
Caffeic Acid-PLGA Conjugate to Design Protein Drug Delivery Systems Stable to Irradiation


 and
and
Abstract
:
1. Introduction
2. Results and Discussion
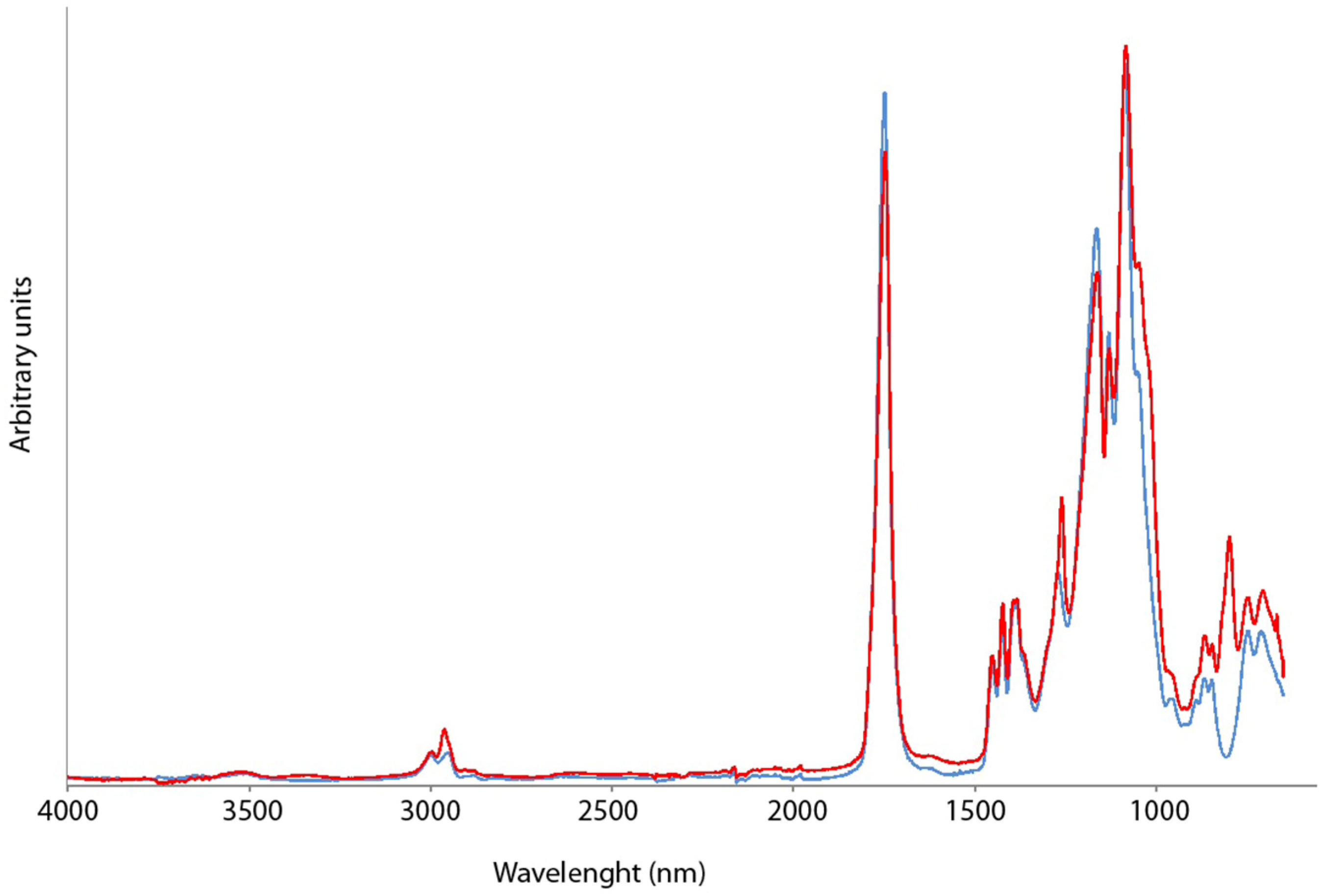
2.1. Chemical and Physico-Chemical Characterization of g-CA-PLGA

{kind=link}
{kind=link}
{kind=link}
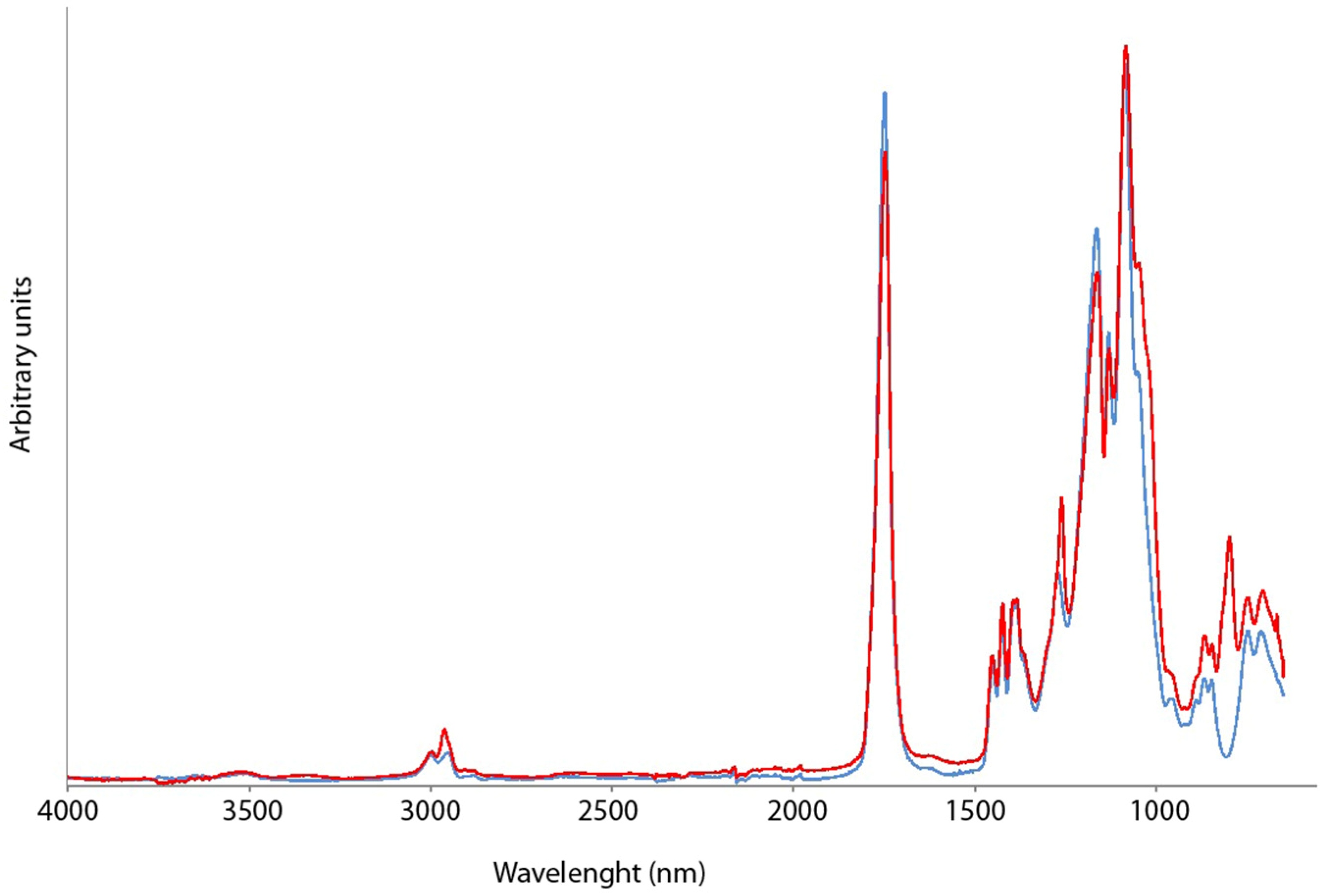
{kind=link}
{kind=link}
| Polymer | Mw (kDa) | Mn (kDa) | PI | Tg (°C) | DPPH inhibition (%) | |||
|---|---|---|---|---|---|---|---|---|
| 1 h | 2 h | 3 h | 24 h | |||||
| PLGA | 26.3 ± 1.3 | 17.3 ± 1.5 | 1.5 ± 0.0 | 47.1 ± 0.5 | 0 ± 0.3 | 0 ± 0.3 | 0 ± 0.5 | 0 ± 0.4 |
| g-CA-PLGA | 22.9 ± 0.7 | 8.4 ± 0.4 | 2.7 ± 0.0 | 45.0 ± 0.7 | 92 ± 0.6 | 93 ± 0.4 | 93 ± 0.5 | 98 ± 0.3 |
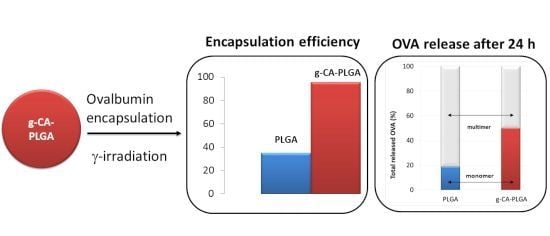
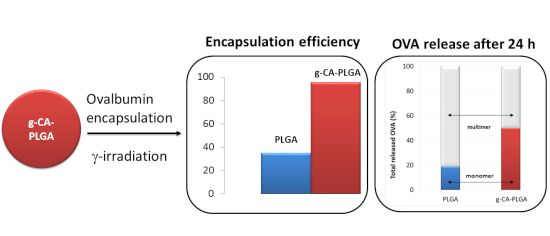
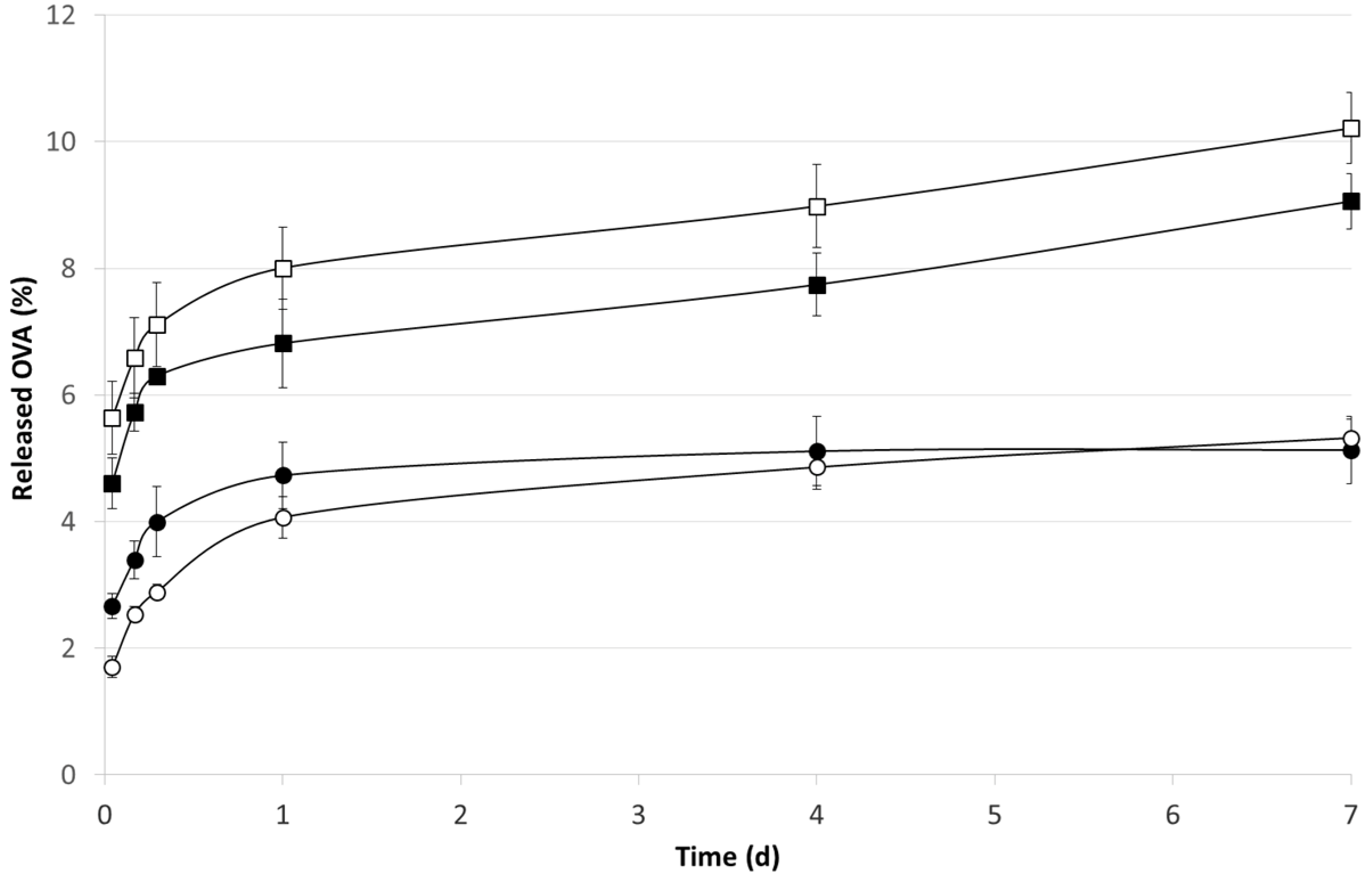
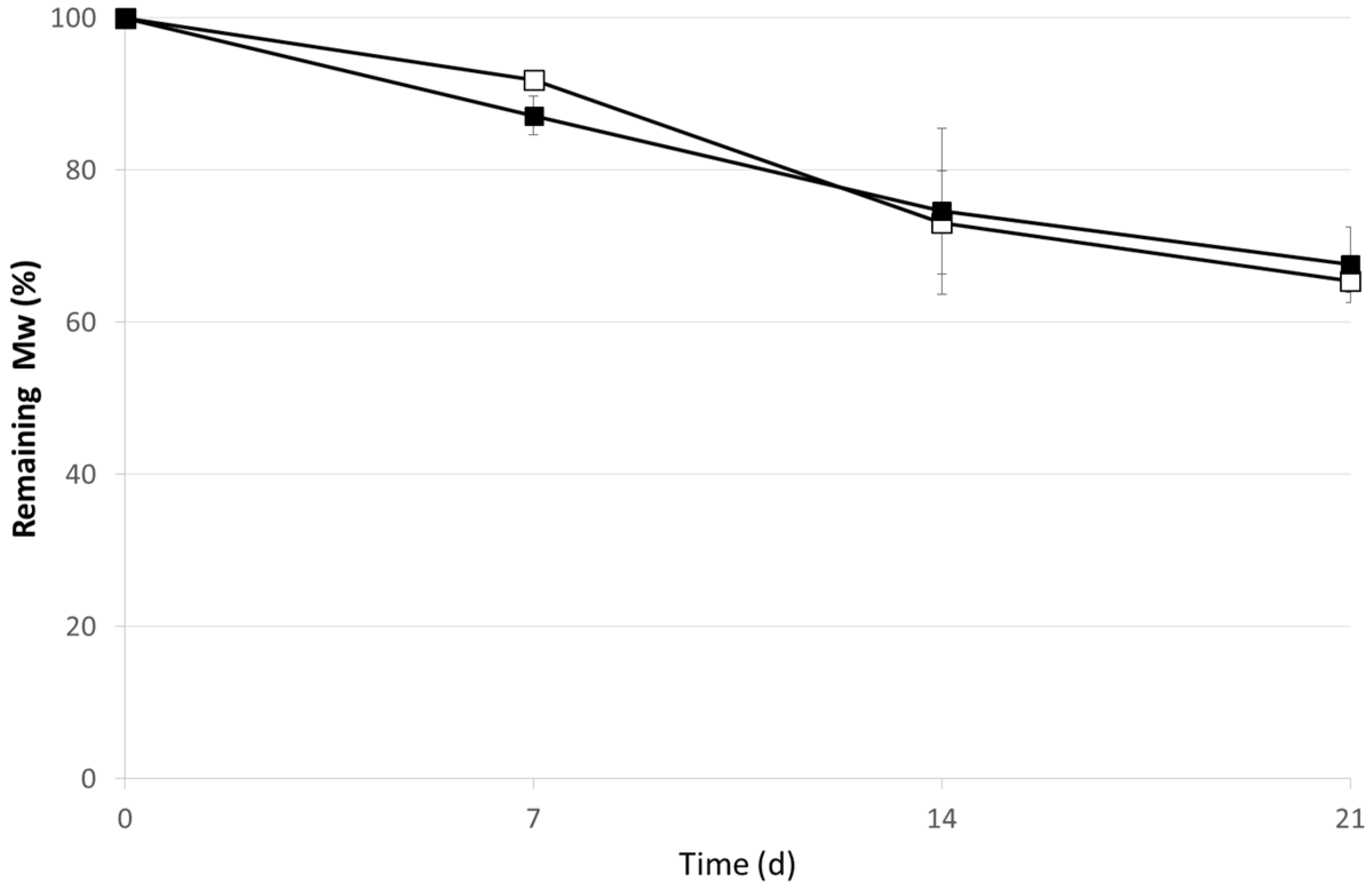
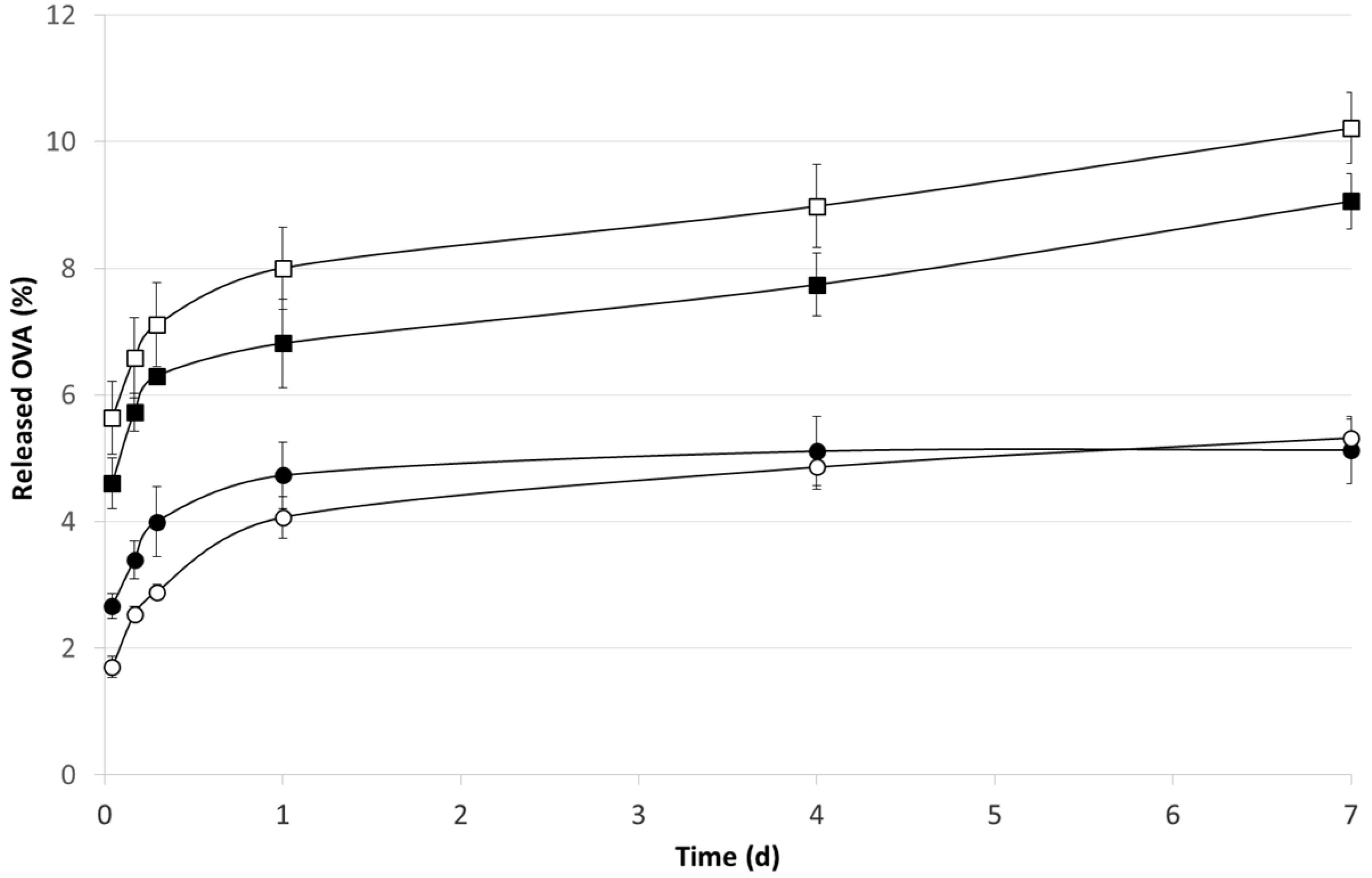
2.2. OVA Loaded Microspheres
| Polymer | EE% | Particle Size (µm) | Span | ζ (mV) | ||
|---|---|---|---|---|---|---|
| d10 | d50 | d90 | ||||
| PLGA | – | 6.4 ± 0.8 | 13.8 ± 0.0 | 38.5 ± 0.0 | 2.3 ± 0.1 | −29.8 ± 1.3 |
| g-CA-PLGA | – | 5.8 ± 0.0 | 13.8 ± 1.1 | 40.7 ± 13.7 | 2.5 ± 0.8 | −24.8 ± 1.4 |
| PLGA | 35.0 ± 0.7 | 5.5 ± 1.2 | 13.1 ± 0.9 | 29.4 ± 8.2 | 1.8 ± 0.3 | −11.2 ± 3.0 |
| g-CA-PLGA | 95.6 ± 2.7 | 4.5 ± 0.1 | 15.4 ± 1.3 | 40.45 ± 1.1 | 2.3 ± 0.7 | −15.1 ± 1.1 |
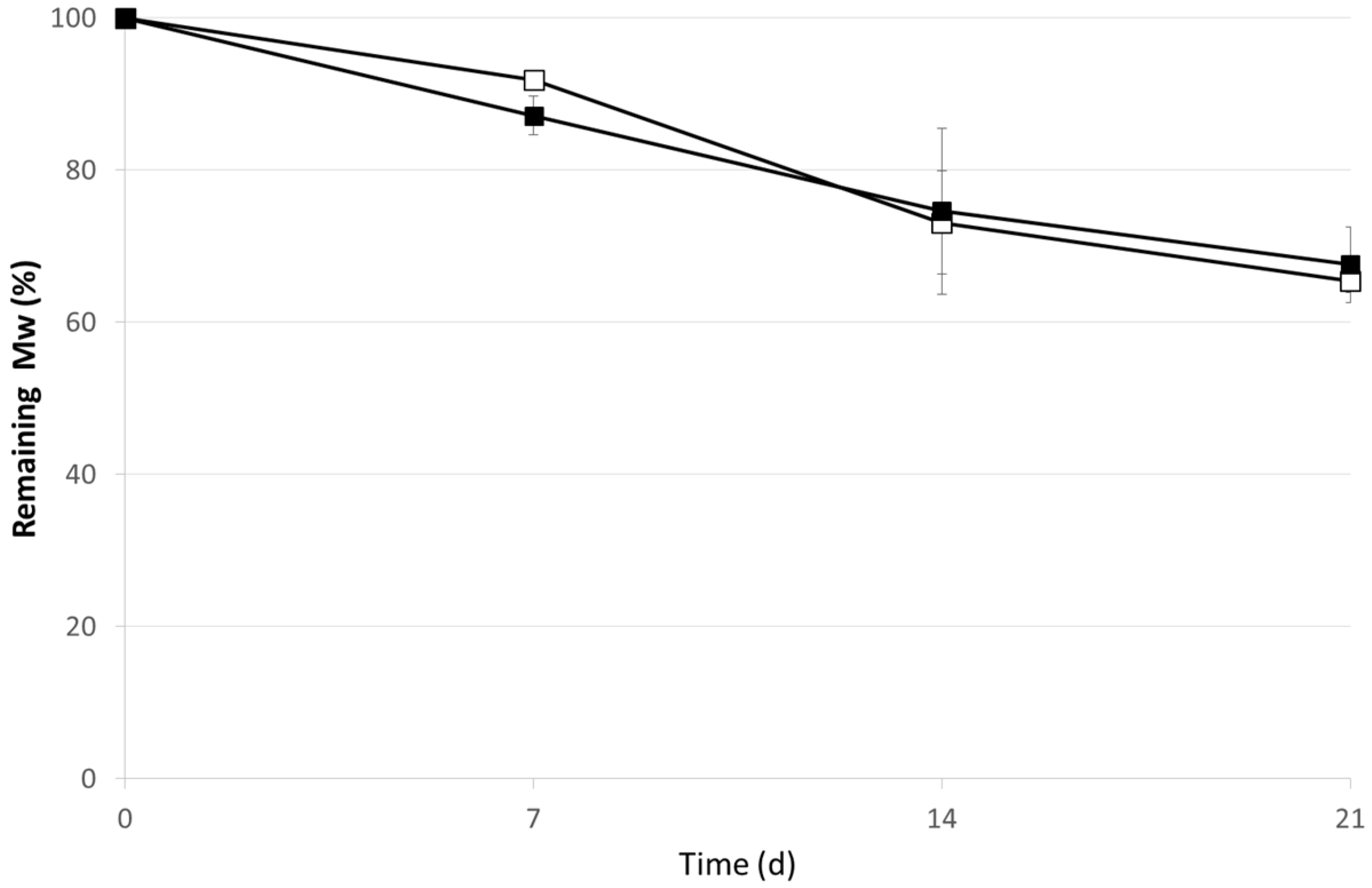
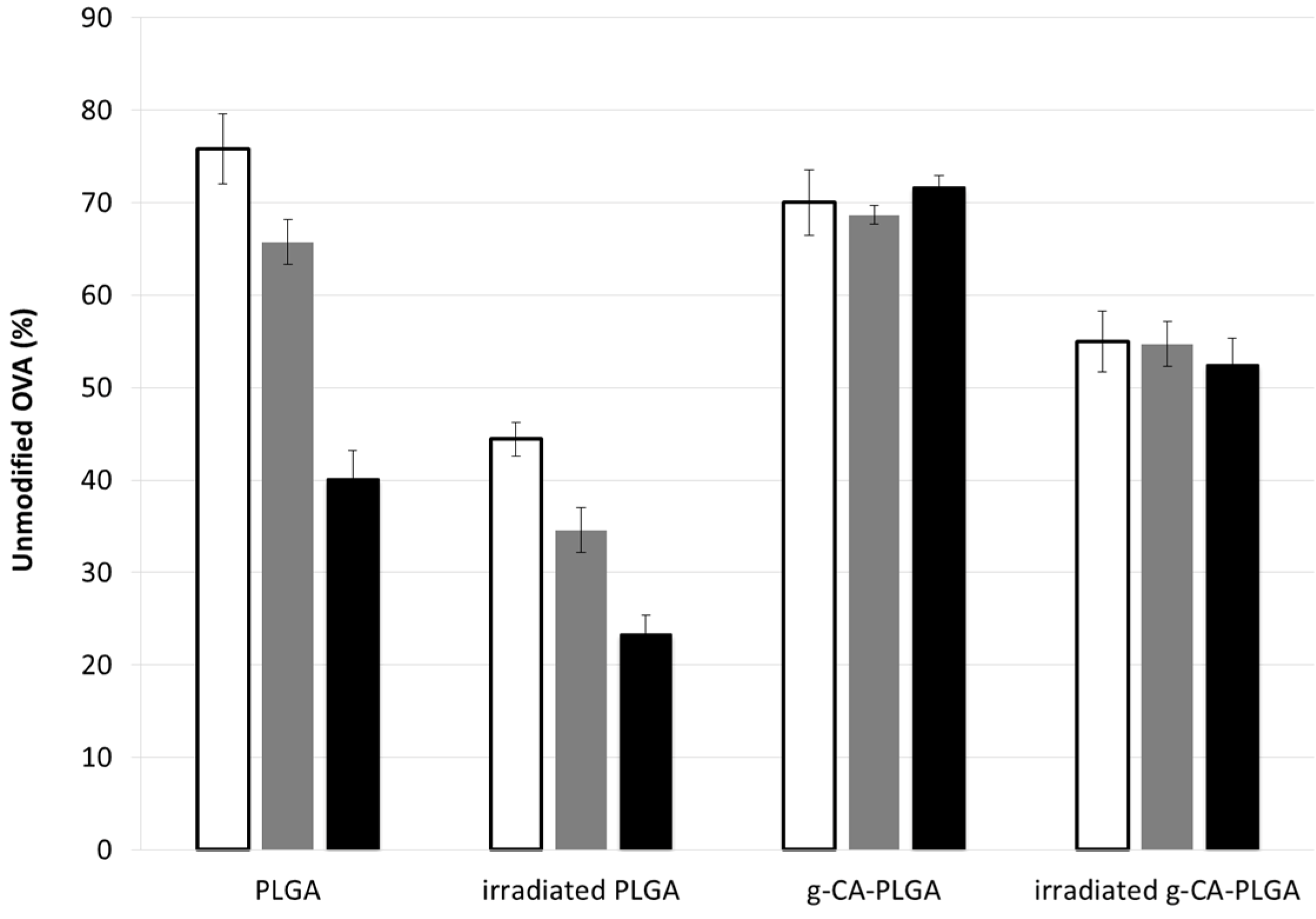
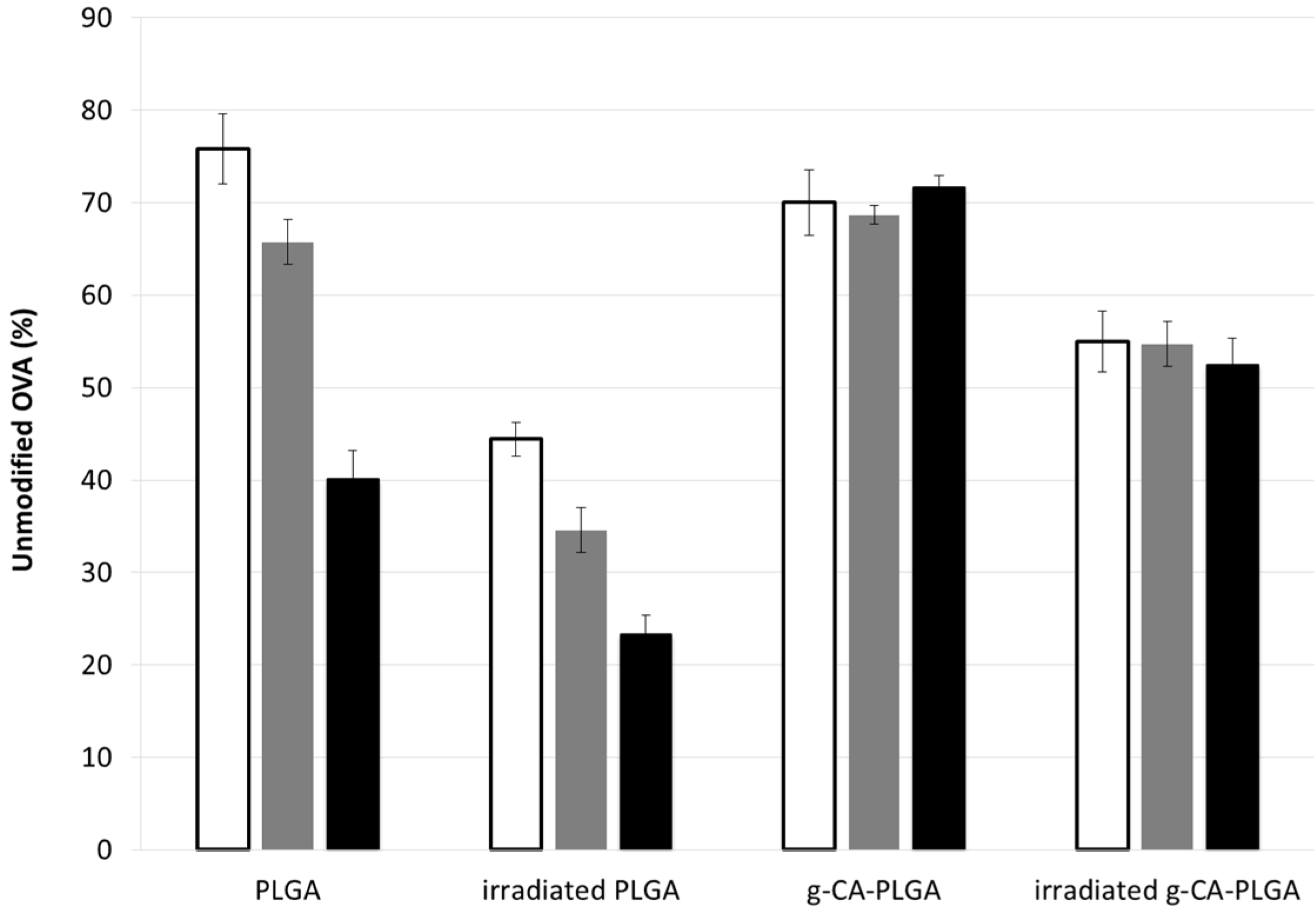
3. Experimental Section
3.1. Synthesis of g-CA-PLGA
3.2. Characterization of g-CA-PLGA
3.2.1. Determination of Scavenging Activity on DPPH Radicals
3.2.2. Evaluation of the Disposable Phenolic Groups by Folin-Ciocalteu Assay
3.3. Molecular Characterization
3.3.1. ATR-FTIR Spectroscopy
3.3.2. Thermal Analysis
3.3.3. GPC Analysis
3.4. Placebo and Ovalbumin Loaded Microspheres
3.4.1. Microspheres Preparation
3.4.2. γ-Irradiation
3.4.3. Microspheres Characterization
Particle Size
Zeta-Potential
Total Protein Content Determination
3.5. In Vitro OVA Release
3.6. Ovalbumin Determination
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Hausberger, A.G.; Kenly, R.A.; DeLuca, P.P. Gamma irradiation effects on molecular weight and in vitro degradation of poly(lactide-co-glycolide) microparticles. Pharm. Res. 1995, 12, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Montanari, L.; Constantini, E.C.; Signoretti, L.; Valvo, L.; Santucci, M.; Bartolomei, P.; Fattibene, P.; Onori, S.; Faucitano, A.; Conti, B.; Genta, I. Gamma irradiation effects on poly(d,l-lactide-co-glycolide) microspheres. J. Control. Release 1998, 56, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Montanari, L.; Cilurzo, F.; Valvo, L.; Faucitano, A.; Buttafava, A.; Groppo, A.; Genta, I.; Conti, B. Gamma irradiation effects on stability of poly(lactide-co-glycolide) microspheres containing clonazepam. J. Control. Release 2001, 75, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Faucitano, A.; Buttafava, A.; Montanari, L.; Cilurzo, F.; Conti, B.; Genta, I.; Valvo, L. Radiation-induced free radical reactions in polymer/drug systems for controlled release: An EPR investigation. Rad. Phys. Chem. 2003, 67, 61–72. [Google Scholar] [CrossRef]
- Montanari, L.; Cilurzo, F.; Conti, B.; Genta, I.; Groppo, A.; Valvo, L.; Faucitano, A.; Buttafava, A. Gamma irradiation effects and EPR investigation on poly(lactide-co-glycolide) microspheres containing bupivacaine. Farmaco 2002, 57, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Mohr, D.; Wolff, M.; Kissel, T. Gamma irradiation for terminal sterilization of 17β-estradiol loaded poly(d,l-lactide-co-glycolide) microparticles. J. Control. Release 1999, 61, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Dorati, R.; Genta, I.; Montanari, L.; Cilurzo, F.; Buttafava, A.; Faucitano, A.; Conti, B. The effect of γ-irradiation on PLGA/PEG microspheres containing ovalbumin. J. Control. Release 2005, 107, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Montanari, L.; Cilurzo, F.; Selmin, F.; Conti, B.; Genta, I.; Poletti, G.; Orsini, F.; Valvo, L. Poly(lactide-co-glycolide) microspheres containing bupivacaine: Comparison between gamma and beta irradiation effects. J. Control. Release 2003, 90, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carballido, A.; Puebla, P.; Herrero-Vanrell, R.; Pastoriza, P. Radiosterilisation of indomethacin PLGA/PEG-derivative microspheres: Protective effects of low temperature during gamma-irradiation. Int. J. Pharm. 2006, 313, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Malyala, P.; Palloro, M.; Giuliani, M.; Petersen, H.; O’Hagan, D.T.; Singh, M. A two-stage strategy for sterilization of poly(lactide-co-glycolide) particles by gamma-irradiation does not impair their potency for vaccine delivery. J. Pharm. Sci. 2011, 100, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Cilurzo, F.; Selmin, F.; Minghetti, P.; Montanari, L. Design of methylprednisolone biodegradable microspheres intended for intra-articular administration. AAPS PharmSciTech. 2008, 9, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Cilurzo, F.; Puoci, F.; Selmin, F.; Iemma, F.; Minghetti, P. Pyrogallic acid-PLGA conjugate as new biodegradable material suitable for final sterilization by irradiation. Pol. Adv. Tech. 2011, 22, 2201–2205. [Google Scholar] [CrossRef]
- Mohanan, D.; Gander, B.; Kündig, T.M.; Johansen, P. Encapsulation of antigen in poly(d,l-lactide-co-glycolde) microspheres protects from harmful effects of γ-irradiation as assessed in mice. Eur. J. Pharm. Biopharm. 2012, 80, 272–281. [Google Scholar] [CrossRef]
- Witt, C.; Kissel, T. Morphological characterization of microspheres, films and implants prepared from poly(lactide-co-glycolide) and ABA triblock copolymers: Is the erosion controlled by degradation, swelling or diffusion? Eur. J. Pharm. Biopharm. 2001, 51, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Floyd, A.G. Injectable emulsions and suspensions. In Pharmaceutical Dosage Forms—Disperse Systems; Lieberman, H.A., Rieger, M.M., Banker, G.S., Eds.; Marcel Dekker: New York, NY, USA, 1996. [Google Scholar]
- Cilurzo, F.; Selmin, F.; Minghetti, P.; Adami, M.; Bertoni, E.; Lauria, S.; Montanari, L. Injectability: An open issue. AAPS PharmSciTech. 2011, 12, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Panusa, A.; Selmin, F.; Rossoni, G.; Carini, M.; Cilurzo, F.; Aldini, G. Methylprednisolone-loaded PLGA microspheres: A new formulation for sustained release via intra-articular administration. A comparison study with methylprednisolone acetate in rats. J. Pharm. Sci. 2011, 100, 4580–4586. [Google Scholar] [CrossRef] [PubMed]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.; Groppo, A.; Genta, I.; Dacarro, C.; Valvo, L.; Cilurzo, F.; Montanari, L. Comparison among different preparation methods of ovalbumin loaded PLGA microspheres. In Proceedings of 27th International Symposium on Controlled Release of Bioactive Materials, Paris, France, 7–13 July 2000.
- The Use of Ionizing Radiation in the Manufacture of Medicinal Products; European Guideline 3AQ4a; European Medicines Agency: London, UK, December 1991.
- Audette-Stuart, M.; Houée-Levin, C.; Potier, M. Radiation-induced protein fragmentation and inactivation in liquid and solid aqueous solutions. Role of OH and electrons. Radiat. Phys. Chem. 2005, 72, 301–306. [Google Scholar] [CrossRef]
- Terryn, H.; Deridder, V.; Sicard-Roselli, C.; Tilquin, B.; Houée-Levin, C. Radiolysis of proteins in the solid state: An approach by EPR and product analysis. J. Synchrotron Radiat. 2005, 12, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Selmin, F.; Blasi, P.; DeLuca, P.P. Accelerated polymer biodegradation of risperidone poly(d,l-lactide-co-glycolide) microspheres. AAPS PharmSciTech. 2012, 13, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selmin, F.; Puoci, F.; Parisi, O.I.; Franzé, S.; Musazzi, U.M.; Cilurzo, F. Caffeic Acid-PLGA Conjugate to Design Protein Drug Delivery Systems Stable to Irradiation. J. Funct. Biomater. 2015, 6, 1-13. https://doi.org/10.3390/jfb6010001
Selmin F, Puoci F, Parisi OI, Franzé S, Musazzi UM, Cilurzo F. Caffeic Acid-PLGA Conjugate to Design Protein Drug Delivery Systems Stable to Irradiation. Journal of Functional Biomaterials. 2015; 6(1):1-13. https://doi.org/10.3390/jfb6010001
Chicago/Turabian StyleSelmin, Francesca, Francesco Puoci, Ortensia I. Parisi, Silvia Franzé, Umberto M. Musazzi, and Francesco Cilurzo. 2015. "Caffeic Acid-PLGA Conjugate to Design Protein Drug Delivery Systems Stable to Irradiation" Journal of Functional Biomaterials 6, no. 1: 1-13. https://doi.org/10.3390/jfb6010001