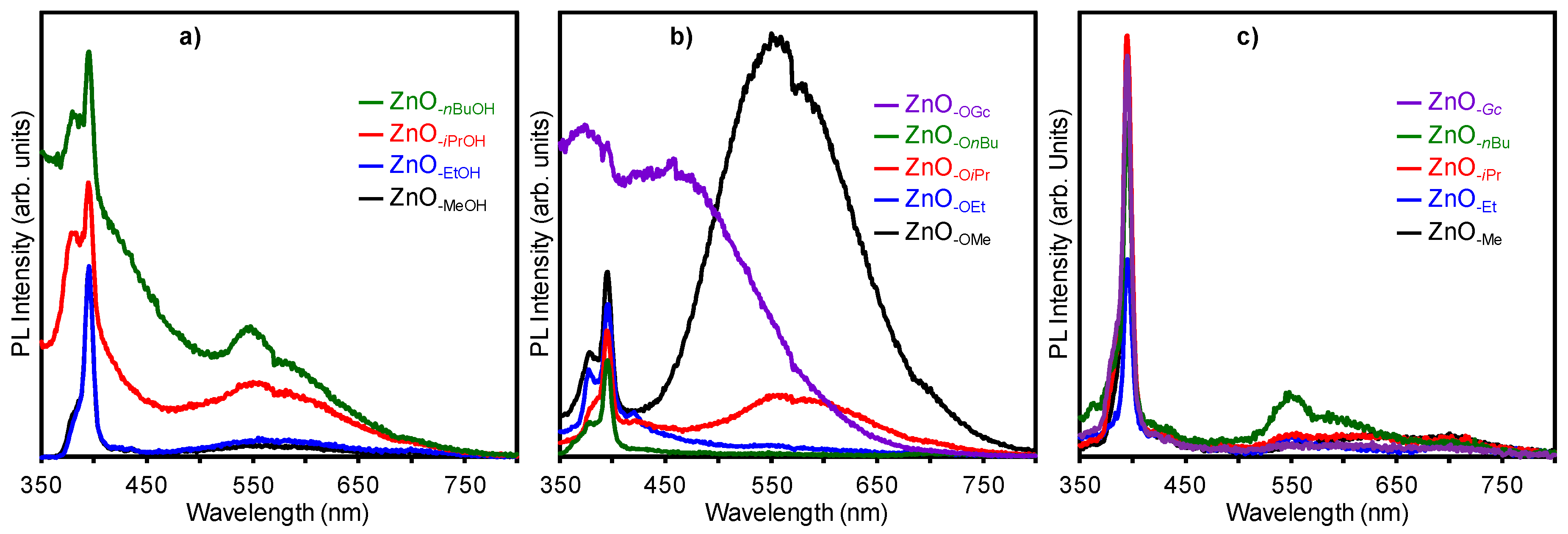
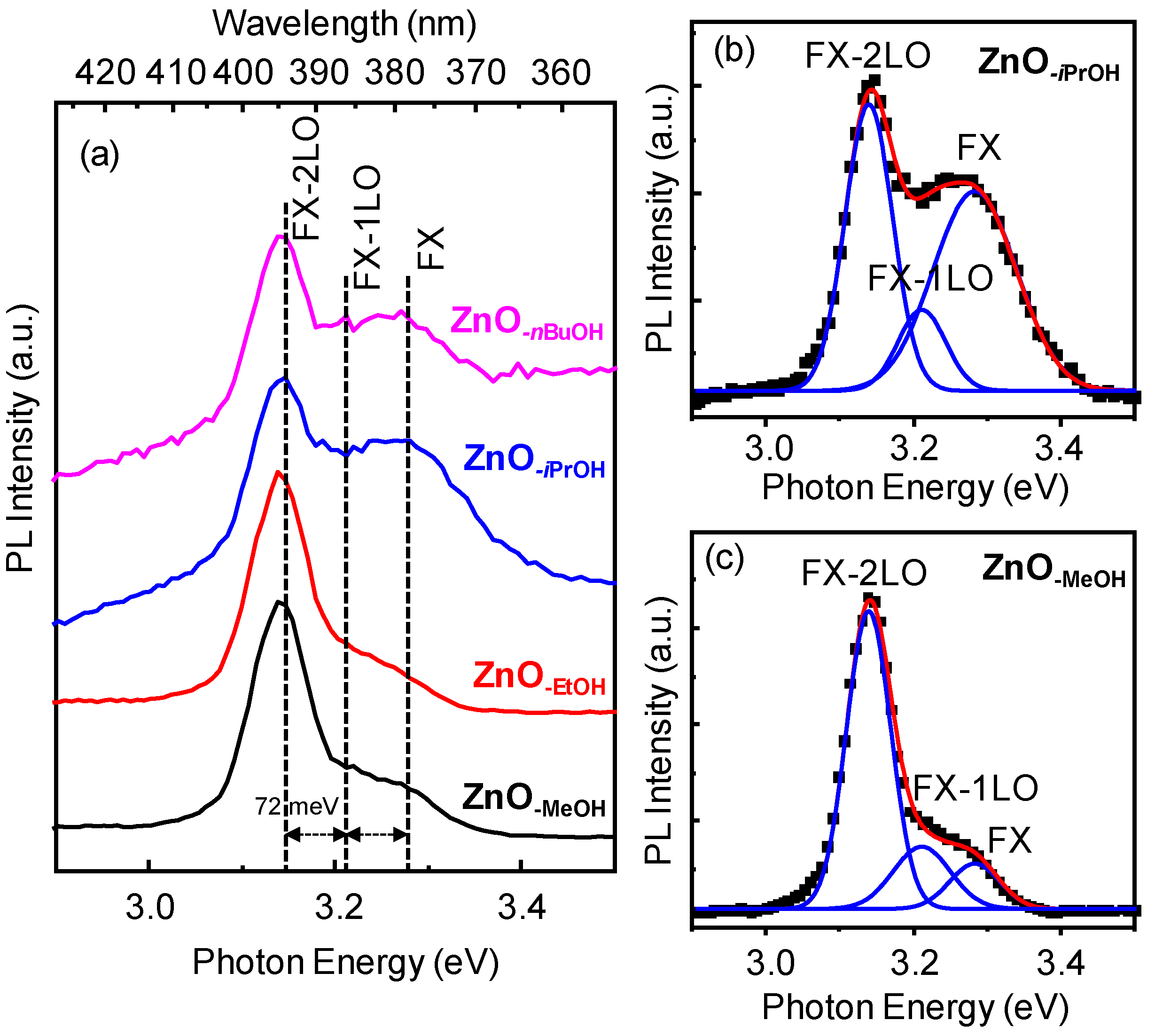
2.3. Optical Properties of a Series of Nano-Structured Zinc Oxide Particles ZnO-ROH, ZnO-OR and ZnO-R (R = Me, Et, iPr, nBu, Gc)
2.3.1. UV-Vis Spectra
2.3.2. Photoluminescence (PL) Property
3. Materials and Methods
3.1. General Consideration
3.2. Synthesis of Crystalline Zinc Oxide NPs
3.2.1. The Preparation of ZnO-ROH (R = Me, Et, iPr, nBu, Gc) Nanoparticles
3.2.2. The Preparation of ZnO-OR (R = Me, Et, iPr, nBu, Gc) Nanoparticles
3.2.3. The Preparation of ZnO-R (R = Me, Et, iPr, nBu, Gc) Nanoparticles
3.3. Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Comini, E.; Sberveglieri, G. Metal Oxides Nanowires as Chemical Sensors. Mater. Today 2014, 13, 36–44. [Google Scholar] [CrossRef]
- Wu, W.-Q.; Chen, D.; Caruso, R.A.; Cheng, Y.-B. Recent Progress in Hybrid Perovskite Solar Cells Based on n-Type Materials. J. Mater. Chem. A 2017, 5, 10092–10109. [Google Scholar] [CrossRef]
- Xue, N.; Zhang, Q.; Zhang, S.; Zong, P.; Yang, F. Highly Sensitive and Selective Hydrogen Gas Sensor Using the Mesoporous SnO2 Modified Layers. Sensors 2017, 17, 2351. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Ma, S.Y.; Li, X.B.; Luo, J.; Li, W.Q.; Li, F.M.; Mao, Y.Z.; Wang, T.T.; Li, Y.F. Highly Sensitive Acetone Sensors Based on Y-Doped SnO2 Prismatic Hollow Nanofibers Synthesized by Electrospinning. Sens. Actuators B Chem. 2014, 200, 181–190. [Google Scholar] [CrossRef]
- Zhai, T.; Li, L.; Wang, X.; Fang, X.; Bando, Y.; Golberg, D. Recent Developments in One-Dimensional Inorganic Nanostructures for Photodetectors. Adv. Funct. Mater. 2010, 20, 4233–4248. [Google Scholar] [CrossRef]
- Yoshida, T.; Zhang, J.; Komatsu, D.; Sawatani, S.; Minoura, H.; Pauporté, T.; Lincot, D.; Oekermann, T.; Schlettwein, D.; Tada, H.; et al. Electrodeposition of Inorganic/Organic Hybrid Thin Films. Adv. Funct. Mater. 2009, 19, 17–43. [Google Scholar] [CrossRef]
- Nikoobakht, B.; Wang, X.; Herzing, A.; Shi, J. Scalable Synthesis and Ddevice Integration of Self-Registered One-Dimensional Zinc Oxide Nanostructures and Related Materials. Chem. Soc. Rev. 2013, 42, 342–365. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhang, S.; Sun, S. High-Temperature Solution-Phase Syntheses of Metal-Oxide Nanocrystals. Chem. Mater. 2013, 25, 1293–1304. [Google Scholar] [CrossRef]
- Bonomo, M.; Naponiello, G.; Venditti, I.; Zardetto, V.; Carlo, A.D.; Dini, D. Electrochemical and Photoelectrochemical Properties of Screen-Printed Nickel Oxide Thin Films Obtained from Precursor Pastes with Different Compositions. J. Electrochem. Soc. 2017, 164, H137–H147. [Google Scholar] [CrossRef]
- Naponiello, G.; Venditti, I.; Zardetto, V.; Saccone, D.; Carlo, A.D.; Fratoddi, I.; Barolo, C.; Dini, D. Photoelectrochemical Characterization of Squaraine-Sensitized Nickel Oxide Cathodes Deposited via Screen-Printing for p-Type Dye-Sensitized Solar Cells. Appl. Surf. Sci. 2015, 356, 911–920. [Google Scholar] [CrossRef]
- Lu, L.T.; Dung, N.T.; Tung, L.D.; Thanh, C.T.; Quy, O.K.; Chuc, N.V.; Maenosono, S.; Thanh, N.T.K. Synthesis of Magnetic Cobalt Ferrite Nanoparticles with Controlled Morphology, Monodispersity and Composition: the Influence of Solvent, Surfactant, Reductant and Synthetic Conditions. Nanoscale 2015, 7, 19596–19610. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhai, T.; Bando, Y.; Golberg, D. Recent Progress of One-Dimensional ZnO Nanostructures Solar Cells. Nano Energy 2012, 1, 91–96. [Google Scholar] [CrossRef]
- Venditti, I.; Barbero, N.; Russo, M.V.; Carlo, A.D.; Decker, F.; Fratoddi, I.; Barolo, C.; Dini, D. Electrodeposited ZnO with Squaraine Sentisizers as Photoactive Anode of DSCs. Mater. Res. Express 2014, 1, 015040. [Google Scholar] [CrossRef]
- Wang, X. Piezoelectric Nanogenerators—Harvesting Ambient Mechanical Energy at the Nanometer Scale. Nano Energy 2012, 1, 13–24. [Google Scholar] [CrossRef]
- Briscoe, J.; Dunnn, S. Piezoelectric Nanogenerators—A Review of Nanostructured Piezoelectric Energy Harvesters. Nano Energy 2015, 14, 15–29. [Google Scholar] [CrossRef]
- Lu, M.-P.; Lu, M.-Y.; Chen, L.-J. p-Type ZnO Nanowires: From Synthesis to Nanoenergy. Nano Energy 2012, 1, 247–258. [Google Scholar] [CrossRef]
- Kumar, B.; Kim, S.-W. Energy Harvesting Based on Semiconducting Piezoelectric ZnO Nanostructures. Nano Energy 2012, 1, 342–355. [Google Scholar] [CrossRef]
- Xiong, H.-M. ZnO Nanoparticles Applied to Bioimaging and Drug Delivery. Adv. Mater. 2013, 25, 5329–5335. [Google Scholar] [CrossRef] [PubMed]
- Dhahri, R.; Leonardi, S.G.; Hjiri, M.; El Mir, L.; Bonavita, A.; Donato, N.; Iannazzo, D.; Neri, G. Enhanced Performance of Novel Calcium/Aluminum co-Doped Zinc Oxide for CO2 Sensors. Sens. Actuators B Chem. 2017, 239, 36–44. [Google Scholar] [CrossRef]
- Özhür, Ü.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Doğan, S.; Avrutin, V.; Cho, S.-J.; Morkoç, H. A Comprehensive Review of ZnO Materials and Devices. J. Appl. Phys. 2005, 98, 041301. [Google Scholar]
- Djurišić, A.B.; Leung, Y.H. Optical Properties of ZnO Nanostructures. Small 2006, 2, 944–961. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, Z.L. One-Dimensional ZnO Nanostructures: Solution Growth and Functional Properties. Nano Res. 2011, 4, 1013–1098. [Google Scholar] [CrossRef]
- Qu, J.; Ge, Y.; Zu, B.; Li, Y.; Dou, X. Transition-Metal-Doped p-Type ZnO Nanoparticle-Based Sensory Array for Instant Discrimination of Explosive Vapors. Small 2016, 12, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- De la Rosa, E.; Sepúlveda-Guzman, S.; Reeja-Jayan, B.; Torres, A.; Salas, P.; Elizondo, N.; Jose Yacaman, M. Controlling the Growth and Luminescence Properties of Well-Faceted ZnO Nanorods. J. Phys. Chem. C 2007, 111, 8489–8495. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, L.; Wu, D. Fabrication of Superhydrophobic Surfaces from Microstructured ZnO-Based Surfaces via a Wet-Chemical Route. Langmuir 2005, 21, 2665–2667. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.M.; Muoz-Esp, R.; Lieberwirth, I.; Wegner, G. Precipitation of Monodisperse ZnO Nanocrystals via Acid-Catalyzed Esterification of Zinc Acetate. J. Mater. Chem. 2006, 16, 2940–2947. [Google Scholar] [CrossRef]
- Wu, J.-J.; Liu, S.-C. Low-Temperature Growth of Well-Aligned ZnO Nanorods by Chemical Vapor Deposition. Adv. Mater. 2002, 14, 215–218. [Google Scholar] [CrossRef]
- Izaki, M.; Omi, T. Transparent Zinc Oxide Films Prepared by Electrochemical Reaction. Appl. Phys. Lett. 1996, 68, 2439–2440. [Google Scholar] [CrossRef]
- Wu, K.; Sun, Z.; Cui, J. Unique Approach toward ZnO Growth with Tunable Properties: Influence of Methanol in an Electrochemical Process. Cryst. Growth Des. 2012, 12, 2864–2871. [Google Scholar] [CrossRef]
- Elias, J.; Lvy-Clment, C.; Bechelany, M.; Michler, J.; Wang, G.-Y.; Wang, Z.; Philippe, L. Hollow Urchin-like ZnO Thin Films by Electrochemical Deposition. Adv. Mater. 2010, 22, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Manzano, C.V.; Caballero-Calero, O.; Hormeño, S.; Penedo, M.; Luna, M.; Martín-González, M.S. ZnO Morphology Control by Pulsed Electrodeposition. J. Phys. Chem. C 2013, 117, 1502–1508. [Google Scholar] [CrossRef]
- Minch, R.; Es-Souni, M. A Versatile Approach to Processing of High Active Area Pillar Coral- and Sponge-Like Pt-Nanostructures. Application to Electrocatalysis. J. Mater. Chem. 2011, 21, 4182–4188. [Google Scholar] [CrossRef]
- Sun, Y.; Fuge, G.M.; Ashfold, M.N.R. Growth of Aligned ZnO Nanorod Arrays by Catalyst-Free Pulsed Laser Deposition Methods. Chem. Phys. Lett. 2004, 396, 21–26. [Google Scholar] [CrossRef]
- Hong, J.I.; Bae, J.; Wang, Z.L.; Snyder, R.L. Room-Temperature, Texture-Controlled Growth of ZnO Thin Ffilms and Their Application for Growing Aligned ZnO Nanowire Arrays. Nanotechnology 2009, 20, 085609. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.W.; Varadarajan, V.; Kaufman, M.; Kim, K.; Norton, D.P.; Ren, F.; Fleming, P.H. Site-Specific Growth of Zno Nanorods Using Catalysis-Driven Molecular-Beam Epitaxy. Appl. Phys. Lett. 2002, 81, 3046–3048. [Google Scholar] [CrossRef]
- Lin, D.; Wu, H.; Pan, W. Photoswitches and Memories Assembled by Electrospinning Aluminum-Doped Zinc Oxide Single Nanowires. Adv. Mater. 2007, 19, 3968–3972. [Google Scholar] [CrossRef]
- Schneider, J.J.; Hoffmann, R.C.; Engstler, J.; Klyszcz, A.; Erdem, E.; Jakes, P.; Eichel, R.-A.; Pitta-Bauermann, L.; Bill, J. Synthesis, Characterization, Defect Chemistry, and FET Properties of Microwave-Derived Nanoscaled Zinc Oxide. Chem. Mater. 2010, 22, 2203–2212. [Google Scholar] [CrossRef]
- Bilecka, I.; Djerdj, I.; Niederberger, M. One-Minute Synthesis of Crystalline Binary and Ternary Metal Oxide Nanoparticles. Chem. Commun. 2008, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Bilecka, I.; Elser, P.; Niederberger, M. Kinetic and Thermodynamic Aspects in the Microwave-Assisted Synthesis of ZnO Nanoparticles in Benzyl Alcohol. ACS Nano 2009, 3, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Gong, J.; Zhang, L.; Yu, J.C. Continuous Size Tuning of Monodisperse ZnO Colloidal Nanocrystal Clusters by a Microwave-Polyol Process and Their Application for Humidity Sensing. Adv. Mater. 2008, 20, 4845–4850. [Google Scholar] [CrossRef]
- Tang, J.; Cui, X.; Liu, Y.; Yang, X. Morphology-Controlled Synthesis of Monodisperse ZnO Troughs at the Air-Water Interface under Mild Conditions. J. Phys. Chem. B 2005, 109, 22244–22249. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Xu, S.; Zhuang, Z.; Wang, X.; Li, Y. A General Chemical Conversion Method to Various Semiconductor Hollow Structures. Small 2005, 1, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.-P.; Zhang, Y.-W.; Sun, L.-D.; Yan, C.-H. Efficient Energy Transfer in Monodisperse Eu-Doped ZnO Nanocrystals Synthesized from Metal Acetylacetonates in High-Boiling Solvents. J. Phys. Chem. C 2008, 112, 12234–12241. [Google Scholar] [CrossRef]
- Joo, J.; Kwon, S.G.; Yu, J.H.; Hyeon, T. Synthesis of ZnO Nanocrystals with Cone, Hexagonal Cone, and Rod Shapes via Non-Hydrolytic Ester Elimination Sol-Gel Reactions. Adv. Mater. 2005, 17, 1873–1877. [Google Scholar] [CrossRef]
- Kwon, S.G.; Hyeon, T. Colloidal Chemical Synthesis and Formation Kinetics of Uniformly Sized Nanocrystals of Metals, Oxides, and Chalcogenides. Acc. Chem. Res. 2008, 41, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Sarkara, D.; Tikkub, S.; Thaparb, V.; Srinivasac, R.S.; Khilar, K.C. Formation of Zinc Oxide Nanoparticles of Different Shapes in Water-in-Oil Microemulsion. Colloids Surf. A Physicochem. Eng. Asp. 2011, 381, 123–129. [Google Scholar] [CrossRef]
- Lin, C.-C.; Li, Y.-Y. Synthesis of ZnO Nanowires by Thermal Decomposition of Zinc Acetate Dihydrate. Mater. Chem. Phys. 2009, 113, 334–337. [Google Scholar] [CrossRef]
- Audebrand, N.; Auffrédic, J.-P.; Louër, D. X-ray Diffraction Study of the Early Stages of the Growth of Nanoscale Zinc Oxide Crystallites Obtained from Thermal Decomposition of Four Precursors. General Concepts on Precursor-Dependent Microstructural Properties. Chem. Mater. 1998, 10, 2450–2461. [Google Scholar] [CrossRef]
- Shim, M.; Guyot-Sionnest, P. Organic-Capped ZnO Nanocrystals: Synthesis and n-Type Character. J. Am. Chem. Soc. 2001, 123, 11651–11654. [Google Scholar] [CrossRef] [PubMed]
- Carnes, C.L.; Klabunde, K.J. Synthesis, Isolation, and Chemical Reactivity Studies of Nanocrystalline Zinc Oxide. Langmuir 2000, 16, 3764–3772. [Google Scholar] [CrossRef]
- Monge, M.; Kahn, M.L.; Maisonnat, A.; Chaudret, B. Room-Temperature Organometallic Synthesis of Soluble and Crystalline ZnO Nanoparticles of Controlled Size and Shape. Angew. Chem. Int. Ed. 2003, 42, 5321–5324. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.L.; Monge, M.; Collière, V.; Senocq, F.; Maisonnat, A.; Chaudret, B. Size- and Shape-Control of Crystalline Zinc Oxide Nanoparticles: A New Organometallic Synthetic Method. Adv. Funct. Mater. 2005, 15, 458–468. [Google Scholar] [CrossRef]
- Auld, J.; Houlton, D.J.; Jones, A.C.; Rushworth, S.A.; Malik, M.A.; O’Brien, P.; Critchlow, G.W. Growth of ZnO by MOCVD using Alkylzinc Alkoxides as Singlesource Precursors. J. Mater. Chem. 1994, 4, 1249–1253. [Google Scholar] [CrossRef]
- Polarz, S.; Roy, A.; Merz, M.; Halm, S.; Schröder, D.; Schneider, L.; Bacher, G.; Kruis, F.E.; Driess, M. Chemical Vapor Synthesis of Size-Selected Zinc Oxide Nanoparticles. Small 2005, 1, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Polarz, S.; Orlov, A.; Hoffmann, A.; Wagner, M.R.; Rauch, C.; Kirste, R.; Gehlhoff, W.; Aksu, Y.; Driess, M.; van den Berg, M.W.E.; et al. A Systematic Study on Zinc Oxide Materials Containing Group I Metals (Li, Na, K)-Synthesis from Organometallic Precursors, Characterization, and Properties. Chem. Mater. 2009, 21, 3889–3897. [Google Scholar] [CrossRef]
- Kim, C.G.; Sung, K.; Chung, T.-M.; Jung, D.Y.; Kim, Y. Monodispersed ZnO Nanoparticles from a Single Molecular Precursor. Chem. Commun. 2003, 2068–2069. [Google Scholar] [CrossRef]
- Polarz, S.; Regenspurger, R.; Hartmann, J. Self-Assembly of Methylzinc-Polyethylene Glycol Amphiphiles and Their Application to Materials Synthesis. Angew. Chem. Int. Ed. 2007, 46, 2426–2430. [Google Scholar] [CrossRef] [PubMed]
- Polarz, S.; Neues, F.; van den Berg, M.W.E.; Grünert, W.; Khodeir, L. Mesosynthesis of ZnO-Silica Composites for Methanol Nanocatalysis. J. Am. Chem. Soc. 2005, 127, 12028–12034. [Google Scholar] [CrossRef] [PubMed]
- Bury, W.; Krajewska, E.; Dutkiewicz, M.; Sokołowski, K.; Justyniak, I.; Kaszkur, Z.; Kurzydłowski, K.J.; Płocińskic, T.; Lewiński, J. tert-Butylzinc Hydroxide as an Efficient Predesigned Precursor of ZnO Nanoparticles. Chem. Commun. 2011, 47, 5467–5469. [Google Scholar] [CrossRef] [PubMed]
- Sokołowski, K.; Justyniak, I.; Bury, W.; Grzonka, J.; Kaszkur, Z.; Mąkolski, Ł.; Dutkiewicz, M.; Lewalska, A.; Krajewska, E.; Kubicki, D.; et al. tert-Butyl(tert-butoxy)zinc Hydroxides: Hybrid Models for Single-Source Precursors of ZnO Nanocrystals. Chem. Eur. J. 2015, 21, 5488–5495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, Y.H. A Systematic Investigation of Decomposition of Nano Zn4O(C8H4O4)3 Metal-Organic Framework. J. Phys. Chem. C 2010, 114, 2566–2572. [Google Scholar] [CrossRef]
- Liu, J.F.; Bei, Y.Y.; Wu, H.P.; Shen, D.; Gong, J.Z.; Li, X.G.; Wang, Y.W.; Jiang, N.P.; Jiang, J.Z. Synthesis of Relatively Monodisperse ZnO Nanocrystals from a Precursor Zinc 2,4-Pentanedionate. Mater. Lett. 2007, 61, 2837–2840. [Google Scholar] [CrossRef]
- Huang, M.H.; Mao, S.; Feick, H.; Yan, H.; Wu, Y.; Kind, H.; Weber, E.; Russo, R.; Yang, P. Room-Temperature Ultraviolet Nanowire Nanolasers. Science 2001, 292, 1897–1899. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.H.; Wu, Y.; Feick, H.; Tran, N.; Weber, E.; Yang, P. Catalytic Growth of Zinc Oxide Nanowires by Vapor Transport. Adv. Mater. 2001, 13, 113–116. [Google Scholar] [CrossRef]
- Andelman, T.; Gong, Y.; Polking, M.; Yin, M.; Kuskovsky, I.; Neumark, G.; O’Brien, S. Morphological Control and Photoluminescence of Zinc Oxide Nanocrystals. J. Phys. Chem. B 2005, 109, 14314–14318. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Gu, Y.; Kuskovsky, I.L.; Andelman, T.; Zhu, Y.; Neumark, G.F.; O’Brien, S. Zinc Oxide Quantum Rods. J. Am. Chem. Soc. 2004, 126, 6206–6207. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, A.; Thareja, R.K. Photoluminescence Study of ZnO Nanowires Grown by Thermal Evaporation on Pulsed Laser Deposited ZnO Buffer Layer. J. Appl. Phys. 2008, 104, 044906. [Google Scholar] [CrossRef]
- Mohanta, A.; Thareja, R.K. Photoluminescence Characteristics of Catalyst Free ZnO Nanowires. Mater. Res. Express 2014, 1, 015023. [Google Scholar] [CrossRef]
- Mohanta, A.; Singh, V.; Thareja, R.K. Photoluminescence from ZnO Nanoparticles in Vapor Phase. J. Appl. Phys. 2008, 104, 064903. [Google Scholar] [CrossRef]
- Mohanta, A.; Kung, P.; Thareja, R.K. Exciton-Exciton Scattering in Vapor Phase ZnO Nanoparticles. Appl. Phys. Lett. 2015, 106, 013108. [Google Scholar] [CrossRef]
- Ma, D.; Huang, J.; Ye, Z.; Wang, L.; Zhao, B. Relationship between Photoluminescence and Structural Properties of the Sputtered Zn1−xCdxO Films on Si Substrates. Opt. Mater. 2004, 25, 367–371. [Google Scholar] [CrossRef]
- Mohanta, A.; Thareja, R.K. Photoluminescence Study of ZnCdO Alloy. J. Appl. Phys. 2008, 103, 024901. [Google Scholar] [CrossRef]
- Mohanta, A.; Thareja, R.K. Temperature-Dependent S-Shaped Photoluminescence in ZnCdO Alloy. J. Appl. Phys. 2010, 107, 084904. [Google Scholar] [CrossRef]
- Boulmaâz, S.; Hubert-Pfalzgraf, L.G. The Quest for Mixed-Metal Alkoxides Based on Zinc: Synthesis and Characterization of Zinc-Tantalum Oxoisopropoxides. J. Sol-Gel Sci. Technol. 1994, 2, 11–15. [Google Scholar] [CrossRef]
- Bochmann, M.; Bwembya, G.; Webb, K.J. Arene Chalrogenolato Complexes of Zinc And Cadmium. Inorg. Synth. 1997, 31, 19–24. [Google Scholar]
- Wang, Y.; Zhang, J.; Zhao, Y. Strength Weakening by Nanocrystals in Ceramic Materials. Nano Lett. 2007, 7, 3196–3199. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Khushalani, D. Nonhydrolytic Route for Synthesis of ZnO and Its Use as a Recyclable Photocatalyst. J. Phys. Chem. C 2010, 114, 2544–2550. [Google Scholar] [CrossRef]
- Dong, H.; Feldmann, C. Porous ZnO Platelets via Controlled Thermal Decomposition of Zinc Glycerolate. J. Alloys Compd. 2012, 513, 125–129. [Google Scholar] [CrossRef]
- Reinoso, D.M.; Damiani, D.E.; Tonetto, G.M. Zinc Glycerolate as a Novel Heterogeneous Catalyst for the Synthesis Offatty Acid Methyl Esters. Appl. Catal. B Environ. 2014, 144, 308–316. [Google Scholar] [CrossRef]
- Polarz, S.; Strunk, J.; Ischenko, V.; van den Berg, M.W.E.; Hinrichsen, O.; Muhler, M.; Driess, M. On the Role of Oxygen Defects in the Catalytic Performance of Zinc Oxide. Angew. Chem. Int. Ed. 2006, 45, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, V.; Polarz, S.; Grote, D.; Stavarache, V.; Fink, K.; Driess, M. Zinc Oxide Nanoparticles with Defects. Adv. Funct. Mater. 2005, 15, 1945–1954. [Google Scholar] [CrossRef]
- Parashar, S.K.S.; Murty, B.S.; Repp, S.; Weber, S.; Erdem, E. Investigation of Intrinsic Defects in Core-Shell Structured ZnO Nanocrystals. J. Appl. Phys. 2012, 111, 113712. [Google Scholar] [CrossRef]
- Vanheusden, K.; Seager, C.H.; Warren, W.L.; Tallant, D.R.; Voigt, J.A. Correlation Between Photoluminescence and Oxygen Vacancies in ZnO Phosphors. Appl. Phys. Lett. 1996, 68, 403–405. [Google Scholar] [CrossRef]
- Wang, X.Y.; Vlasenko, L.S.; Pearton, S.J.; Chen, W.M.; Buyanova, I.A. Oxygen and Zinc Vacancies in As-Grown ZnO Single Crystals. J. Phys. D Appl. Phys. 2009, 42, 175411. [Google Scholar] [CrossRef]
- Janotti, A.; Van de Walle, C.G. Oxygen Vacancies in ZnO. Appl. Phys. Lett. 2005, 87, 122102. [Google Scholar] [CrossRef]
- Drouilly, C.; Krafft, J.-M.; Averseng, F.; Casale, S.; Bazer-Bachi, D.; Chizallet, C.; Lecocq, V.; Vezin, H.; Lauron-Pernot, H.; Costentin, G. ZnO Oxygen Vacancies Formation and Filling Followed by in Situ Photoluminescence and in Situ EPR. J. Phys. Chem. C 2012, 116, 21297–21307. [Google Scholar] [CrossRef]
- Stavale, F.; Nilius, N.; Freund, H.-J. STM Luminescence Spectroscopy of Intrinsic Defects in ZnO(0001) Thin Films. J. Phys. Chem. Lett. 2013, 4, 3972–3976. [Google Scholar] [CrossRef]
- Kaftelen, H.; Ocakoglu, K.; Thomann, R.; Tu, S.; Weber, S.; Erdem, E. EPR and Photoluminescence Spectroscopy Studies on the Defect Structure of ZnO Nanocrystals. Phys. Rev. B 2012, 86, 014113. [Google Scholar] [CrossRef]
- Galland, D.; Herve, A. ESR Spectra of the Zinc Vacancy in ZnO. Phys. Lett. A 1970, 33, 1–2. [Google Scholar] [CrossRef]
- Taylor, A.L.; Filipovi, G.; Lindeber, G. Electron Paramagnetic Resonance Associated with Zn Vacancies in Neutron-Irradiated ZnO. Solid State Commun. 1970, 8, 1359–1361. [Google Scholar] [CrossRef]
- Yu, B.; Zhu, C.; Gan, F.; Huang, Y. Electron Spin Resonance Properties of ZnO Microcrystallites. Mater. Lett. 1998, 33, 247–250. [Google Scholar] [CrossRef]
- Jing, L.Q.; Xu, Z.L.; Shang, J.; Sun, X.J.; Cai, W.M.; Guo, H.C. The Preparation and Characterization of ZnO Ultrafine Particles. Mater. Sci. Eng. A 2002, 332, 356–361. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Yin, L.W.; Wang, C.X.; Lun, N.; Qi, Y.X.; Xiang, D. Origin of Visible Photoluminescence of ZnO Quantum Dots: Defect-Dependent and Size-Dependent. J. Phys. Chem. C 2010, 114, 9651–9658. [Google Scholar] [CrossRef]
- Kakazev, N.G.; Sreckovic, T.V.; Ristic, M.M. Electronic Paramagnetic Resonance Investigation of the Evolution of Defects in Zinc Oxide During Tribophysical Activation. J. Mater. Sci. 1997, 32, 4619–4622. [Google Scholar] [CrossRef]
- Schulz, M. ESR Experiments on Ga Donors in ZnO Crystals. Phys. Status Solidi A 1975, 27, K5–K8. [Google Scholar] [CrossRef]
- Block, D.; Hereve, A.; Cox, R.T. Optically Detected Magnetic Resonance and Optically Detected ENDOR of Shallow Indium Donors in ZnO. Phys. Rev. B 1982, 25, 6049. [Google Scholar] [CrossRef]
- Gonzales, C.; Block, D.; Cox, R.T.; Herve, A. Magnetic Resonance Studies of Shallow Donors in Zinc Oxide. J. Cryst. Growth 1982, 59, 357–362. [Google Scholar] [CrossRef]
- Garces, N.Y.; Wang, L.; Bai, L.; Giles, N.C.; Halliburton, L.E.; Cantwell, G. Role of Copper in the Green Luminescence from ZnO Crystals. Appl. Phys. Lett. 2002, 81, 622–624. [Google Scholar] [CrossRef]
- La Porta, F.A.; Andrés, J.; Vismara, M.V.G.; Graeff, C.F.O.; Sambrano, J.R.; Li, M.S.; Varela, J.A.; Longo, E. Correlation between Structural and Electronic Order-Disorder Effects and Optical Properties in ZnO Nanocrystals. J. Mater. Chem. C 2014, 2, 10164–10174. [Google Scholar] [CrossRef]
- Mondal, O.; Pal, M. Strong and Unusual Violet-Blue Emission in Ring Shaped ZnO Nanocrystals. J. Mater. Chem. 2011, 21, 18354–18358. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Xiang, Q.; Li, H.; Pan, Q.; Xu, P. Brush-Like Hierarchical ZnO Nanostructures: Synthesis, Photoluminescence and Gas Sensor Properties. J. Phys. Chem. C 2009, 113, 3430–3435. [Google Scholar] [CrossRef]
- Koch, U.; Fojtik, A.; Weller, H.; Henglein, A. Photochemistry of Semiconductor Colloids. Preparation of Extremely Small ZnO Particles, Fluorescence Phenomena and Size Quantization Effects. Chem. Phys Lett. 1982, 122, 507–510. [Google Scholar] [CrossRef]
- Wang, L.; Giles, N.C. Temperature Dependence of the Free-Exciton Transition Energy in Zinc Oxide by Photoluminescence Excitation Spectroscopy. J. Appl. Phys. 2003, 94, 973–978. [Google Scholar] [CrossRef]
- Hsu, H.-C.; Hsieh, W.-F. Excitonic Polaron and Phonon Assisted Photoluminescence of ZnO Nanowires. Solid State Commun. 2004, 131, 371–375. [Google Scholar] [CrossRef]
- Liu, R.; Pan, A.; Fan, H.; Wang, F.; Shen, Z.; Yang, G.; Xie, S.; Zou, B. Phonon-Assisted Stimulated Emission in Mn-Doped ZnO Nanowires. J. Phys. Condens. Matter 2007, 19, 136206. [Google Scholar] [CrossRef]
- Yang, S.; Tian, X.; Wang, L.; Wei, J.; Qi, K.; Li, X.; Xu, Z.; Wang, W.; Zhao, J.; Bai, X.; et al. In-situ Optical Transmission Electron Microscope Study of Exciton Phonon Replicas in ZnO Nanowires by Cathodoluminescence. Appl. Phys. Lett. 2014, 105, 071901. [Google Scholar] [CrossRef]
- Wang, X.; Ding, Y.; Summers, C.J.; Wang, Z.L. Large-Scale Synthesis of Six-Nanometer-Wide ZnO Nanobelts. J. Phys. Chem. B 2004, 108, 8773–8777. [Google Scholar] [CrossRef]
- Fonoberov, V.A.; Balandin, A.A. Origin of Ultraviolet Photoluminescence in ZnO Quantum Dots: Confined Excitons Versus Surface-Bound Impurity Exciton Complexes. Appl. Phys. Lett. 2004, 85, 5971–5973. [Google Scholar] [CrossRef]
- Li, J.W.; Yang, L.W.; Zhou, Z.F.; Chu, P.K.; Wang, X.H.; Zhou, J.; Li, L.T.; Sun, C.Q. Bandgap Modulation in ZnO by Size, Pressure, and Temperature. J. Phys. Chem. C 2010, 114, 13370–13374. [Google Scholar] [CrossRef]
- Kohan, A.F.; Ceder, G.; Morgan, D.; Van de Walle, C.G. First-Principles Study of Native Point Defects in ZnO. Phys. Rev. B 2000, 61, 15019. [Google Scholar] [CrossRef]
- Lin, B.; Fu, Z.; Jia, Y. Green Luminescent Center in Undoped Zinc Oxide Films Deposited on Silicon Substrates. Appl. Phys. Lett. 2001, 79, 943–945. [Google Scholar] [CrossRef]
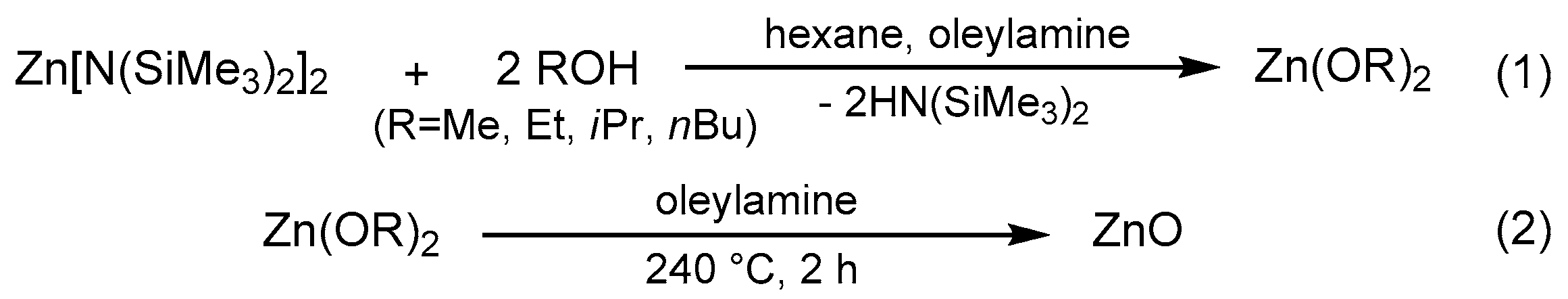







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
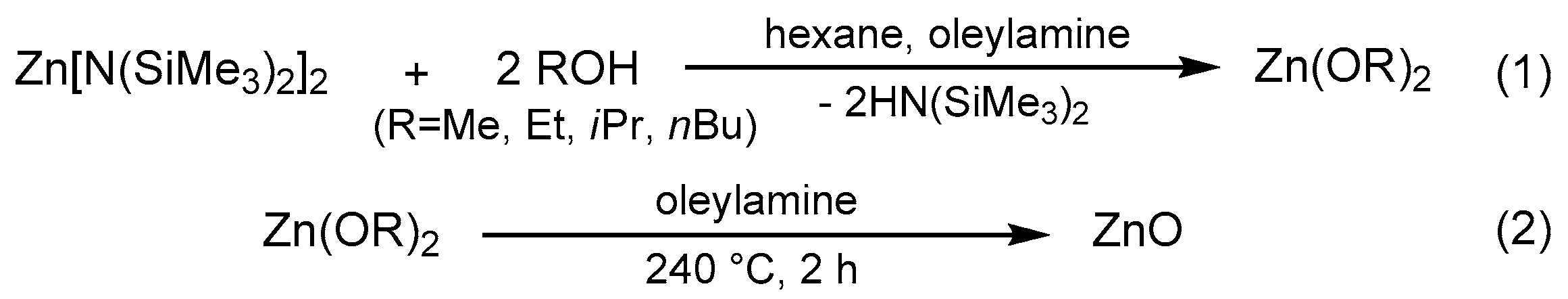{kind=link}
| Sample | Particle Size/nm a | Particle Size/nm b | Electron g-Factor | Absorption Edge/nm |
|---|---|---|---|---|
| ZnO-MeOH | 5.9 ± 0.3 | 4.55; 4.86 | 2.00 | 352 |
| ZnO-EtOH | 11.7 ± 0.6 | 10.7; 10.2 | 2.02, 2.00, 1.99, 1.96 | 358 |
| ZnO-iPrOH | 7.5 ± 0.2 | 9.0; 8.3 | 2.00 | 356 |
| ZnO-nBuOH | - | 4.3 | 2.12, 2.07, 2.00, 1.96 | 351 |
| ZnO-GcOH | - | 3.8 | - | - |
| ZnO-OMe | different sizes | 5.3 | 2.01, 2.00, 1.99 | 364 |
| ZnO-OEt | 5.4 ± 0.1 | 4.8 | 2.12~1.90 | 361 |
| ZnO-OiPr | 4.2 ± 0.6 | 4.8 | 2.12~1.90 | 347 |
| ZnO-OnBu | 4.5 ± 0.2 | 5.1 | - | 346 |
| ZnO-OGc | - | - | 2.00 | 348 |
| ZnO-Me | - | 34.1 | 1.96 | 373 |
| ZnO-Et | - | 7.5 | 1.96 | 358 |
| ZnO-iPr | - | 28.4 | 1.96 | 365 |
| ZnO-nBu | - | 5.4 | 1.96 | 358 |
| ZnO-Gc | - | 21.3 | 2.00, 1.96 | 373 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Wicker, S.; Wang, X.; Erichsen, E.S.; Fu, F. Organozinc Precursor-Derived Crystalline ZnO Nanoparticles: Synthesis, Characterization and Their Spectroscopic Properties. Nanomaterials 2018, 8, 22. https://doi.org/10.3390/nano8010022
Liang Y, Wicker S, Wang X, Erichsen ES, Fu F. Organozinc Precursor-Derived Crystalline ZnO Nanoparticles: Synthesis, Characterization and Their Spectroscopic Properties. Nanomaterials. 2018; 8(1):22. https://doi.org/10.3390/nano8010022
Chicago/Turabian StyleLiang, Yucang, Susanne Wicker, Xiao Wang, Egil Severin Erichsen, and Feng Fu. 2018. "Organozinc Precursor-Derived Crystalline ZnO Nanoparticles: Synthesis, Characterization and Their Spectroscopic Properties" Nanomaterials 8, no. 1: 22. https://doi.org/10.3390/nano8010022