Synthesis of Microspherical LiFePO4-Carbon Composites for Lithium-Ion Batteries

Abstract
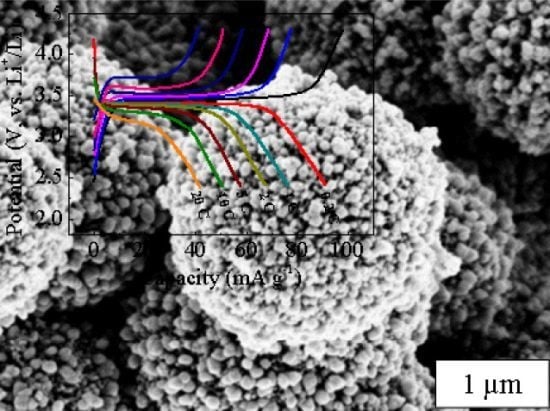
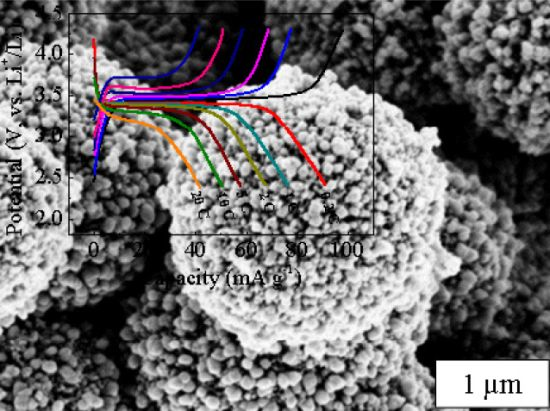
:
{kind=link}
{kind=link}
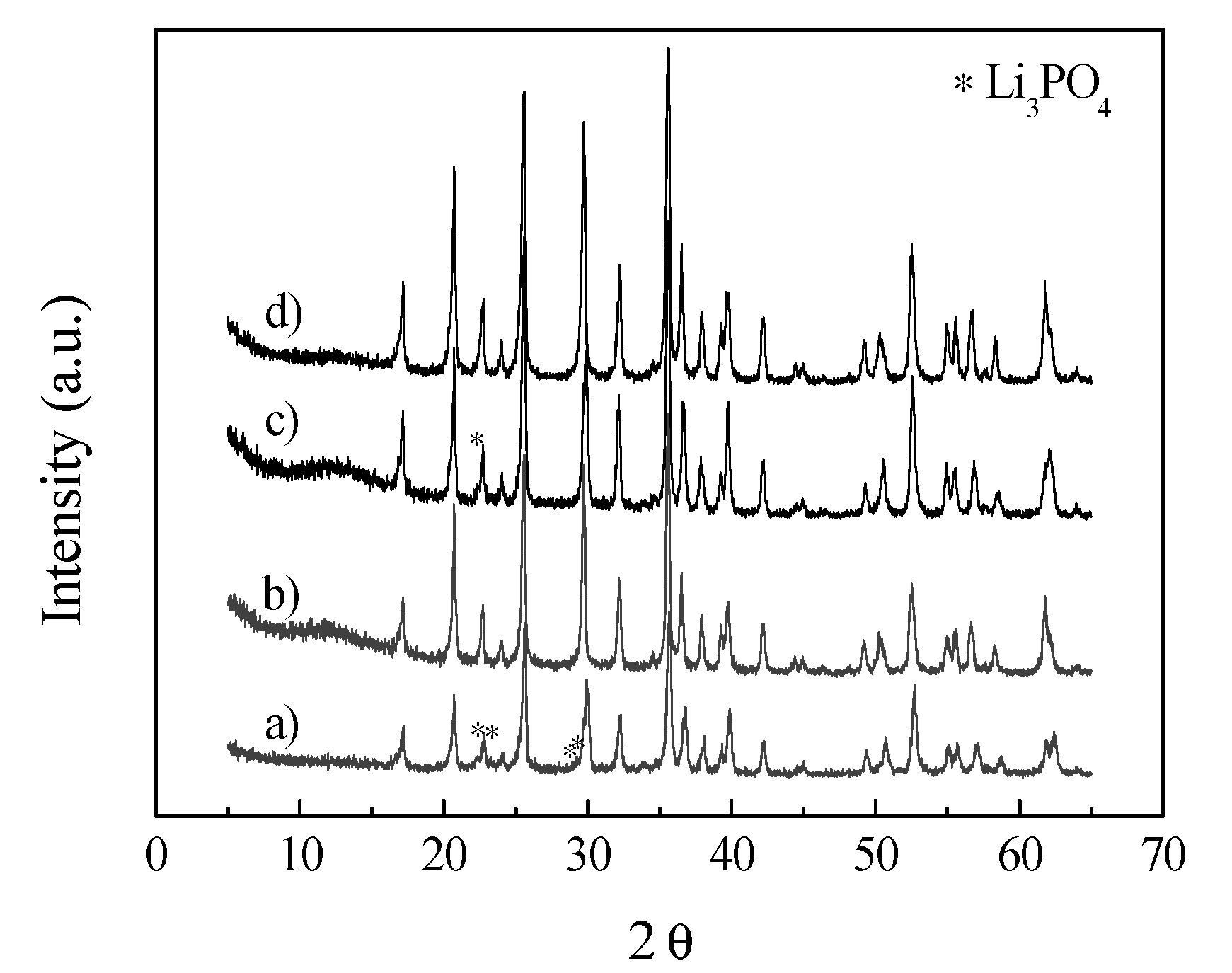{kind=link}
{kind=link}
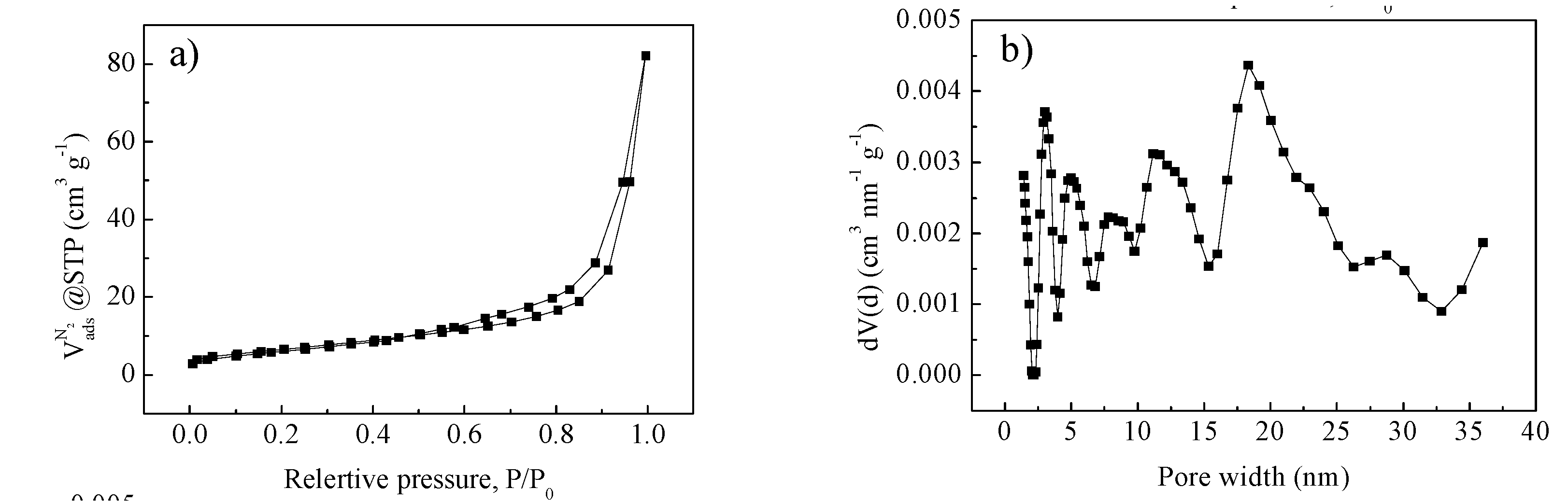{kind=link}
{kind=link}
{kind=link}
1. Introduction
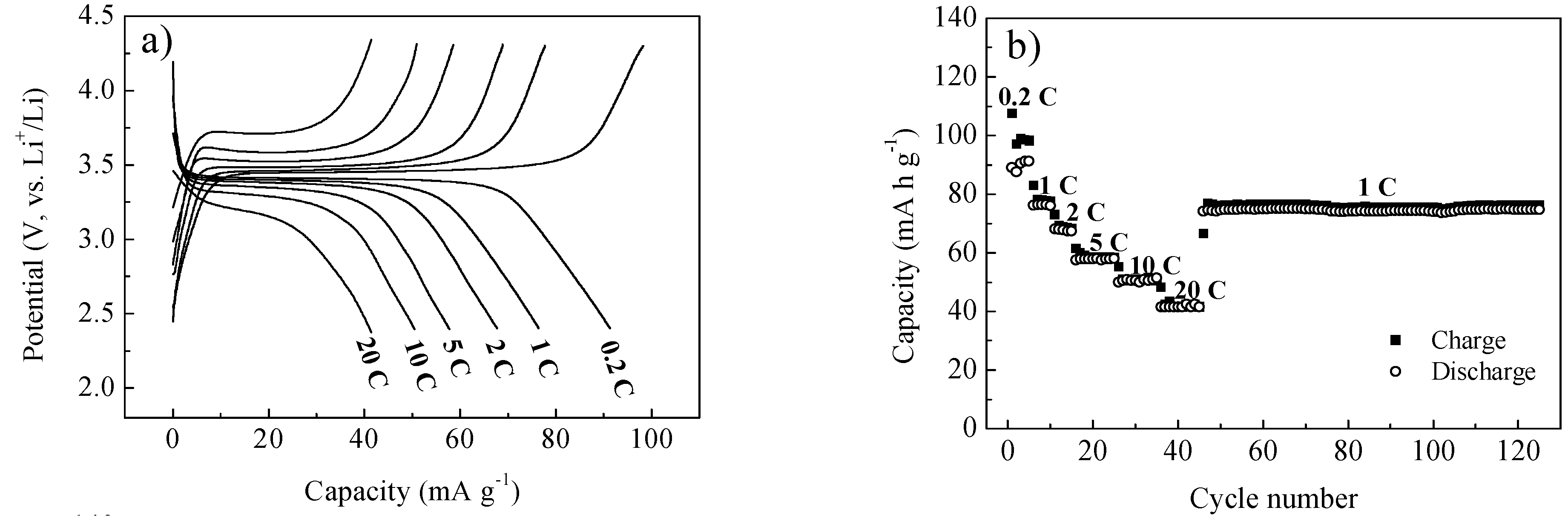
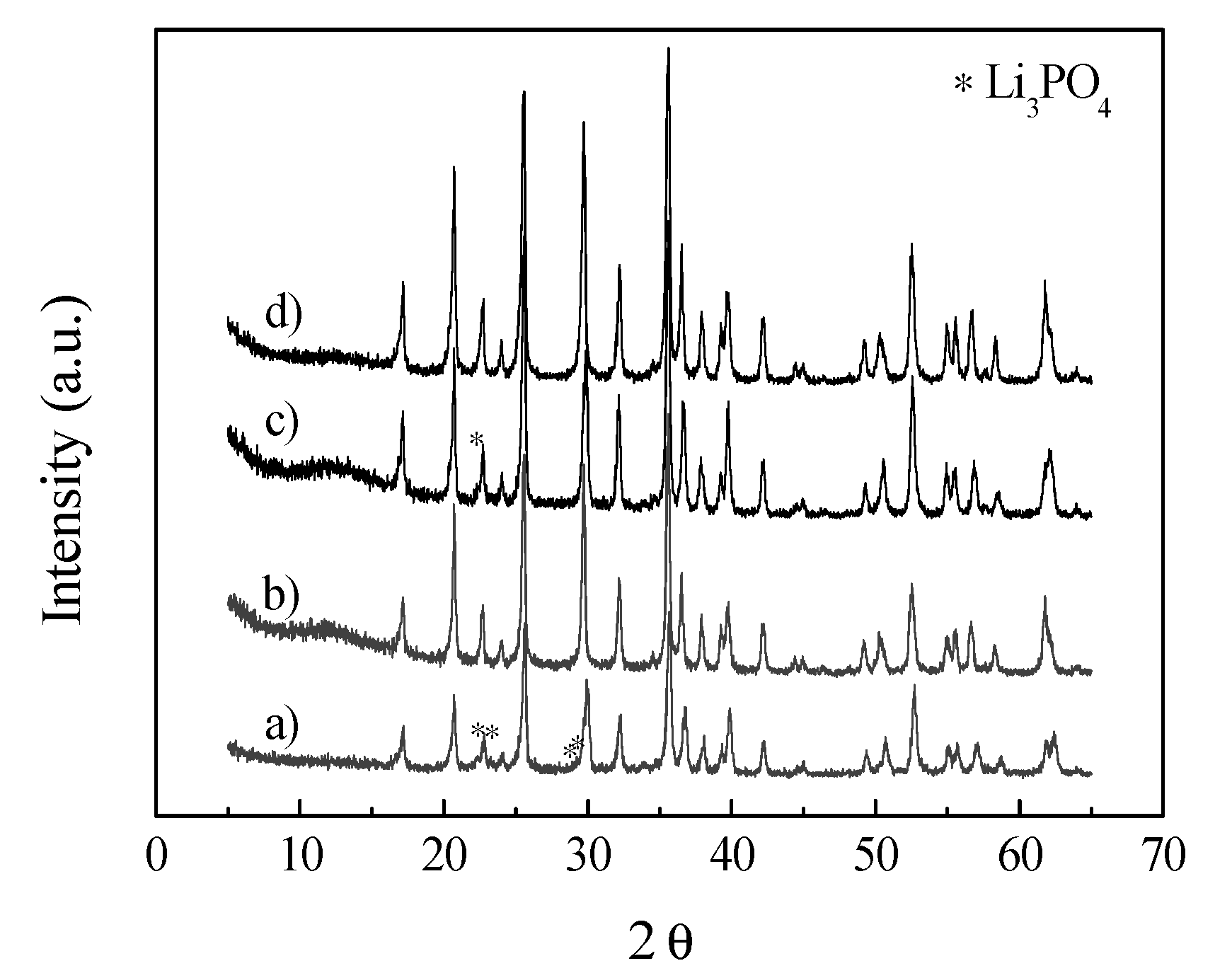
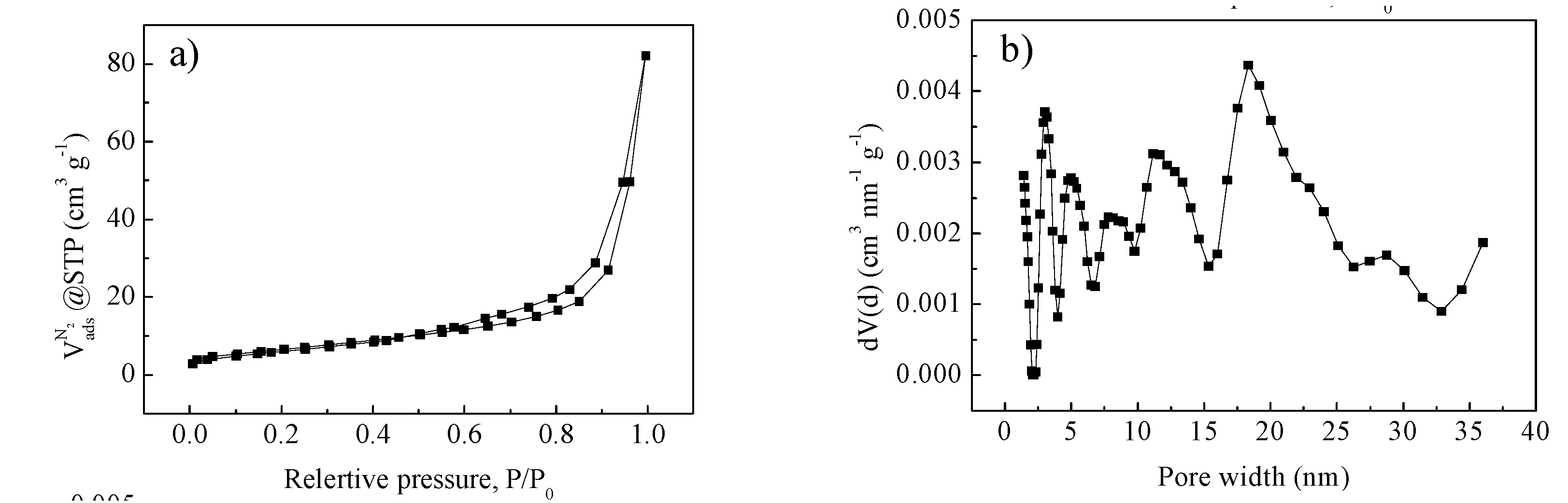
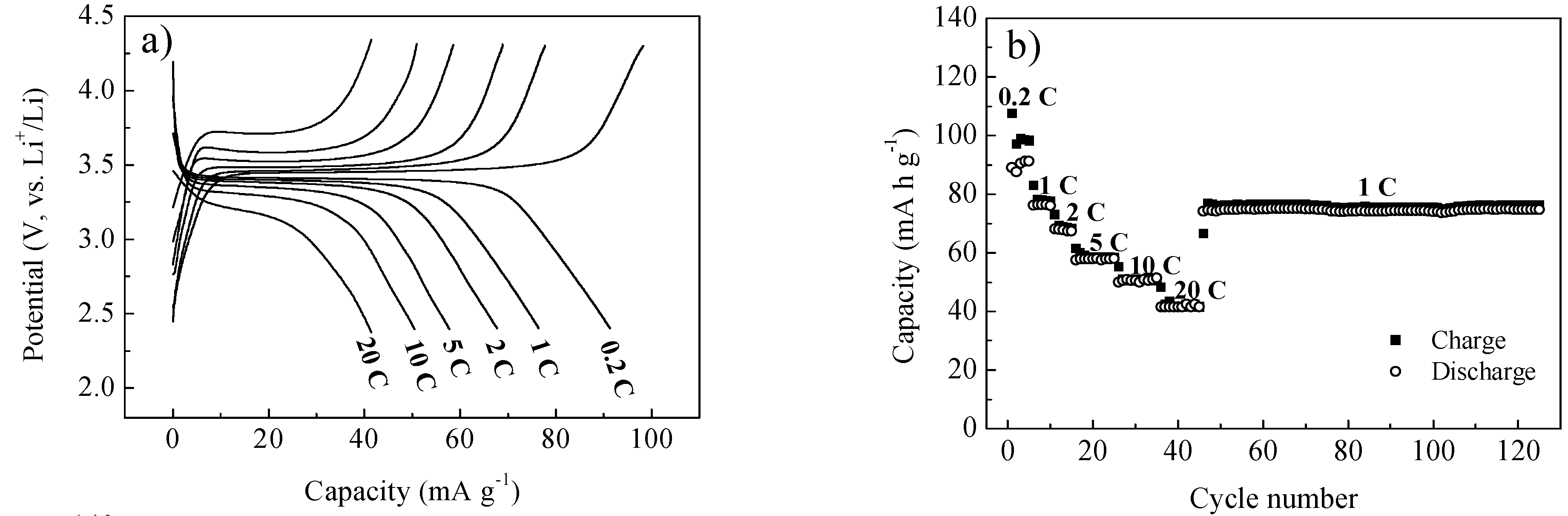
2. Results and Discussion



3. Experimental Section
3.1. Chemicals
3.2. Synthesis of LiFePO4/C Microspheres
3.3. Characterization
3.4. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflict of Interest
References
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Wang, Y.G.; He, P.; Zhou, H.S. Olivine LiFePO(4): Development and future. Energy Environ. Sci. 2011, 4, 805–817. [Google Scholar] [CrossRef]
- Ellis, B.L.; Lee, K.T.; Nazar, L.F. Positive electrode materials for Li-ion and Li-batteries. Chem. Mater. 2010, 22, 691–714. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sour. 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Huang, H.; Faulkner, T.; Barker, J.; Saidi, M.Y. Lithium metal phosphates, power and automotive applications. J. Power Sour. 2009, 189, 748–751. [Google Scholar] [CrossRef]
- Tarascon, J.M.; Recham, N.; Armand, M.; Chotard, J.N.; Barpanda, P.; Walker, W.; Dupont, L. Hunting for better Li-based electrode materials via low temperature inorganic synthesis. Chem. Mater. 2010, 22, 724–739. [Google Scholar] [CrossRef]
- Huang, H.; Yin, S.C.; Nazar, L.F. Approaching theoretical capacity of LiFePO4 at room temperature at high rates. Electrochem. Solid State Lett. 2001, 4, A170–A172. [Google Scholar] [CrossRef]
- Ravet, N.; Chouinard, Y.; Magnan, J.F.; Besner, S.; Gauthier, M.; Armand, M. Electroactivity of natural and synthetic triphylite. J. Power Sour. 2001, 97–98, 503–507. [Google Scholar] [CrossRef]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Chung, S.Y.; Bloking, J.T.; Chiang, Y.M. Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mater. 2002, 1, 123–128. [Google Scholar] [CrossRef]
- Barker, J.; Saidi, M.Y.; Swoyer, J.L. Lithium iron(II) phospho-olivines prepared by a novel carbothermal reduction method. Electrochem. Solid State Lett. 2003, 6, A53–A55. [Google Scholar] [CrossRef]
- Gaberscek, M.; Dominko, R.; Bele, M.; Remskar, M.; Hanzel, D.; Jamnik, J. Porous, carbon-decorated LiFePO4 prepared by sol-gel method based on citric acid. Solid State Ionics 2005, 176, 1801–1805. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, J.L.; Yang, J.; Nuli, Y.N. High-rate LiFePO4 electrode material synthesized by a novel route from FePO4 center dot 4H(2)O. Adv. Funct. Mater. 2006, 16, 2135–2140. [Google Scholar] [CrossRef]
- Hsu, K.F.; Tsay, S.Y.; Hwang, B.J. Synthesis and characterization of nano-sized LiFePO4 cathode materials prepared by a citric acid-based sol-gel route. J. Mater. Chem. 2004, 14, 2690–2695. [Google Scholar] [CrossRef]
- Wagemaker, M.; Ellis, B.L.; Luetzenkirchen-Hecht, D.; Mulder, F.M.; Nazar, L.F. Proof of supervalent doping in olivine LiFePO4. Chem. Mater. 2008, 20, 6313–6315. [Google Scholar] [CrossRef]
- Wang, D.Y.; Li, H.; Shi, S.Q.; Huang, X.J.; Chen, L.Q. Improving the rate performance of LiFePO4 by Fe-site doping. Electrochim. Acta 2005, 50, 2955–2958. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Hosono, E.; Wang, K.; Zhou, H. The design of a LiFePO4/carbon nanocomposite with a core-shell structure and its synthesis by an in situ polymerization restriction method. Angew. Chem. Int. Ed. 2008, 47, 7461–7465. [Google Scholar] [CrossRef]
- Liu, J.; Conry, T.E.; Song, X.; Doeff, M.M.; Richardson, T.J. Nanoporous spherical LiFePO4 for high performance cathodes. Energy Environ. Sci. 2011, 4, 885–888. [Google Scholar] [CrossRef]
- Konarova, M.; Taniguchi, I. Synthesis of carbon-coated LiFePO4 nanoparticles with high rate performance in lithium secondary batteries. J. Power Sour. 2010, 195, 3661–3667. [Google Scholar] [CrossRef]
- Ying, J.R.; Wan, C.R.; Jiang, C.Y.; Li, Y.X. Preparation and characterization of high-density spherical LiNi0.8Co0.2O2 cathode material for lithium secondary batteries. J. Power Sour. 2001, 99, 78–84. [Google Scholar] [CrossRef]
- Qian, J.F.; Zhou, M.; Cao, Y.L.; Ai, X.P.; Yang, H.X. Template-free hydrothermal synthesis of nanoembossed mesoporous LiFePO4 microspheres for high-performance lithium-ion batteries. J. Phys. Chem. C 2010, 114, 3477–3482. [Google Scholar] [CrossRef]
- Yuan, L.X.; Wang, Z.H.; Zhang, W.X.; Hu, X.L.; Chen, J.T.; Huang, Y.H.; Goodenough, J.B. Development and challenges of LiFePO4 cathode material for lithium-ion batteries. Energy Environ. Sci. 2011, 4, 269–284. [Google Scholar] [CrossRef]
- Oh, S.W.; Myung, S.T.; Oh, S.M.; Yoon, C.S.; Amine, K.; Sun, Y.K. Polyvinylpyrrolidone-assisted synthesis of microscale C-LiFePO4 with high tap density as positive electrode materials for lithium batteries. Electrochim. Acta 2010, 55, 1193–1199. [Google Scholar] [CrossRef]
- Lu, A.-H.; Salabas, E.L.; Schueth, F. Magnetic nanoparticles: Synthesis, protection, functionalization, and application. Angew. Chem.-Int. Ed. 2007, 46, 1222–1244. [Google Scholar] [CrossRef]
- Zhao, H.; Qu, Z.-R.; Ye, H.-Y.; Xiong, R.-G. In situ hydrothermal synthesis of tetrazole coordination polymers with interesting physical properties. Chem. Soc. Rev. 2008, 37, 84–100. [Google Scholar]
- Titirici, M.-M.; White, R.J.; Falco, C.; Sevilla, M. Black perspectives for a green future: Hydrothermal carbons for environment protection and energy storage. Energy Environ. Sci. 2012, 5, 6796–6822. [Google Scholar] [CrossRef]
- Hu, B.; Wang, K.; Wu, L.H.; Yu, S.H.; Antonietti, M.; Titirici, M.M. Engineering carbon materials from the hydrothermal carbonization process of biomass. Adv. Mater. 2010, 22, 813–828. [Google Scholar] [CrossRef]
- Sun, C.; Rajasekhara, S.; Goodenough, J.B.; Zhou, F. Monodisperse porous LiFePO4 microspheres for a high power Li-ion battery cathode. J. Am. Chem. Soc. 2011, 133, 2132–2135. [Google Scholar] [CrossRef]
- Su, J.; Wu, X.-L.; Yang, C.-P.; Lee, J.-S.; Kim, J.; Guo, Y.-G. Self-assembled LiFePO4/C nano/microspheres by using phytic acid as phosphorus source. J. Phys. Chem. C 2012, 116, 5019–5024. [Google Scholar]
- Huang, X.J.; Yan, S.J.; Zhao, H.Y.; Zhang, L.; Guo, R.; Chang, C.K.; Kong, X.Y.; Han, H.B. Electrochemical performance of LiFePO4 nanorods obtained from hydrothermal process. Mater. Charact. 2010, 61, 720–725. [Google Scholar] [CrossRef]
- Yang, S.F.; Zavalij, P.Y.; Whittingham, M.S. Hydrothermal synthesis of lithium iron phosphate cathodes. Electrochem. Commun. 2001, 3, 505–508. [Google Scholar] [CrossRef]
- Ellis, B.; Kan, W.H.; Makahnouk, W.R.M.; Nazar, L.F. Synthesis of nanocrystals and morphology control of hydrothermally prepared LiFePO4. J. Mater. Chem. 2007, 17, 3248–3254. [Google Scholar] [CrossRef]
- Xiang, H.; Zhang, D.; Jin, Y.; Chen, C.; Wu, J.; Wang, H. Hydrothermal synthesis of ultra-thin LiFePO4 platelets for Li-ion batteries. J. Mater. Sci. 2011, 46, 4906–4912. [Google Scholar] [CrossRef]
- Murugan, A.V.; Muraliganth, T.; Manthiram, A. Comparison of microwave assisted solvothermal and hydrothermal syntheses of LiFePO4/C nanocomposite cathodes for lithium ion batteries. J. Phys. Chem. C 2008, 112, 14665–14671. [Google Scholar] [CrossRef]
- Devaraju, M.K.; Honma, I. Hydrothermal and solvothermal process towards development of LiMPO4 (M = Fe, Mn) nanomaterials for Lithium-ion batteries. Adv. Energy Mater. 2012, 2, 284–297. [Google Scholar] [CrossRef]
- Meligrana, G.; Gerbaldi, C.; Tuel, A.; Bodoardo, S.; Penazzi, N. Hydrothermal synthesis of high surface LiFePO4 powders as cathode for Li-ion cells. J. Power Sour. 2006, 160, 516–522. [Google Scholar] [CrossRef]
- Dokko, K.; Koizumi, S.; Nakano, H.; Kanamura, K. Particle morphology, crystal orientation, and electrochemical reactivity of LiFePO4 synthesized by the hydrothermal method at 443 K. J. Mater. Chem. 2007, 17, 4803–4810. [Google Scholar] [CrossRef]
- Kim, D.K.; Muralidharan, P.; Lee, H.W.; Ruffo, R.; Yang, Y.; Chan, C.K.; Peng, H.; Huggins, R.A.; Cui, Y. Spinel LiMn2O4 nanorods as lithium ion battery cathodes. Nano Lett. 2008, 8, 3948–3952. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, L.; Cai, D.; Wang, H.; Titirici, M.-M. Synthesis of Microspherical LiFePO4-Carbon Composites for Lithium-Ion Batteries. Nanomaterials 2013, 3, 443-452. https://doi.org/10.3390/nano3030443
Yu L, Cai D, Wang H, Titirici M-M. Synthesis of Microspherical LiFePO4-Carbon Composites for Lithium-Ion Batteries. Nanomaterials. 2013; 3(3):443-452. https://doi.org/10.3390/nano3030443
Chicago/Turabian StyleYu, Linghui, Dandan Cai, Haihui Wang, and Maria-Magdalena Titirici. 2013. "Synthesis of Microspherical LiFePO4-Carbon Composites for Lithium-Ion Batteries" Nanomaterials 3, no. 3: 443-452. https://doi.org/10.3390/nano3030443