Grafting of Polycaprolactone on Oxidized Nanocelluloses by Click Chemistry

Abstract
:1. Introduction
2. Results and Discussion
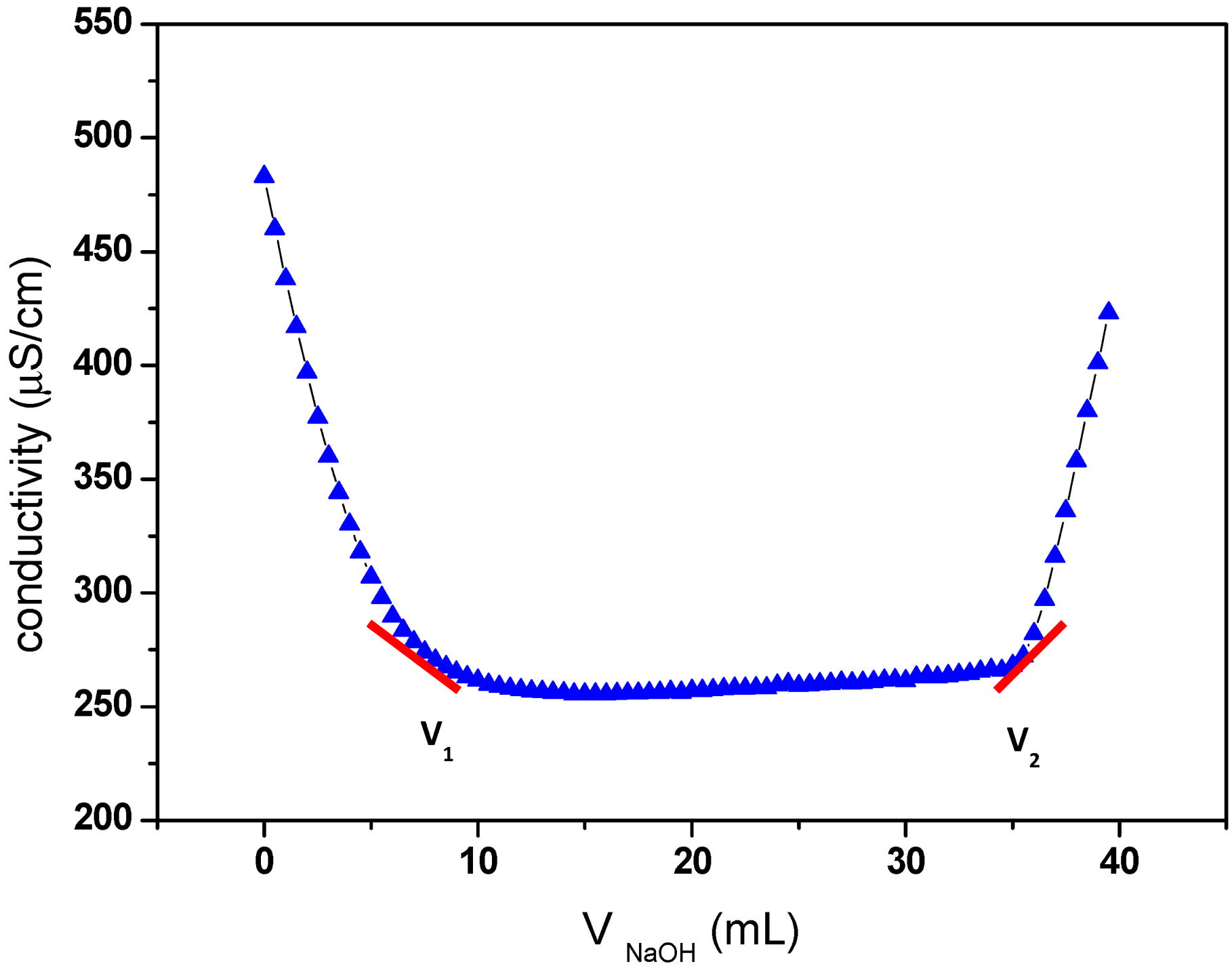
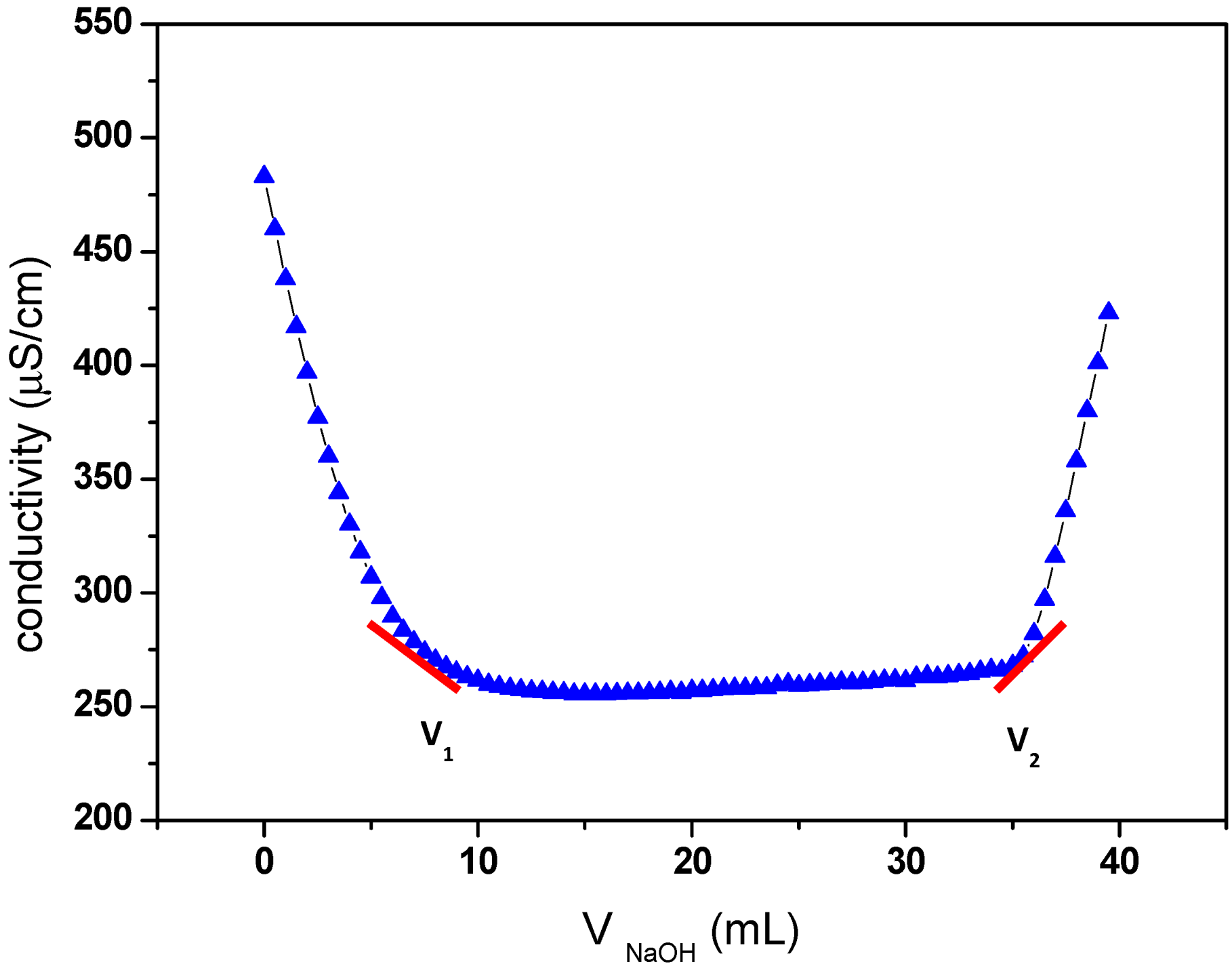
2.1. TEMPO-Mediated Oxidation of Cellulose
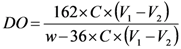
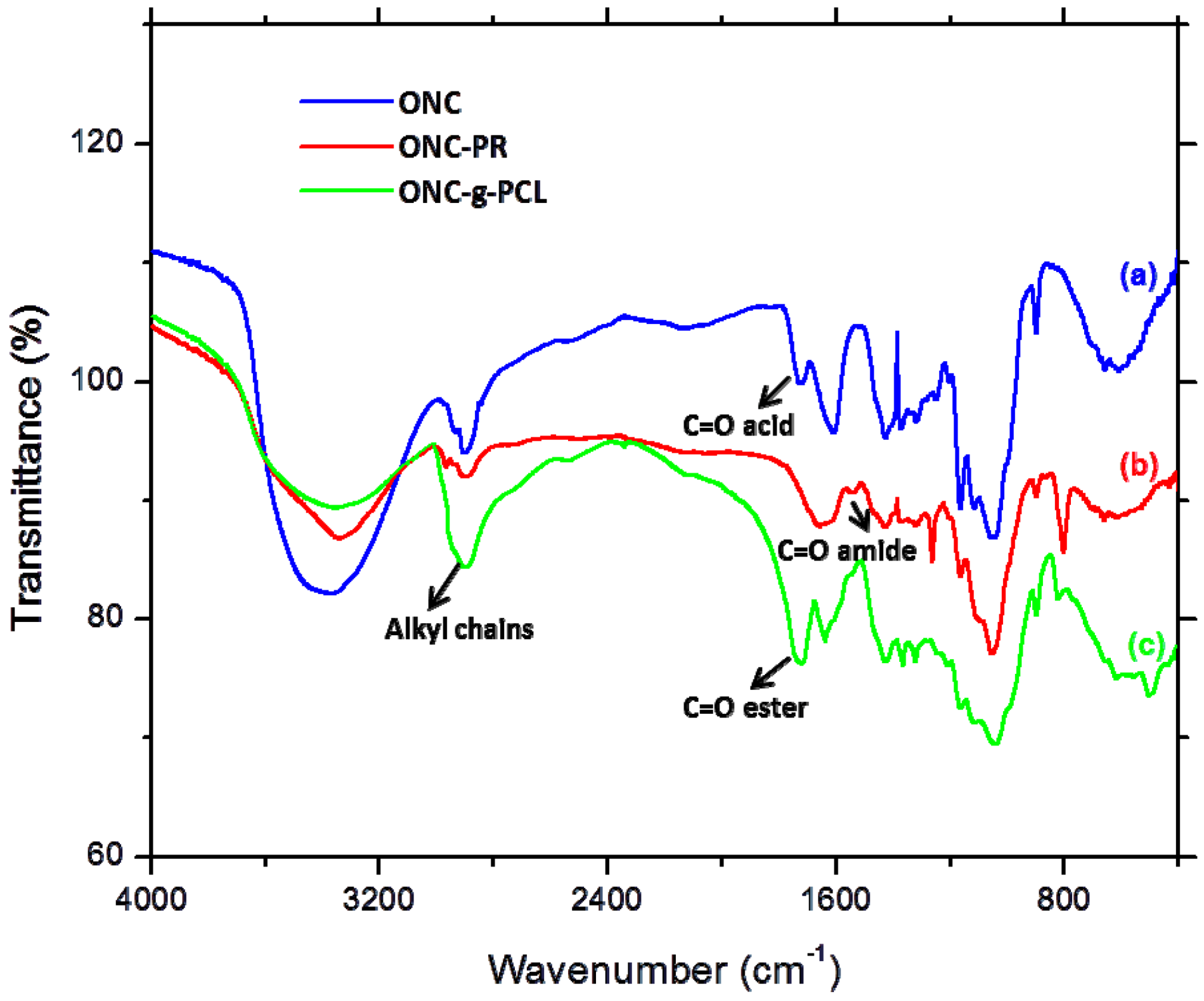
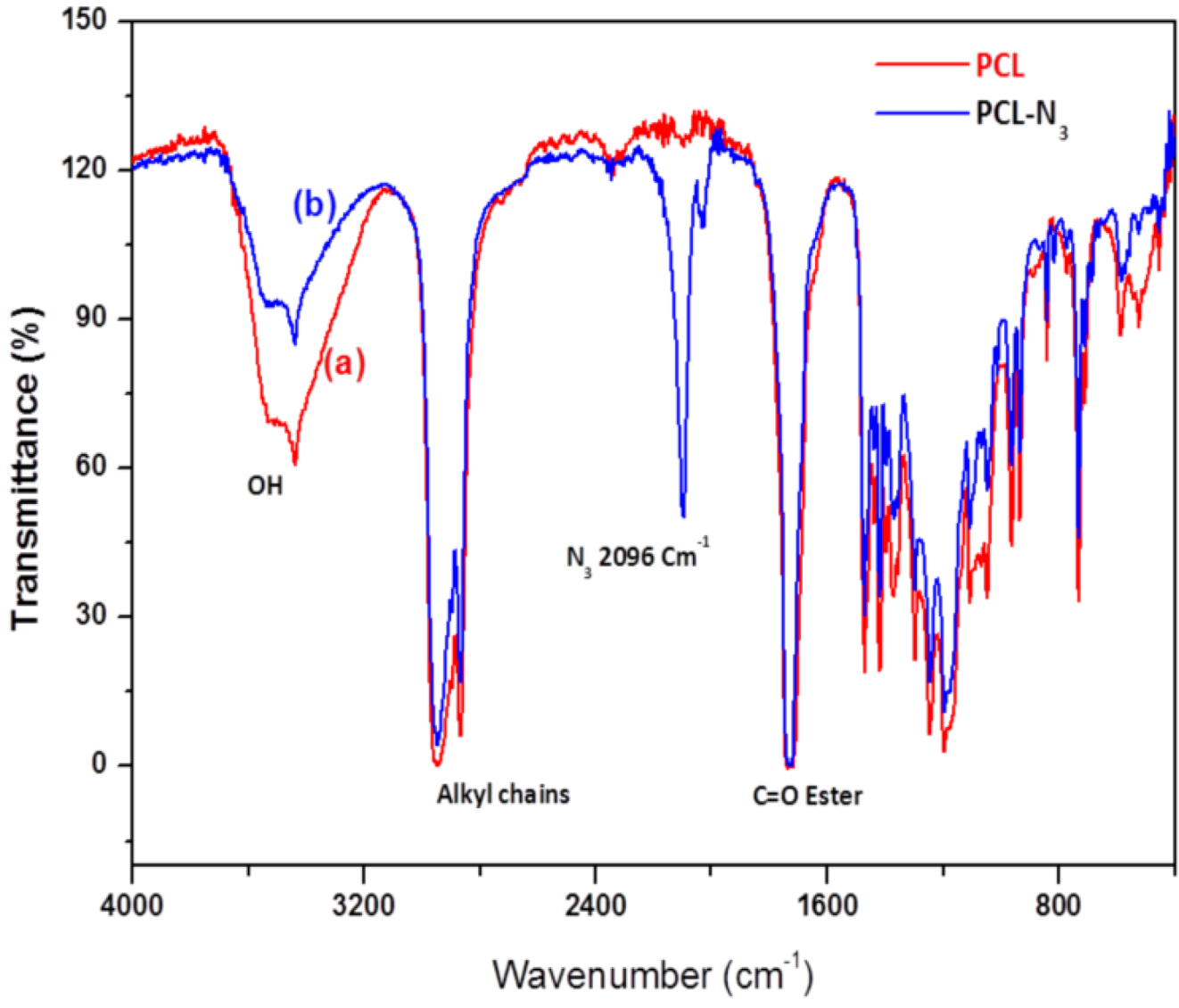
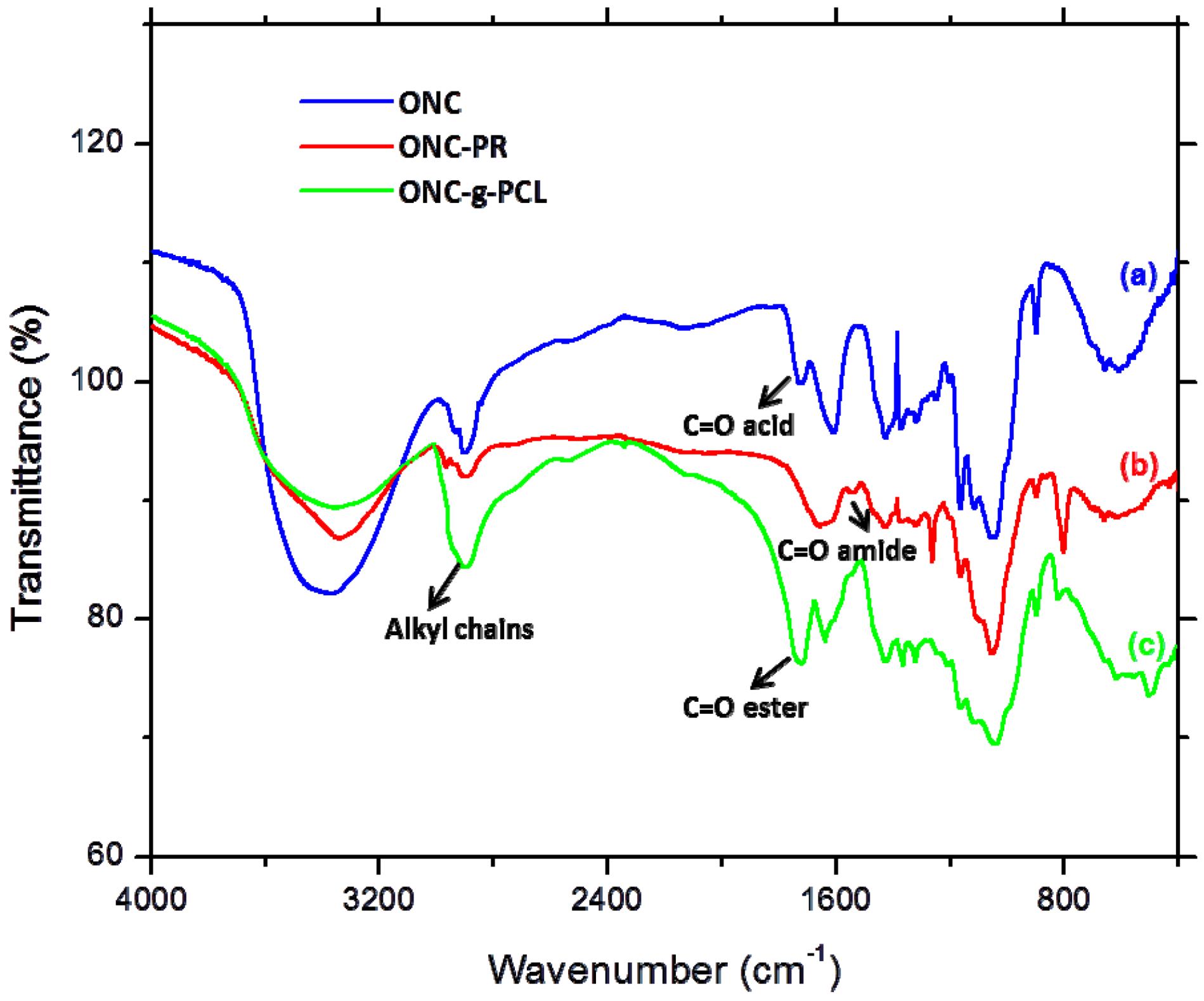
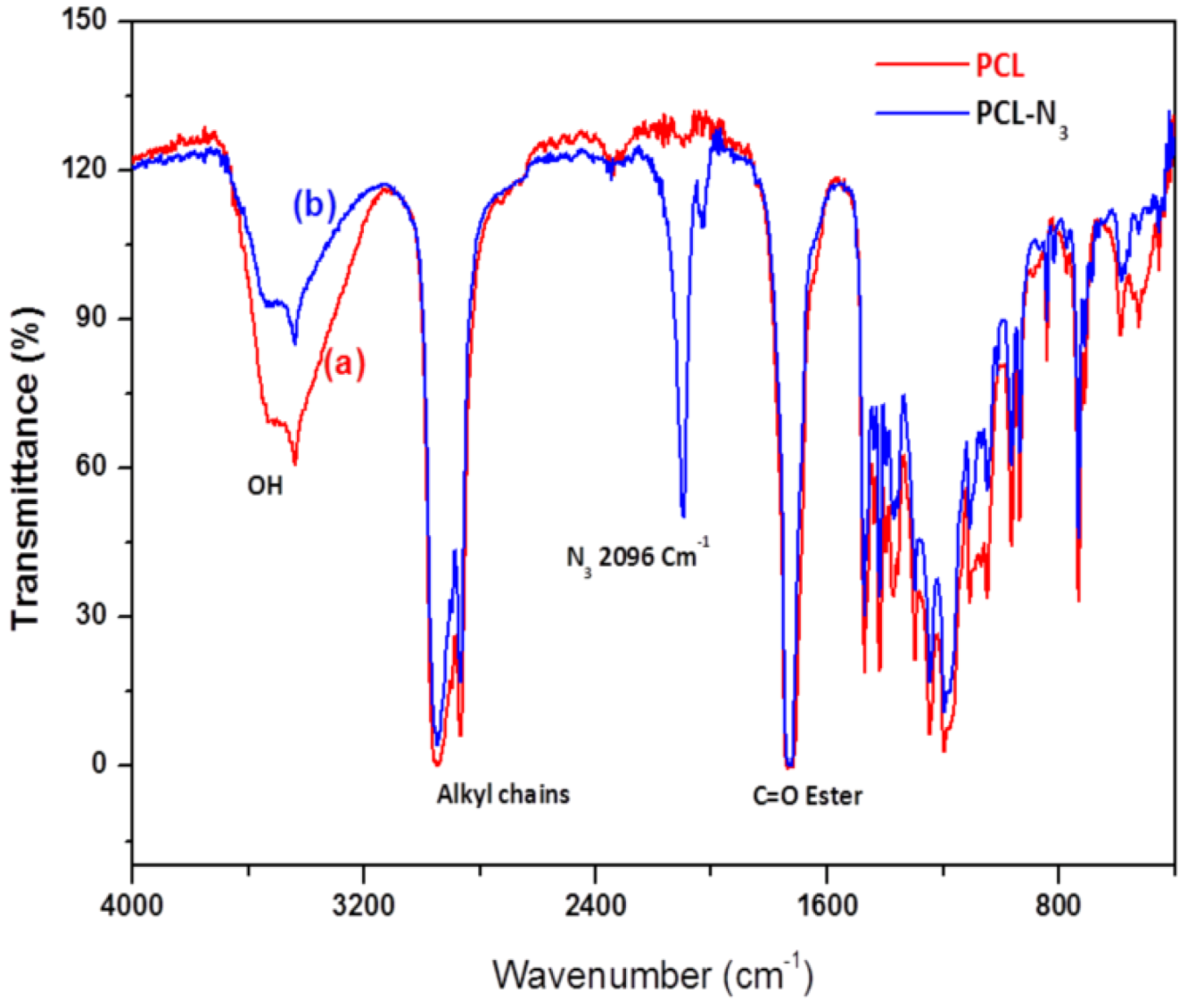
2.2. FTIR Experiments
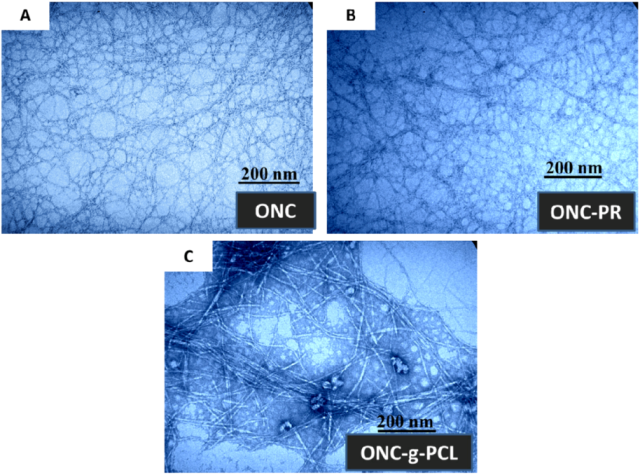
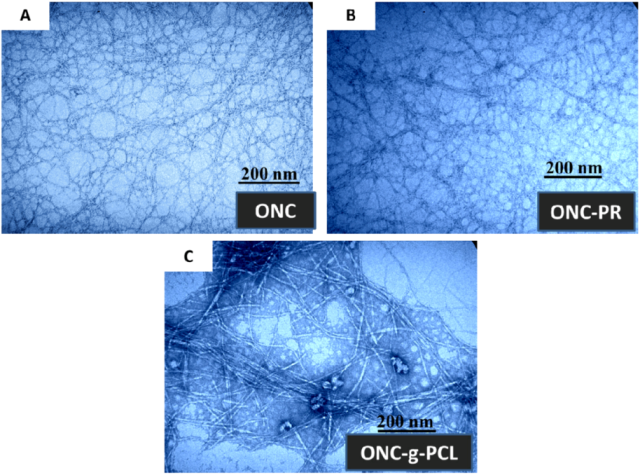
2.3. TEM Images
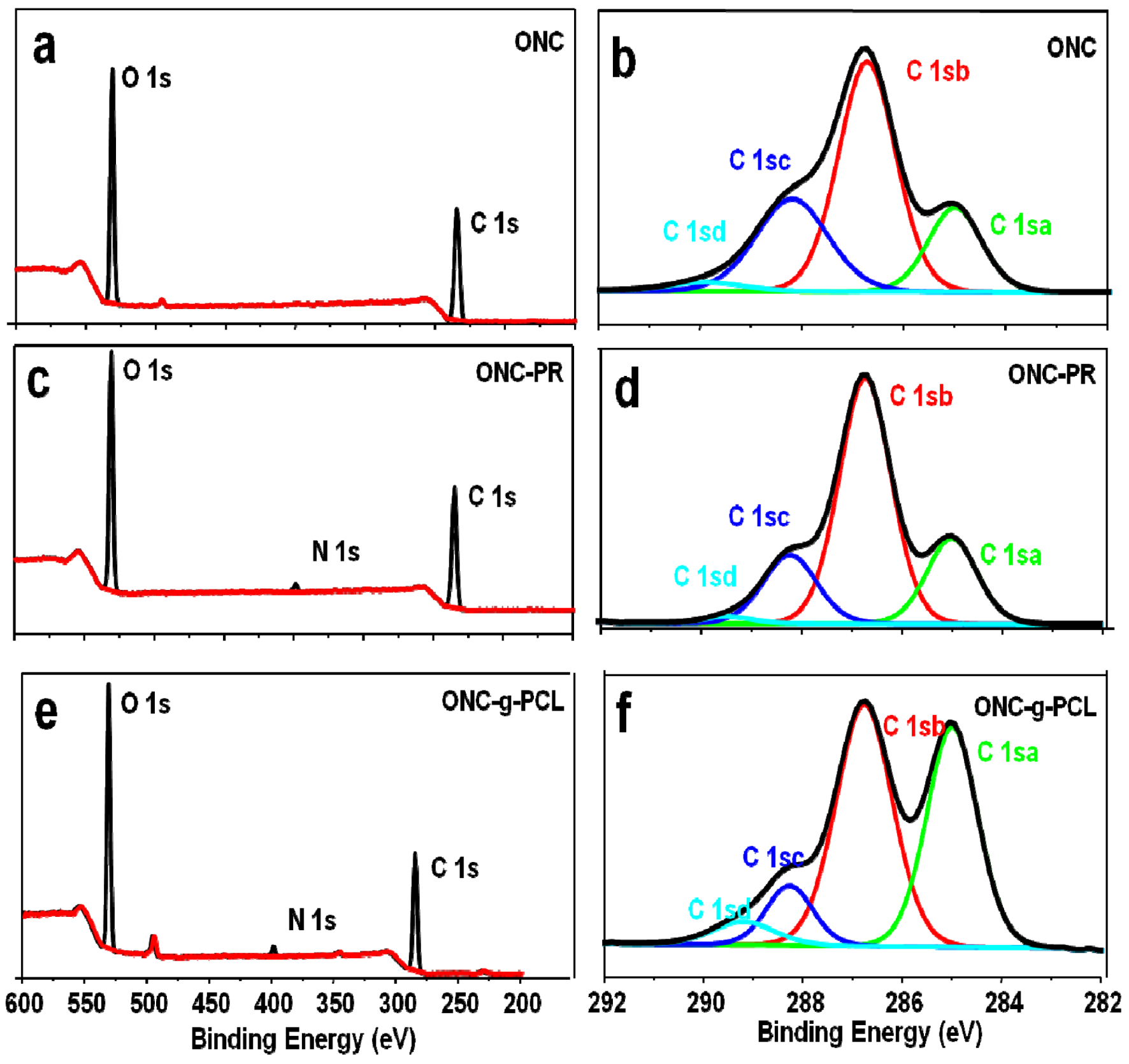
2.4. XPS Results


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding type | C 1sa (C–C/C–H) | C 1sb (C–O) | C 1sc (C=O) | C 1sd (O–C=O) |
|---|---|---|---|---|
| Energy (eV) | 285 | 286.7–8 | 288.2–3 | 289.2–9 |
| ONC | 18.14 | 52.71 | 26.67 | 2.49 |
| ONC-PR | 20.62 | 60.66 | 17.40 | 1.32 |
| ONC-g-PCL | 38.59 | 46.77 | 9.55 | 5.1 |
| Sample | Atomic content % | O/C | ||
|---|---|---|---|---|
| C | O | N | ||
| ONC | 63.16 | 36.84 | 0 | 0.58 |
| ONC-PR | 63.24 | 34.88 | 1.88 | 0.55 |
| ONC-g-PCL | 70.19 | 26.54 | 2.35 | 0.37 |
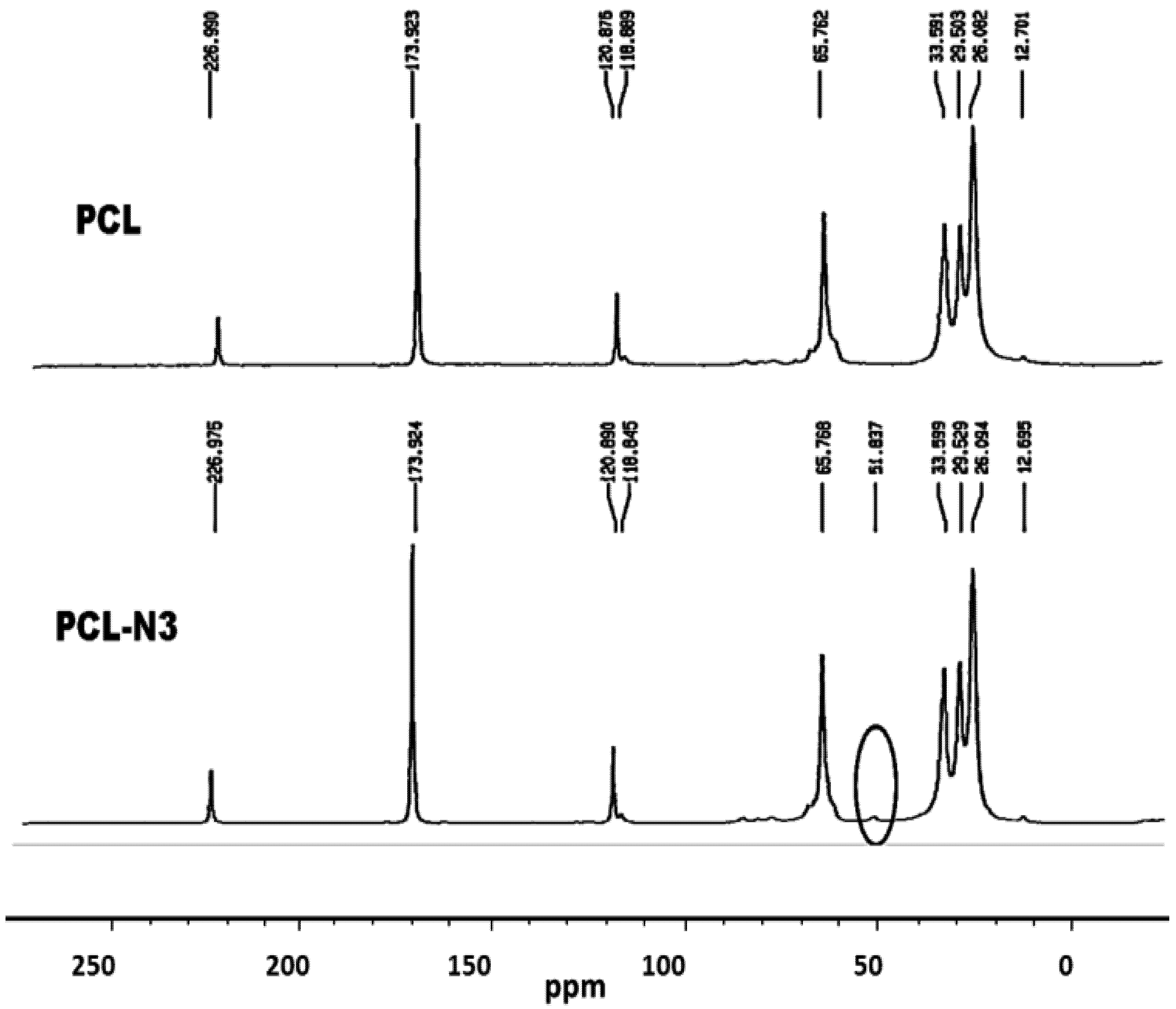
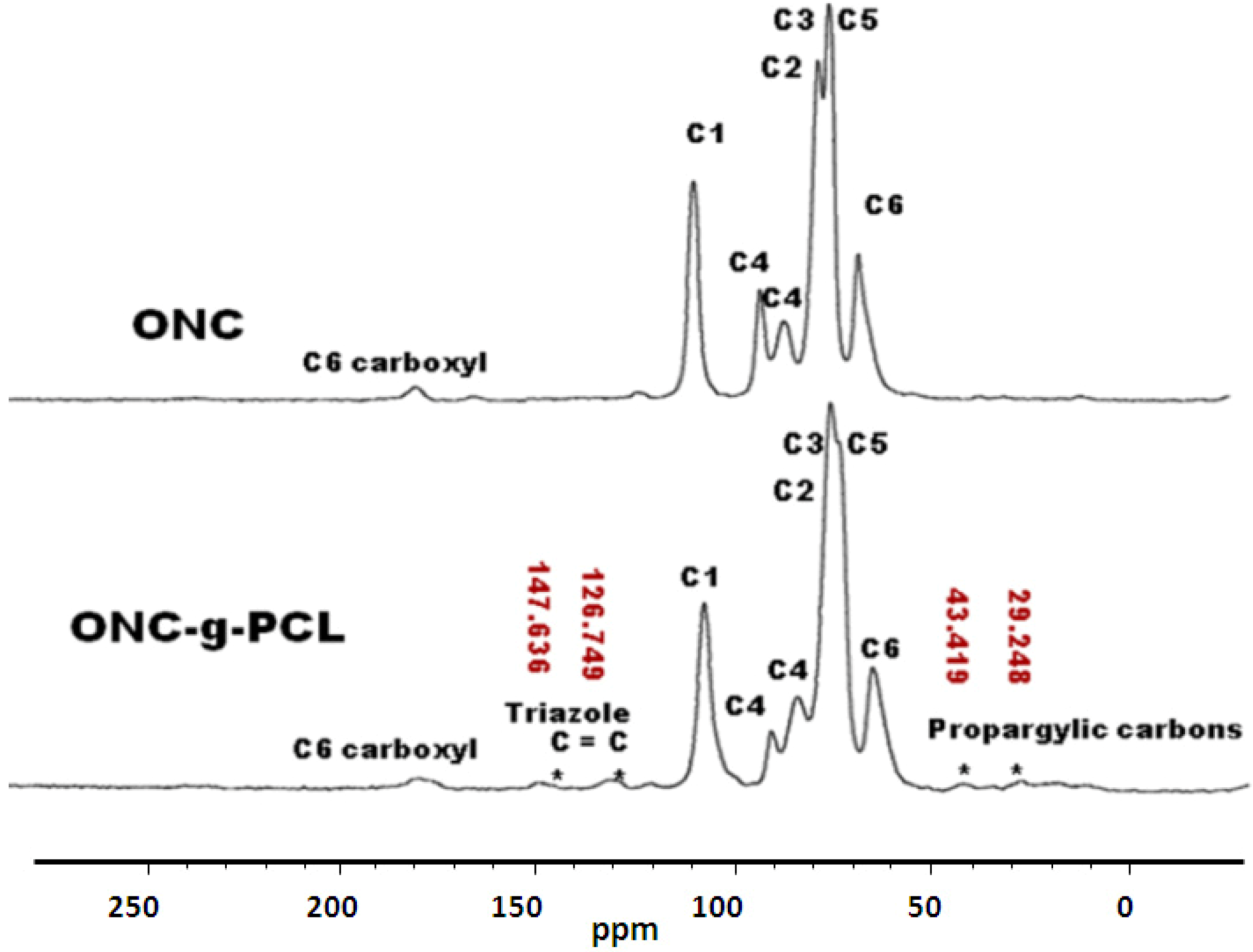
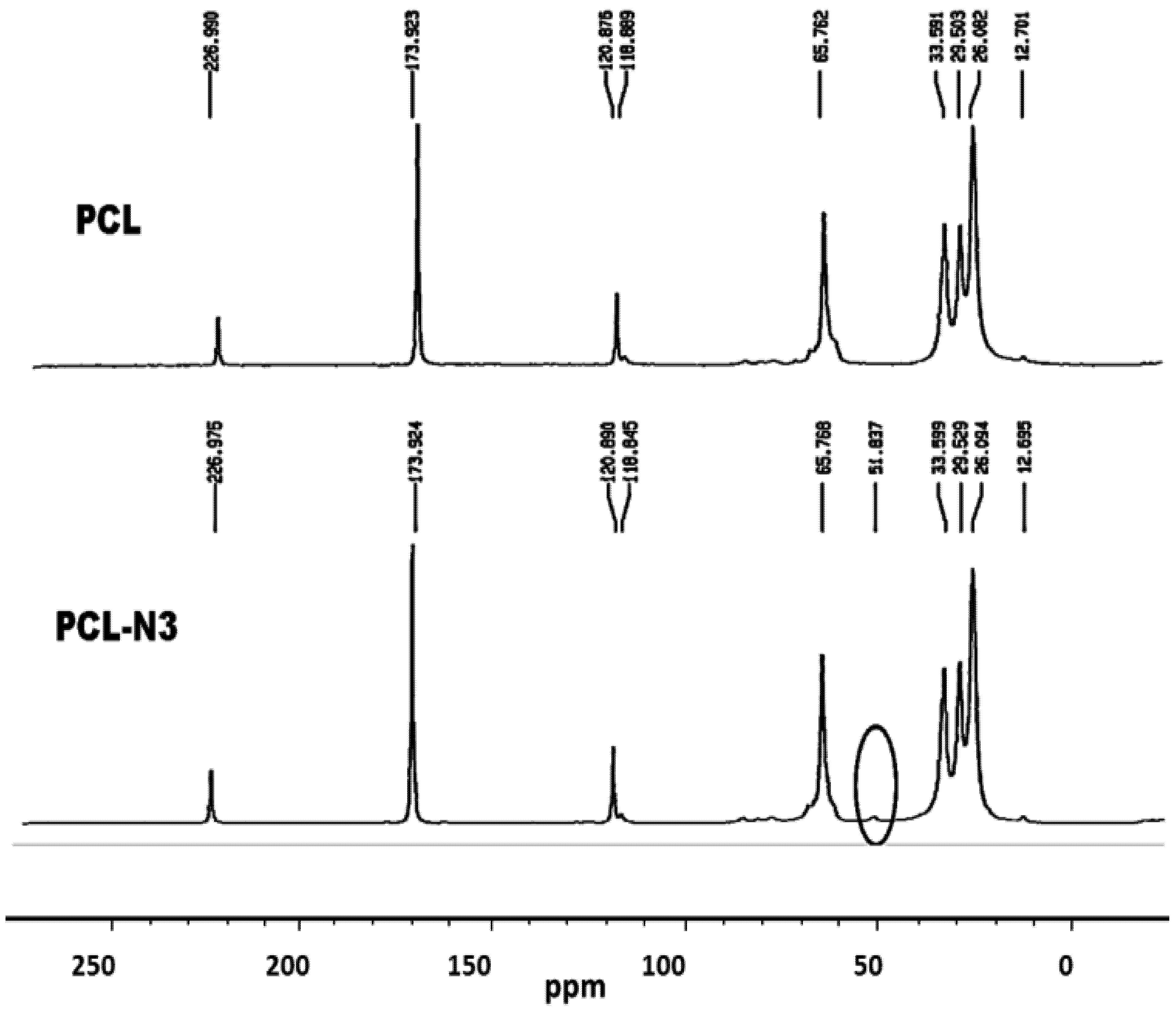
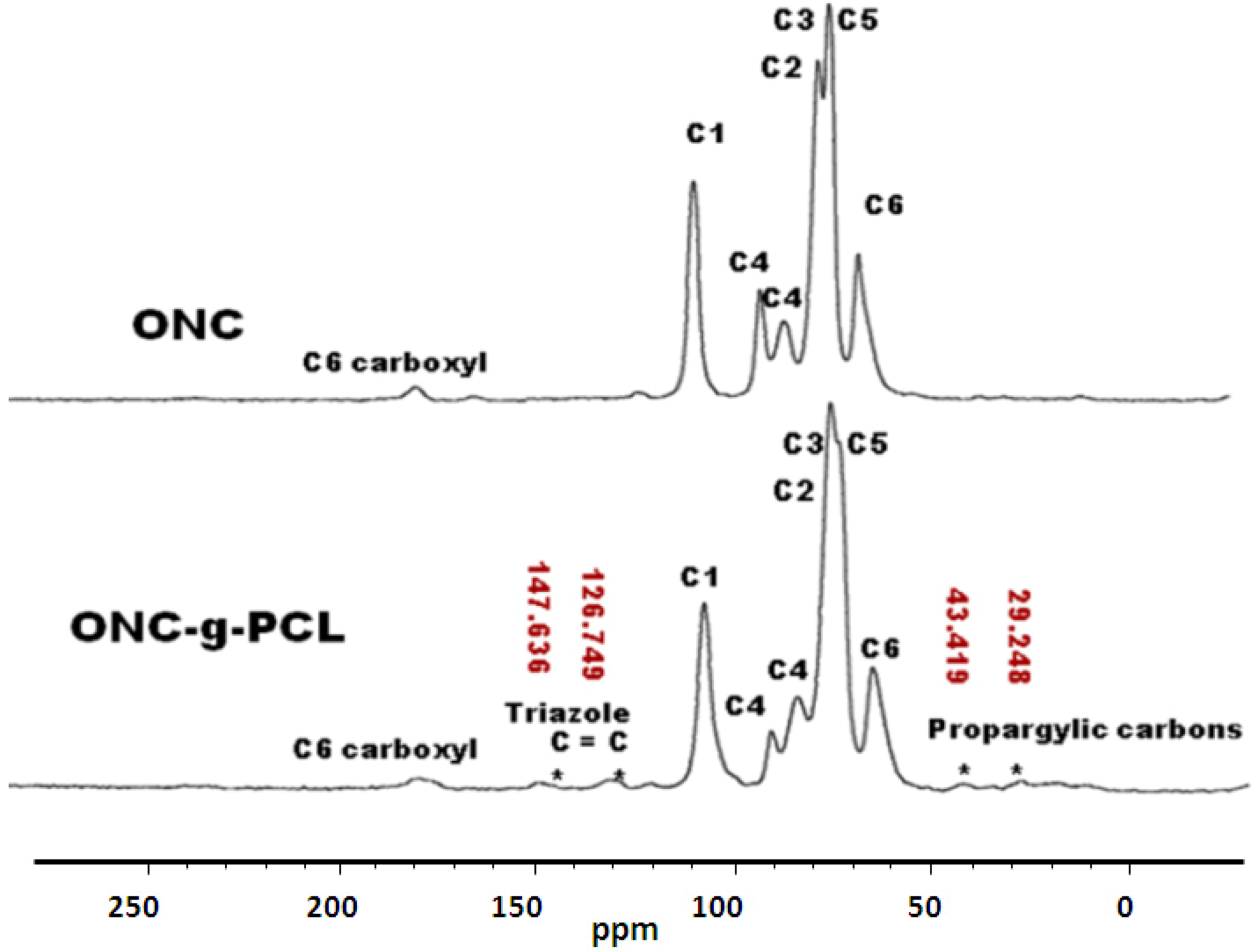
2.5. 13C-NMR Results
3. Experimental Section
3.1. Materials
3.2. Methods
3.2.1. Preparation of Oxidized Nanocelluloses ONC
3.2.2. Measurement of Carboxyl Group Content
3.2.3. Production of Oxidized Nanocelluloses (ONC)
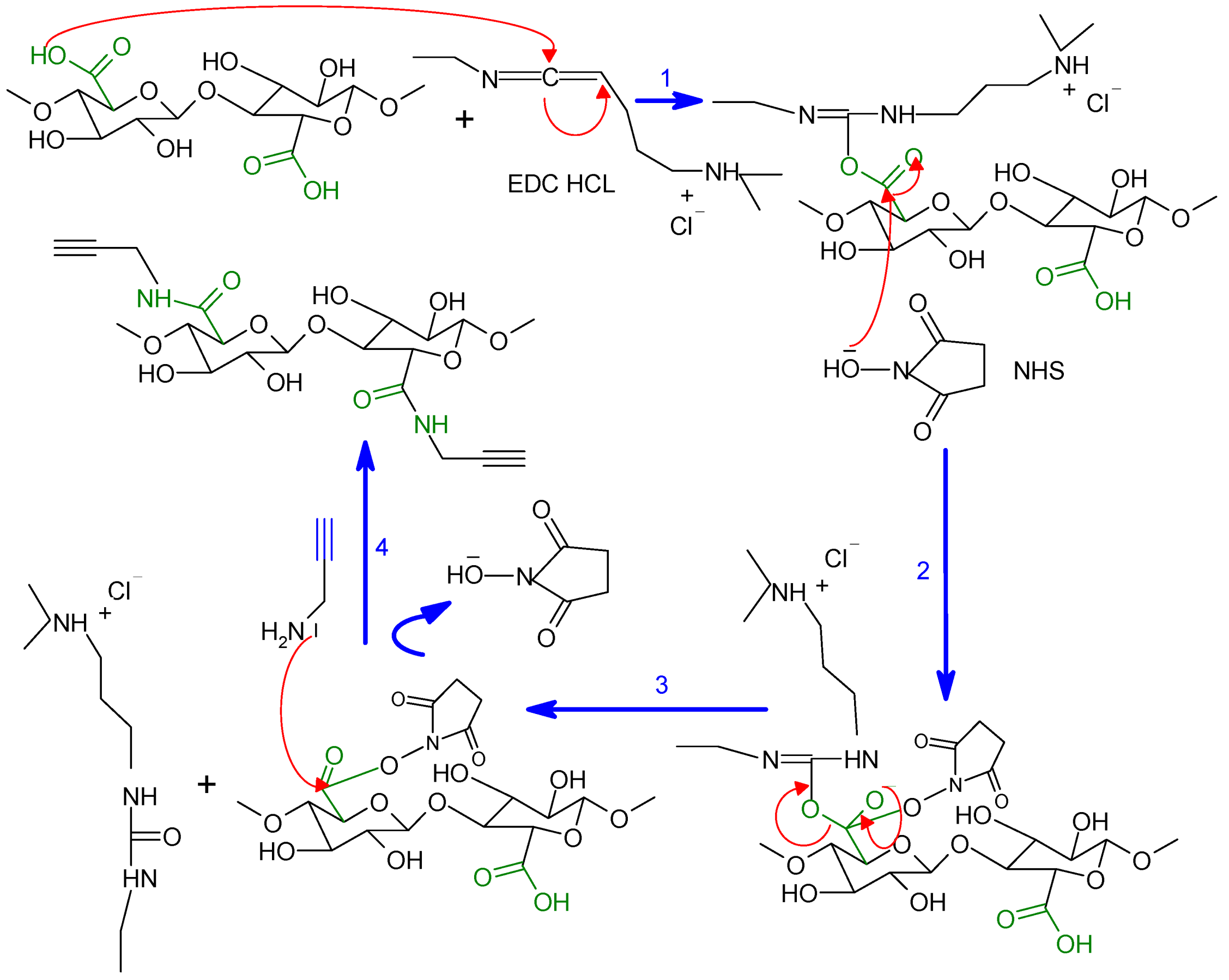
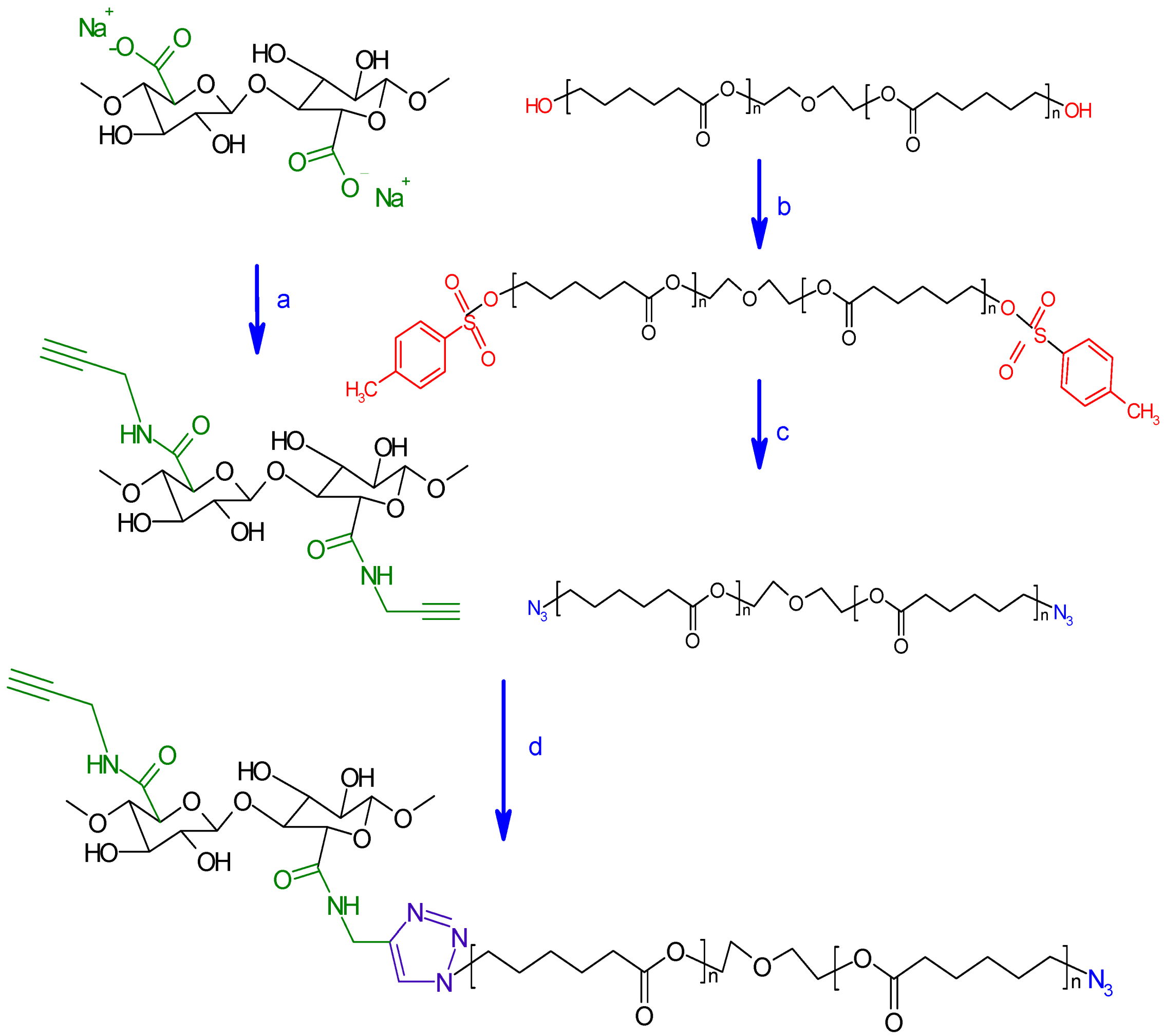
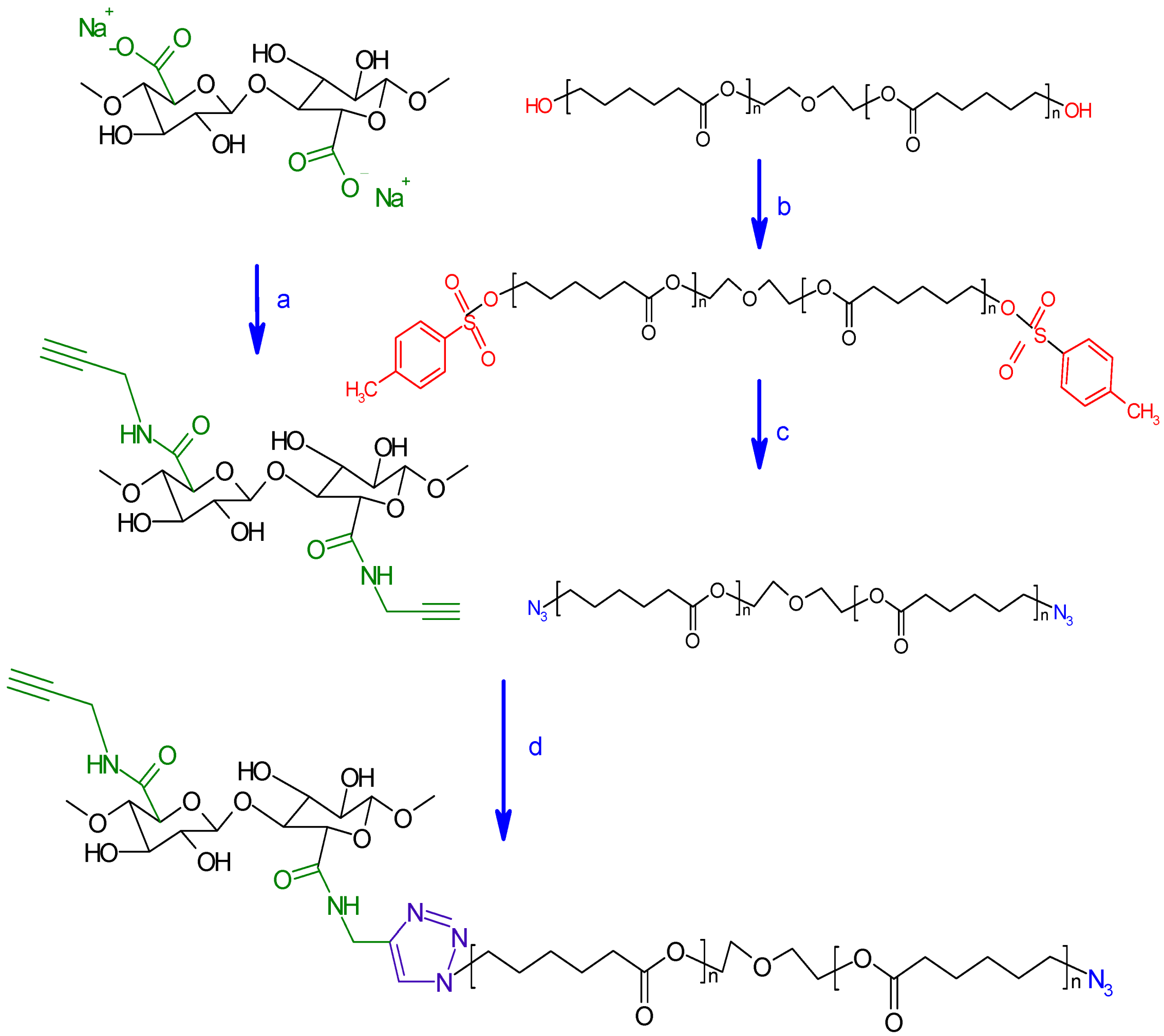
3.2.4. Synthesis of Click Precursor Bearing Alkyne Groups (ONC-PR)

3.2.5. Conversion of Polycaprolactone into Azido-Polycaprolactone (PCL-N3)
3.2.6. Grafting of Azido-Polycaprolactone onto ONC-PR by Click Chemistry
3.3. Instrumentation
3.3.1. Fourier Transform Infrared Spectrometry (FTIR)
3.3.2. X-Ray Photoelectron Spectroscopy (XPS)
3.3.3. Transmission Electron Microscopy (TEM)
3.3.4. 13C-NMR
4. Conclusions
Acknowledgements
References
- Chanzy, H. Aspects of cellulose structure. In Cellulose Sources and Exploitation; Kennedy, J.F., Philips, G.O., William, P.A., Eds.; Ellis Horwood Ltd.: New York, NY, USA, 1990; pp. 3–12. [Google Scholar]
- De Nooy, A.E.; Besemer, A.C.; van Bekkum, H. Highly selective nitrosyl radica-mediated oxidation of primary alcohol groups in water-soluble glucans. Carbohydr. Res. 1995, 69, 89–98. [Google Scholar]
- Chang, P.S.; Robyt, J.F. Oxidation of primary alcohol groups of naturally occurring polysacccharides with 2,2,6,6-tetramethyl-1-piperidine oxoammonium ion. J. Carbohydr. Chem. 1996, 15, 819–830. [Google Scholar]
- Beck-Candanedo, S.; Roman, M.; Gray, D.G. Effect of reaction conditions on the properties and behavior of wood cellulose nanocrystal suspensions. Biomacromolecules 2005, 6, 1048–1054. [Google Scholar] [CrossRef]
- Saito, T.; Isogai, A. Ion-exchange behavior of carboxylate groups in fibrous cellulose oxidized by the TEMPO mediated system. Carbohydr. Polym. 2005, 61, 183–190. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, Y.; Putaux, J.L.; Vignon, M.; Isogai, A. Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 2006, 7, 1687–1691. [Google Scholar]
- Bondeson, D.; Mathew, A.; Oksman, K. Optimization of the isolation of nanocrystals from microcrystalline cellulose by acid hydrolysis. Cellulose 2006, 13, 171–180. [Google Scholar] [CrossRef]
- Elazzouzi-Hafraoui, S.; Nishiyama, Y.; Putaux, J.L.; Heux, L.; Dubreuil, F.; Rochas, C. The shape and size distribution of crystalline nanoparticles prepared by acid hydrolysis of native cellulose. Biomacromolecules 2008, 9, 57–65. [Google Scholar] [CrossRef]
- Revol, J.F.; Bradford, H.; Giasson, J.; Marchessault, R.H.; Gray, D.G. Helicoidal self-ordering of cellulose microfibrils in aqueous suspension. Int. J. Biol. Macromol. 1992, 14, 170–172. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S.; Okano, T. Flow properties of microcrystalline cellulose suspension prepared by acid treatment of native cellulose. Colloids Surf. A 1998, 142, 75–82. [Google Scholar] [CrossRef]
- Azizi Samir, M.A.S.; Alloin, F.; Dufresne, A. Review of recent research into cellulosic whiskers, their properties and their application in nanocomposite field. Biomacromolecules 2005, 6, 612–626. [Google Scholar] [CrossRef]
- Zhang, J.; Elder, T.J.; Pu, Y.; Ragauskas, A.J. Facile synthesis of spherical cellulose nanoparticles. Carbohyd. Polym. 2007, 69, 607–611. [Google Scholar] [CrossRef]
- Sugiyama, J.; Chanzy, H.; Revol, J.F. On the polarity of cellulose in the cell wall of Valonia. Planta 1994, 193, 260–265. [Google Scholar]
- Mishra, S.P.; Thirree, J.; Manent, A.S.; Chabot, B.; Daneault, C. Ultrasound-catalyzed TEMPO-mediated oxydation of native cellulose for the production of nanocellulose: Effect of process variables. BioResources 2011, 6, 121–143. [Google Scholar]
- Baiardo, M.; Frisoni, G.; Scandola, M.; Licciardello, A. Surface chemical modification of natural cellulose fibers. J. Appl. Polym. Sci. 2002, 83, 38–45. [Google Scholar] [CrossRef]
- Belgacem, M.; Gandini, A. The surface modification of cellulose fibres for use as reinforcing elements in composite materials. Compos. Interfaces 2005, 12, 41–75. [Google Scholar] [CrossRef]
- Tingaut, P.; Zimmermann, T.; Lopez-Suevos, F. Synthesis and characterization of bionanocomposites with tunable properties from poly(lactic acid) and acetylated microfibrillated cellulose. Biomacromolecules 2010, 11, 454–464. [Google Scholar]
- Lönnberg, H.; Zhou, Q.; Brumer, H.; Teeri Tuula, T.; Malmström, E.; Hult, A. Grafting of cellulose fibers with poly(e-caprolactone) and poly(llactic acid) via ring-opening polymerization. Biomacromolecules 2006, 7, 2178–2185. [Google Scholar] [CrossRef]
- Hafrén, J.; Zou, W.; Cordova, A. Heterogeneous organoclick derivatization of polysaccharides. Macromol. Rapid Commun. 2006, 27, 1362–1366. [Google Scholar] [CrossRef]
- Optseen, J.A.; van Hest, J.C.M. Modular synthesis of block copolymers via cycloaddition of terminal azide and alkyne functionalized polymers. Chem. Commun. 2005, 1, 57–59. [Google Scholar]
- Vogt, A.P.; Summerlin, B.S. Efficient route to macromonomers via ATRP and click chemistry. Macromolecules 2006, 39, 5286–5292. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Davis, T.P.; Fane, A.G. Honeycomb structured porous films prepared from carbohydrate based polymers synthesised via the RAFT process. J. Mater. Chem. 2003, 13, 2090–2097. [Google Scholar] [CrossRef]
- Hernandez-Guerrero, M.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Polystyrene comb polymers built on cellulose or poly(styrene-co-2-hydroxyethyl-methacrylate) backbones as substrates for the preparation of structured honeycomb films. Eur. Polym. J. 2005, 41, 2264–2277. [Google Scholar] [CrossRef]
- Benoit, D.; Grimaldi, S.; Robin, S.; Finet, J.P.; Tordo, P.; Gnanou, Y. Kinetics and mechaism of controlled free-radical polymerization of styrene and n-butyl acrylate in the presence of an acyclic phosphonylated nitroxide. J. Am. Chem. Soc. 2000, 122, 5929–5939. [Google Scholar]
- Okada, M. Chemical syntheses of biodegradable polymers. Progress Polym. Sci. 2002, 27, 87–133. [Google Scholar] [CrossRef]
- Ikada, Y.; Tsuji, H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 1999, 21, 117–132. [Google Scholar] [CrossRef]
- Habibi, Y.; Goffin, A.-L.; Schiltz, N.; Duquesne, E.; Dubois, P.; Dufresne, A. Bionanocomposites based on poly(epsilon-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. J. Mater. Chem. 2008, 41, 5002–5010. [Google Scholar]
- Habibi, Y.; Dufresne, A. Highly filled bionanocomposites from functionalized polysaccharide nanocrystals. Biomacromolecules 2008, 9, 1974–1980. [Google Scholar] [CrossRef]
- Habibi, Y.; Zoppe, O.; Peresin, S.; Richard, A.; Orlando, J. Reinforcing poly(ε-caprolactone) nanofibers with cellulose nanocrystals. ACS Appl. Mater. Interfaces 2009, 1, 1996–2004. [Google Scholar] [CrossRef]
- Ahmed Said Azizi, S.; Lonnberg, H.; Fogelstrom, L.; Berglund, L.; Malmstrom, E.; Anders, H. Surface grafting of microfibrillated cellulose with poly(caprolactone)–Synthesis and characterization. Eur. Polym. J. 2008, 44, 2991–2997. [Google Scholar] [CrossRef]
- Krouit, M.; Bras, J. Cellulose surface grafting with polycaprolactone by heterogeneous click-chemistry. Eur. Polym. J. 2008, 44, 4074–4081. [Google Scholar] [CrossRef]
- Bulpitt, P.; Aeschlimann, D. New strategy for chemical modification of hyaluronic acid: Preparation of functionalized derivatives and their use in the formation of novel biocompatible hydrogels. J. Biomed. Mater. Res. 1999, 47, 152–169. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S. Steric stabilization of a cellulose microcrystal suspension by poly(ethylene glycol) grafting. Langmuir 2001, 17, 21–27. [Google Scholar] [CrossRef]
- Lasseuguette, E. Grafting onto microfibrils of native cellulose. Cellulose 2008, 15, 571–580. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef]
- Julien, O.; Krouit, M.; Bras, P.; Thielemans, W.; Belgacem, M.N. Surface modification of cellulose by PCL grafts. Acta Mater. 2010, 58, 792–801. [Google Scholar] [CrossRef]
- Enomoto-Rogers, Y.; Kamitakahara, H.; Yoshinaga, A.; Takano, T. Comb-shaped graft copolymers with cellulose side-chains prepared via click chemistry. Carbohydr. Polym. 2012, 87, 2237–2245. [Google Scholar] [CrossRef]
- Filpponen, I.; Kontturi, E.; Nummelin, S.; Rosilo, H.; Kolehmainen, E.; Ikkala, O.; Laine, J. Generic method for modular surface modification of cellulosic materials in aqueous medium by sequential “click” reaction and adsorption. Biomacromolecules 2012, 13, 736–742. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Benkaddour, A.; Jradi, K.; Robert, S.; Daneault, C. Grafting of Polycaprolactone on Oxidized Nanocelluloses by Click Chemistry. Nanomaterials 2013, 3, 141-157. https://doi.org/10.3390/nano3010141
Benkaddour A, Jradi K, Robert S, Daneault C. Grafting of Polycaprolactone on Oxidized Nanocelluloses by Click Chemistry. Nanomaterials. 2013; 3(1):141-157. https://doi.org/10.3390/nano3010141
Chicago/Turabian StyleBenkaddour, Abdelhaq, Khalil Jradi, Sylvain Robert, and Claude Daneault. 2013. "Grafting of Polycaprolactone on Oxidized Nanocelluloses by Click Chemistry" Nanomaterials 3, no. 1: 141-157. https://doi.org/10.3390/nano3010141