Cytocompatibility and Mechanical Properties of Short Phosphate Glass Fibre Reinforced Polylactic Acid (PLA) Composites: Effect of Coupling Agent Mediated Interface

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Reinforcement | Flexural strength | Modulus | Reference |
|---|---|---|---|---|
| Vf (%) | (MPa) | (GPa) | ||
| PLA | Continuous UD and 10mm random Quinternary PGF (15‑20%) | 106‑115 | 6.8‑9 | [9] |
| POE | Short random ternary PGF (0–50%) | 65–103 | 1.5‑9.4 | [12] |
| PLA | 10mm random quaternary PGF 14% | 90 | 5 | [13] |
| methacrylate-modified oligolactide | 50 cm long quinternary PGF (NR) | 115 ± 11.9 | 16 ± 2.4 | [14] |
| PCL ( in situ polymerisation) | Continuous UD quaternary PGF 25% | 105 ± 12 | 5.9 ± 6 | [15] |
| PCL (compression moulding) | Continuous UD quaternary PGF 25% | 55 ± 8 | 2.1 ± 0.3 | [15] |
| PCL | Continuous UD quinternary PGF (10% wt) | 72 | 2.74 | [16] |
| methacrylate-modified oligolactide | 30 cm quaternary PGF | 110–190 | 15–20 | [17] |
| PCL | 10mm random binary PGF (6‑18%) | 30 | 2.5 | [18] |
| PLA | Continuous UD and short random PGF (40‑55%) | 120–350 | 10 to 30 | [19] |
2. Results and Discussion
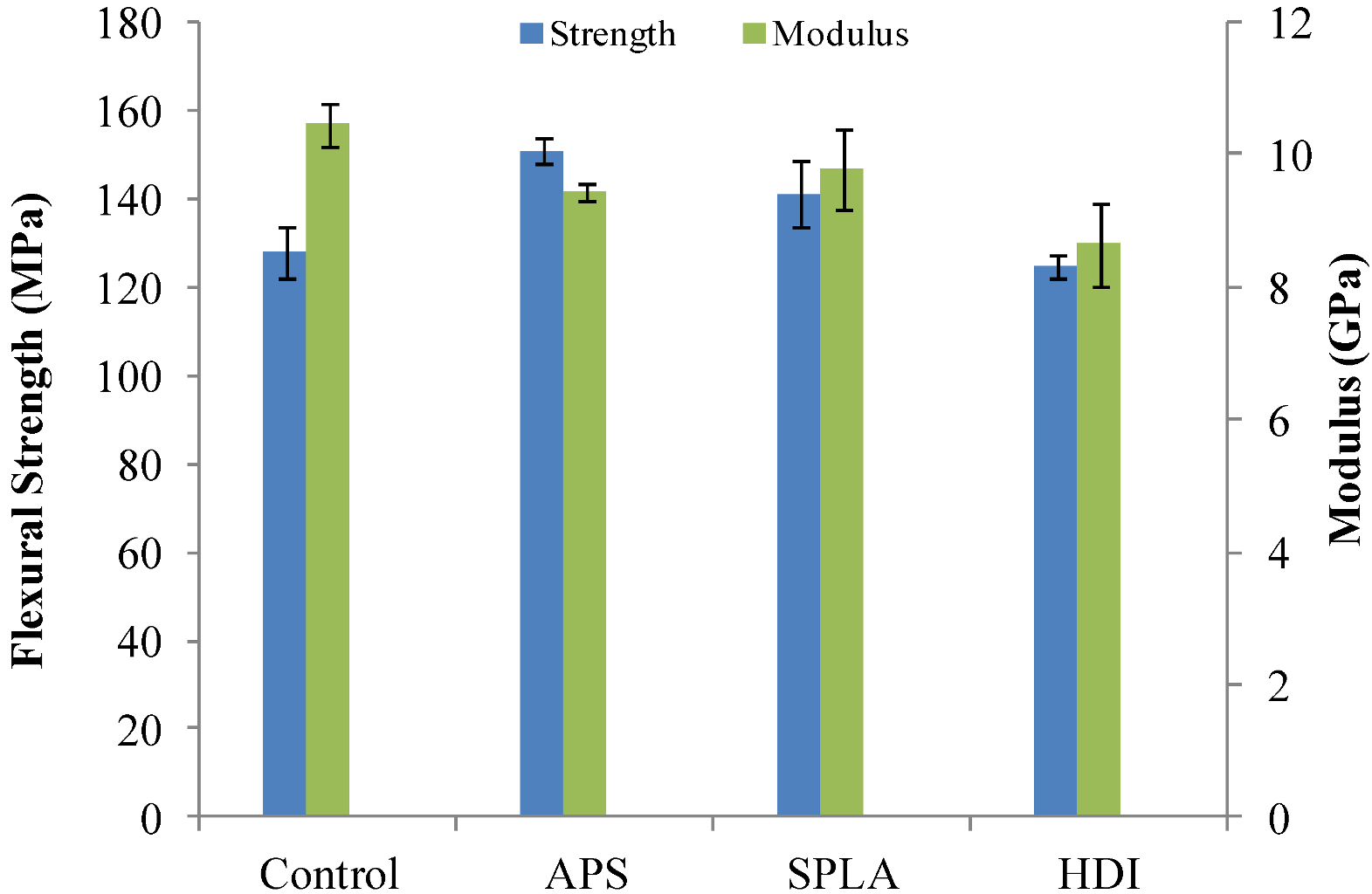
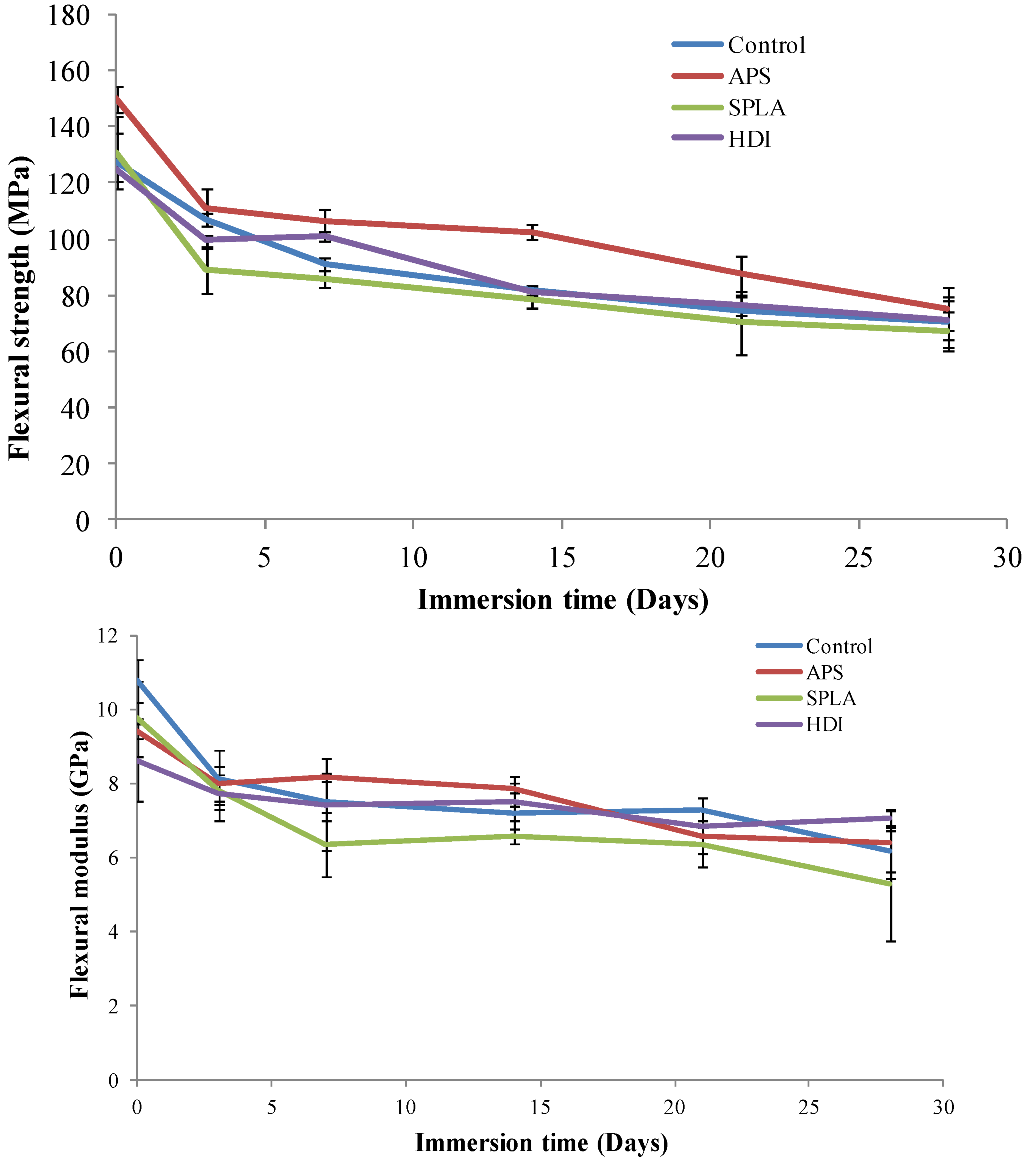
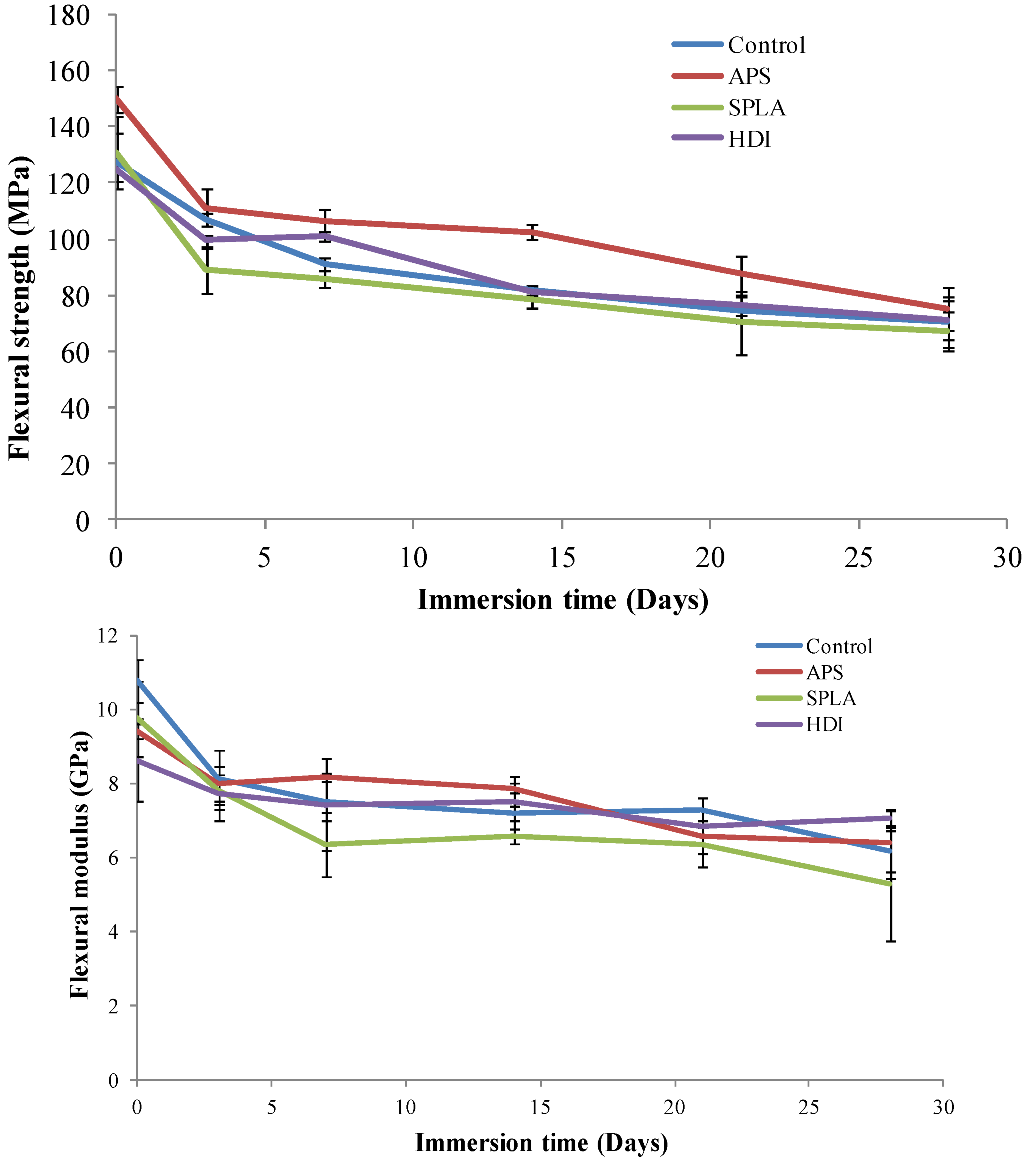
2.1. Flexural Mechanical Properties
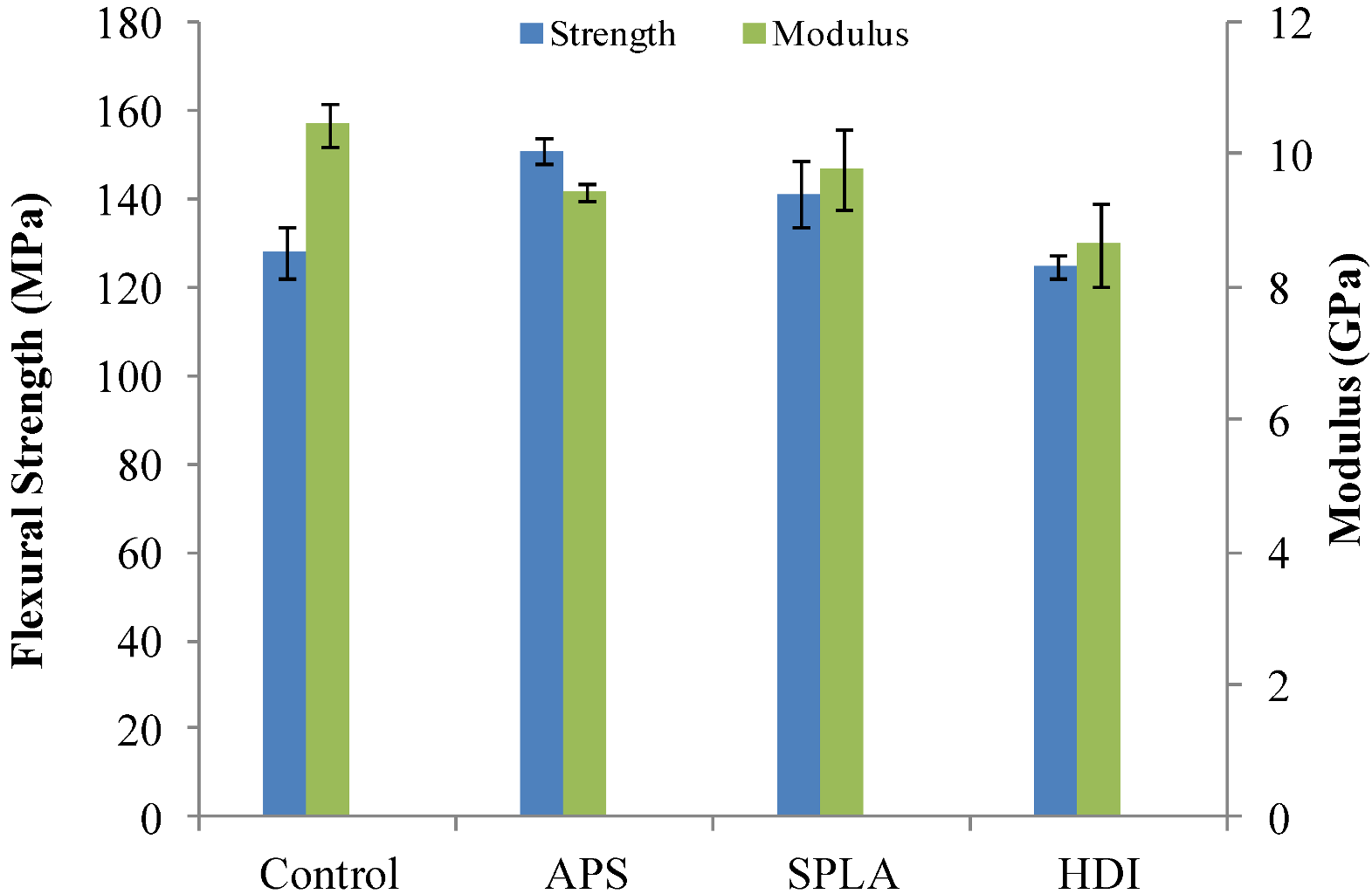
| Sample code | Fibre volume fraction (%) |
|---|---|
| Control | 20 ± 4 |
| APS | 23 ± 6 |
| SPLA | 21 ± 5 |
| HDI | 18 ± 3 |
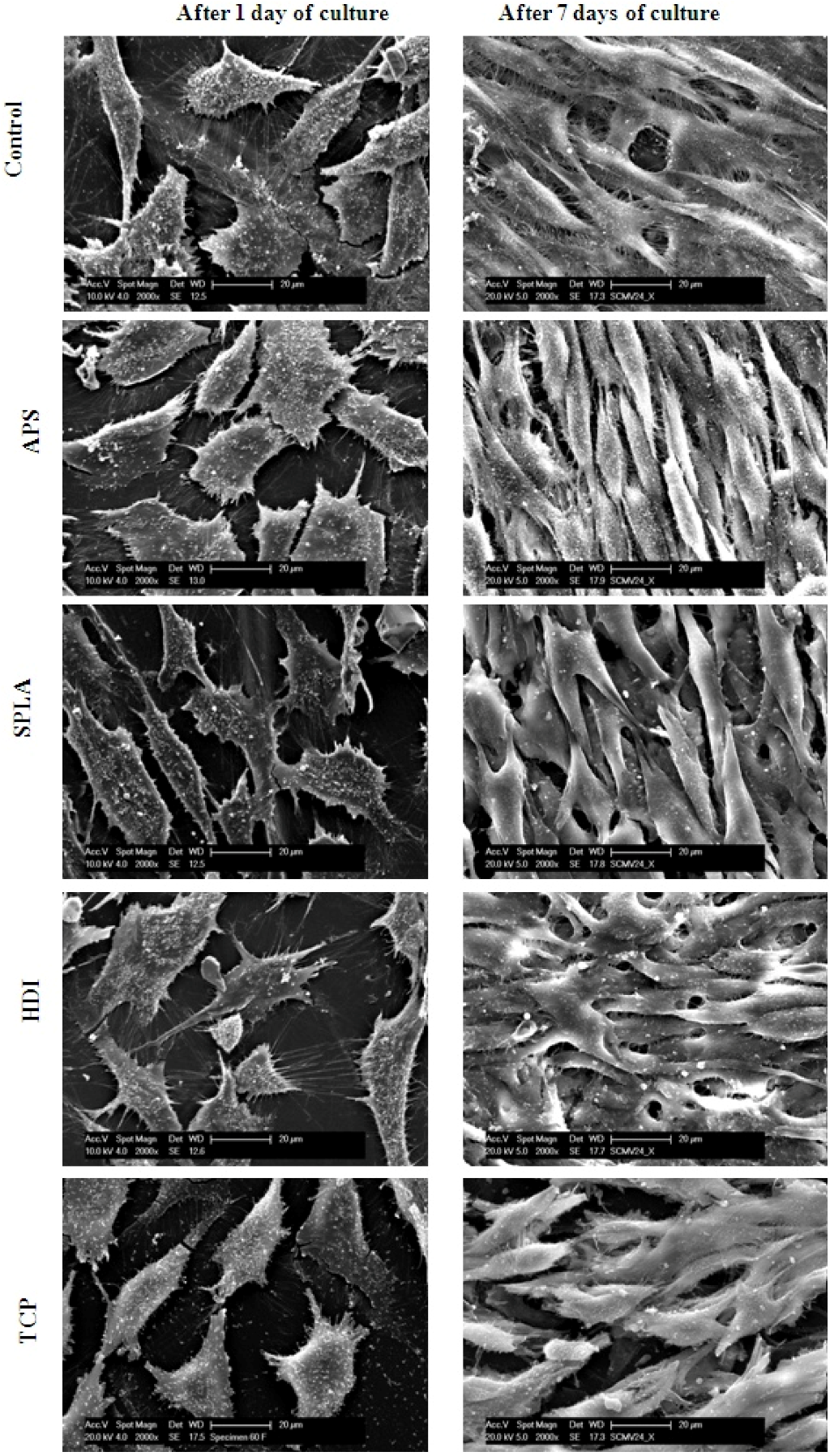
2.2. SEM Analysis of PGF/PLA Interface

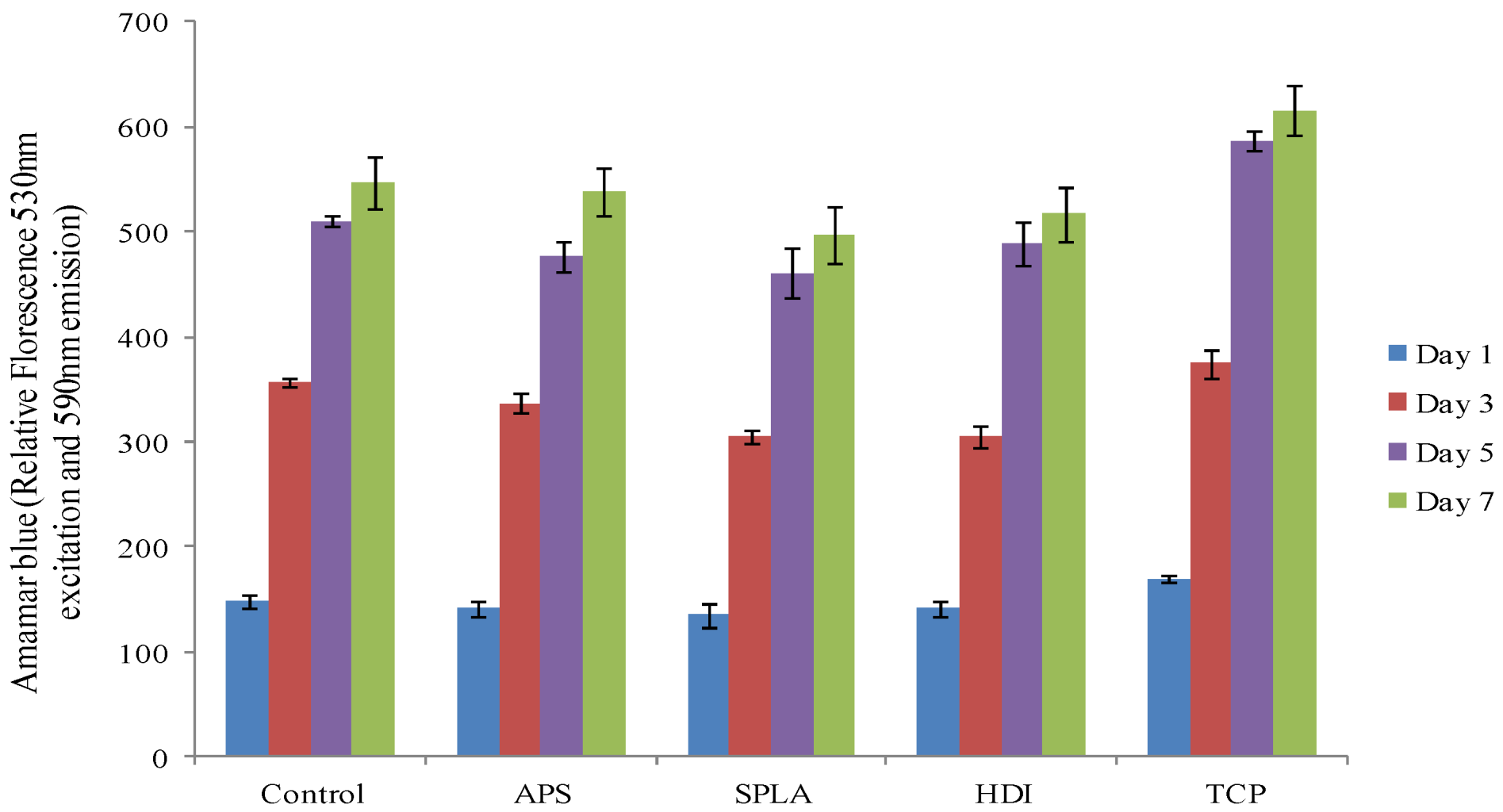
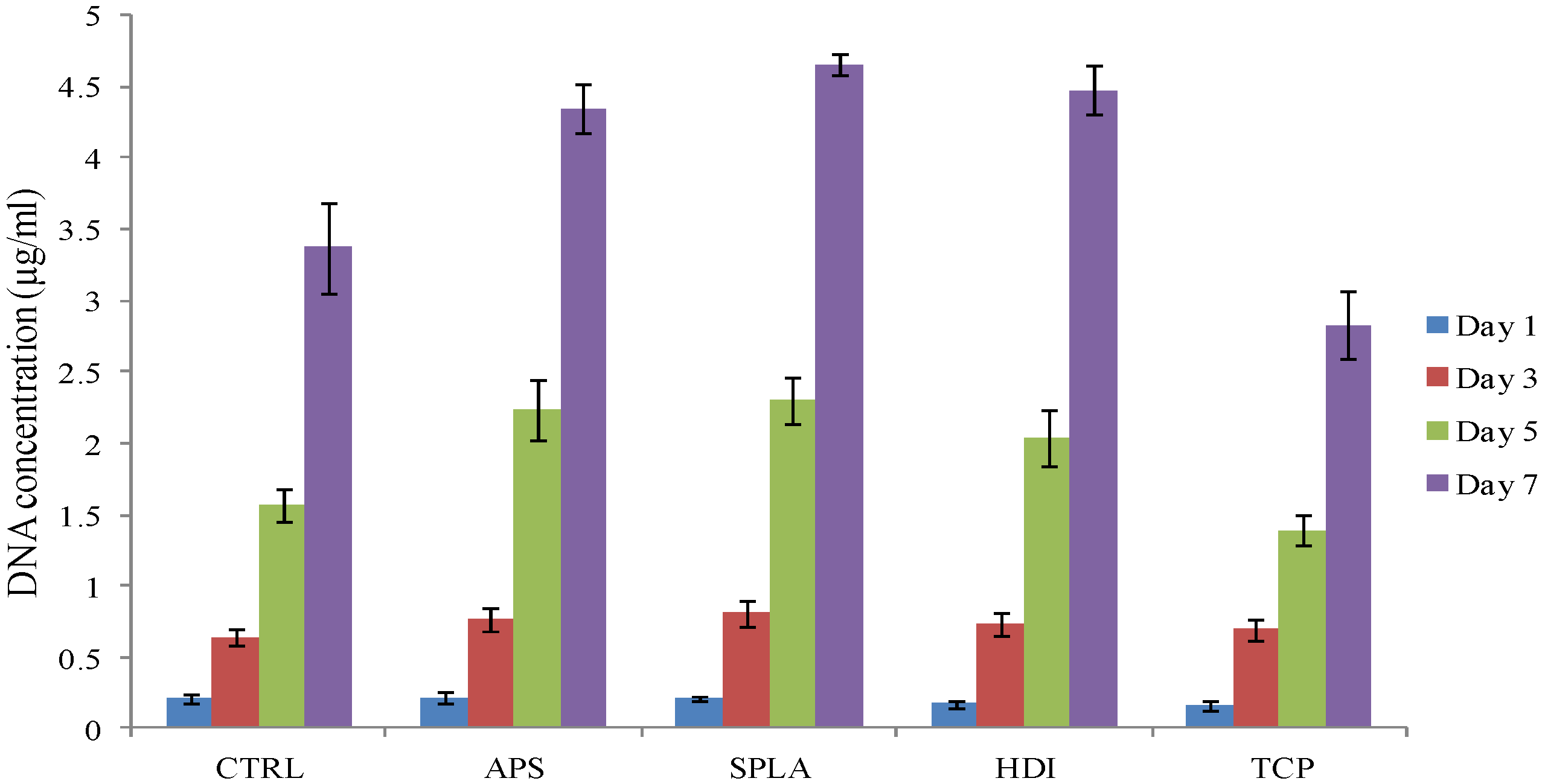
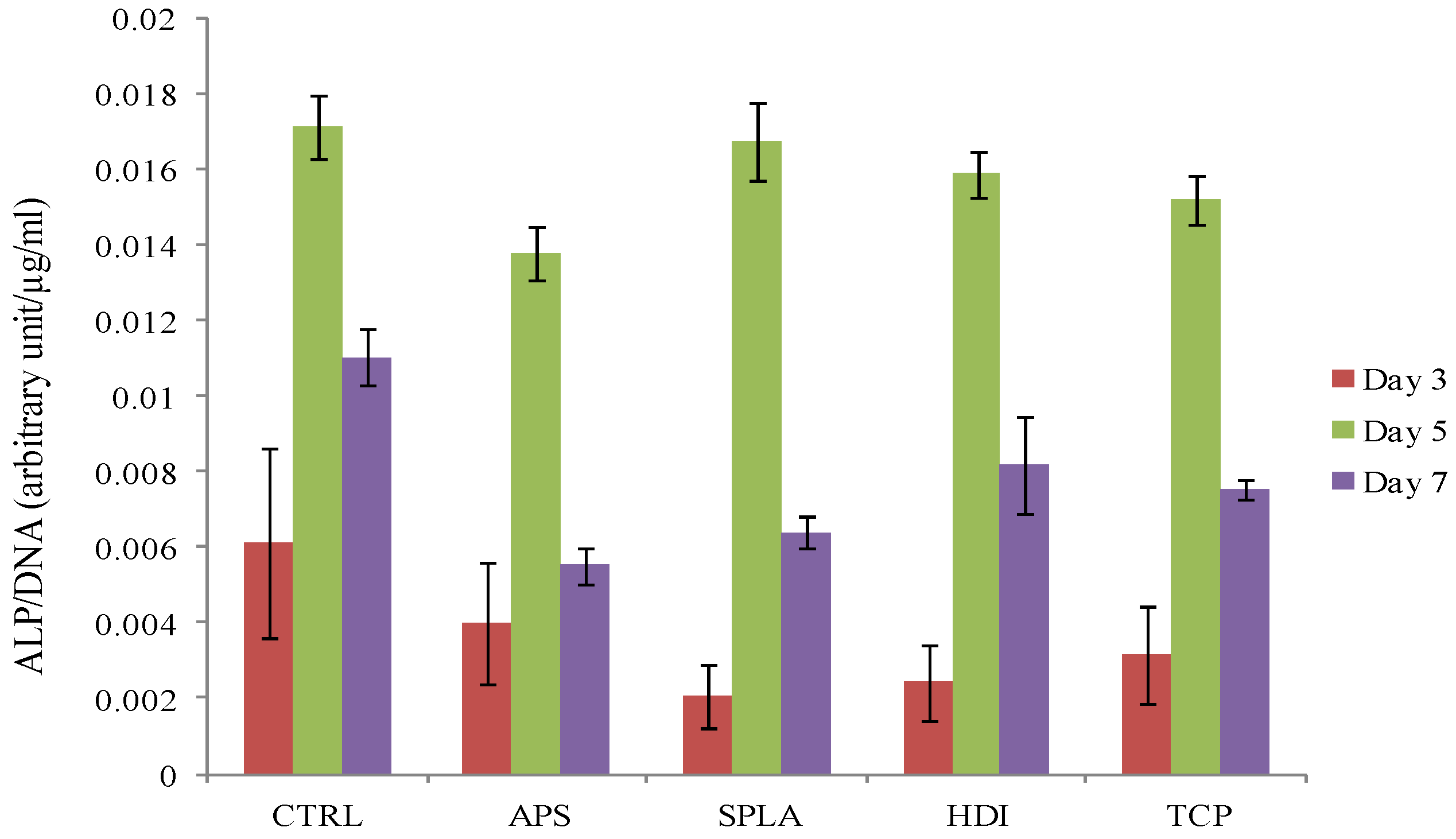
2.3. Cytocompatibility Analysis
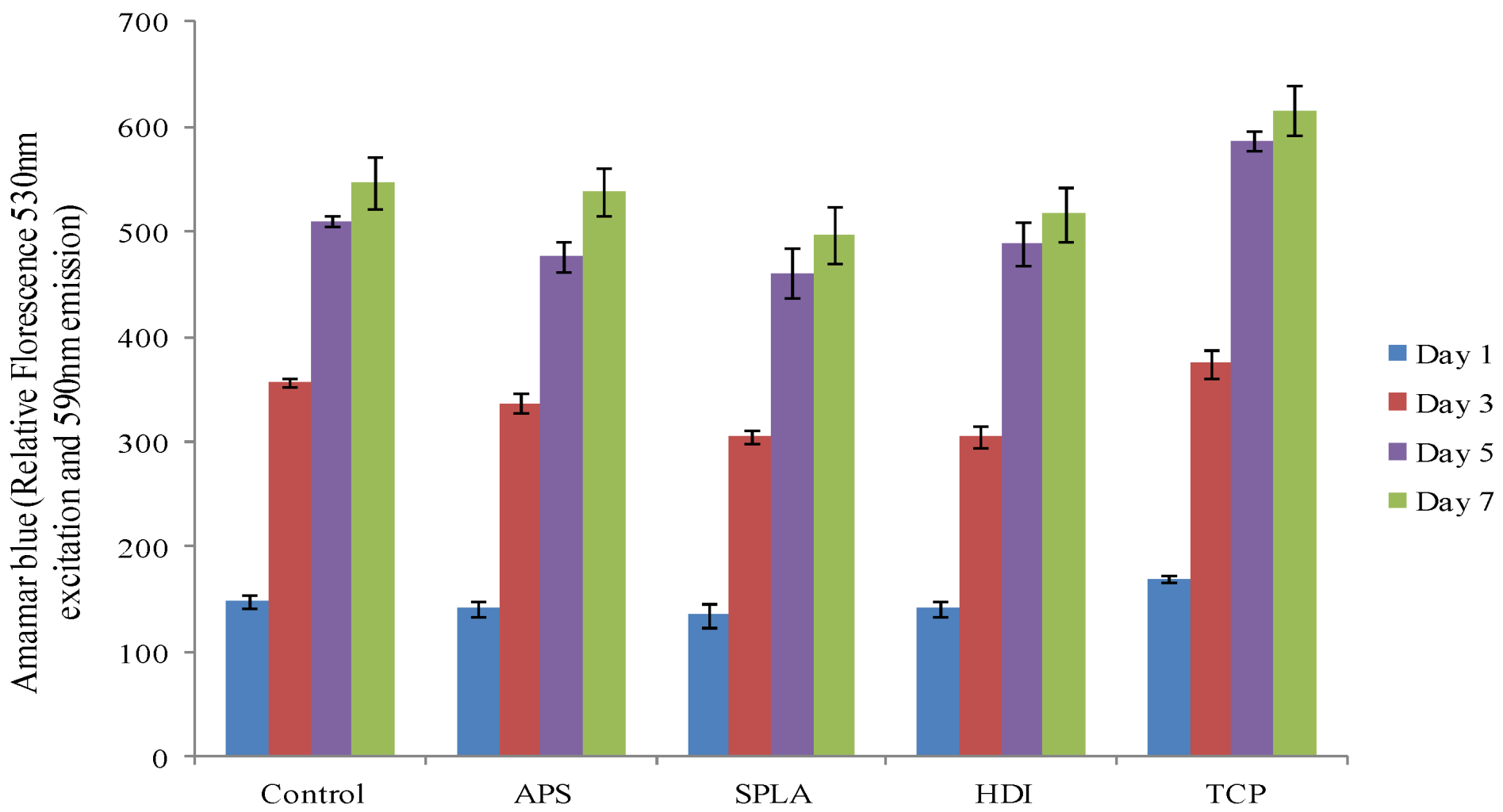
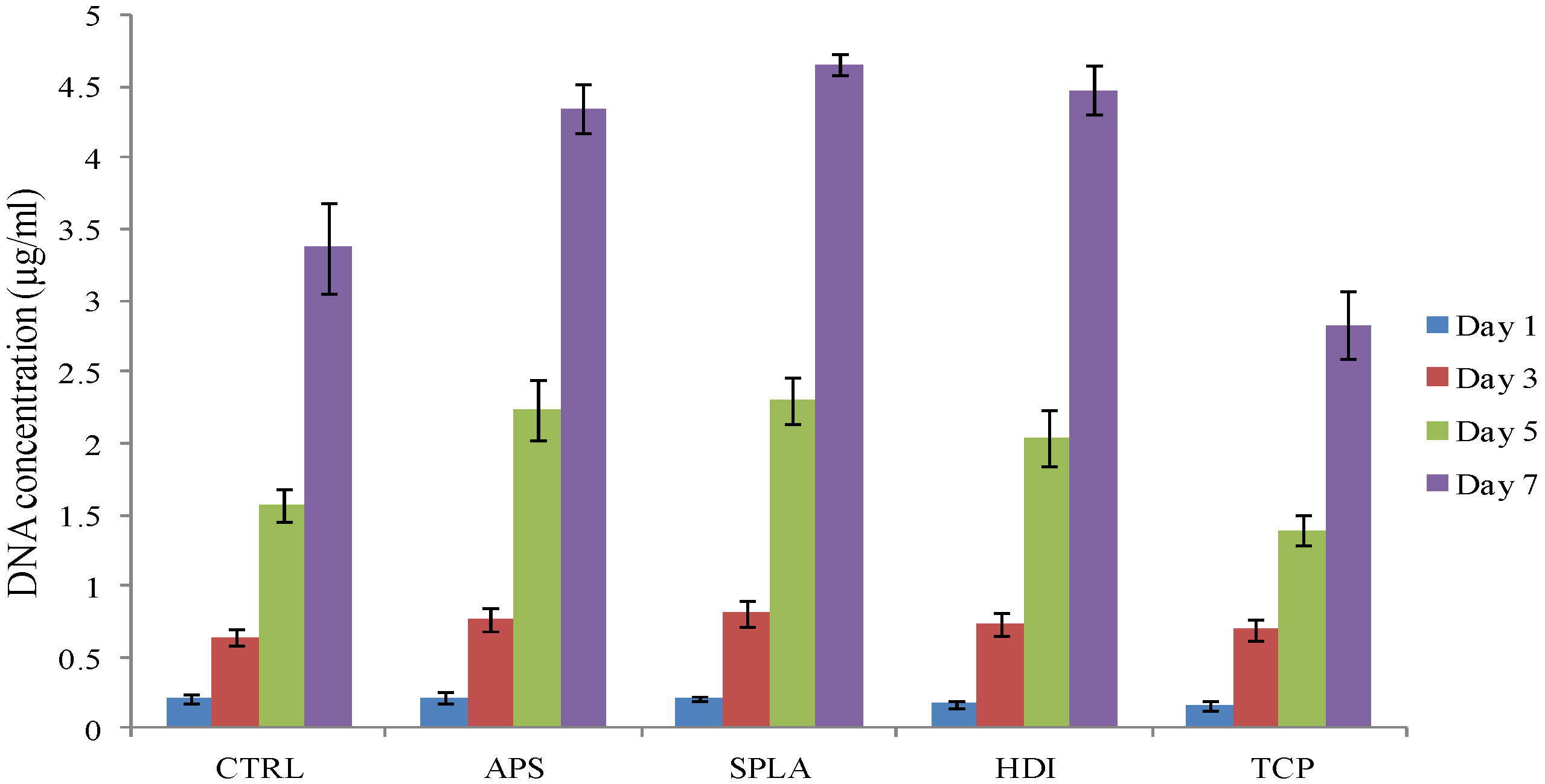
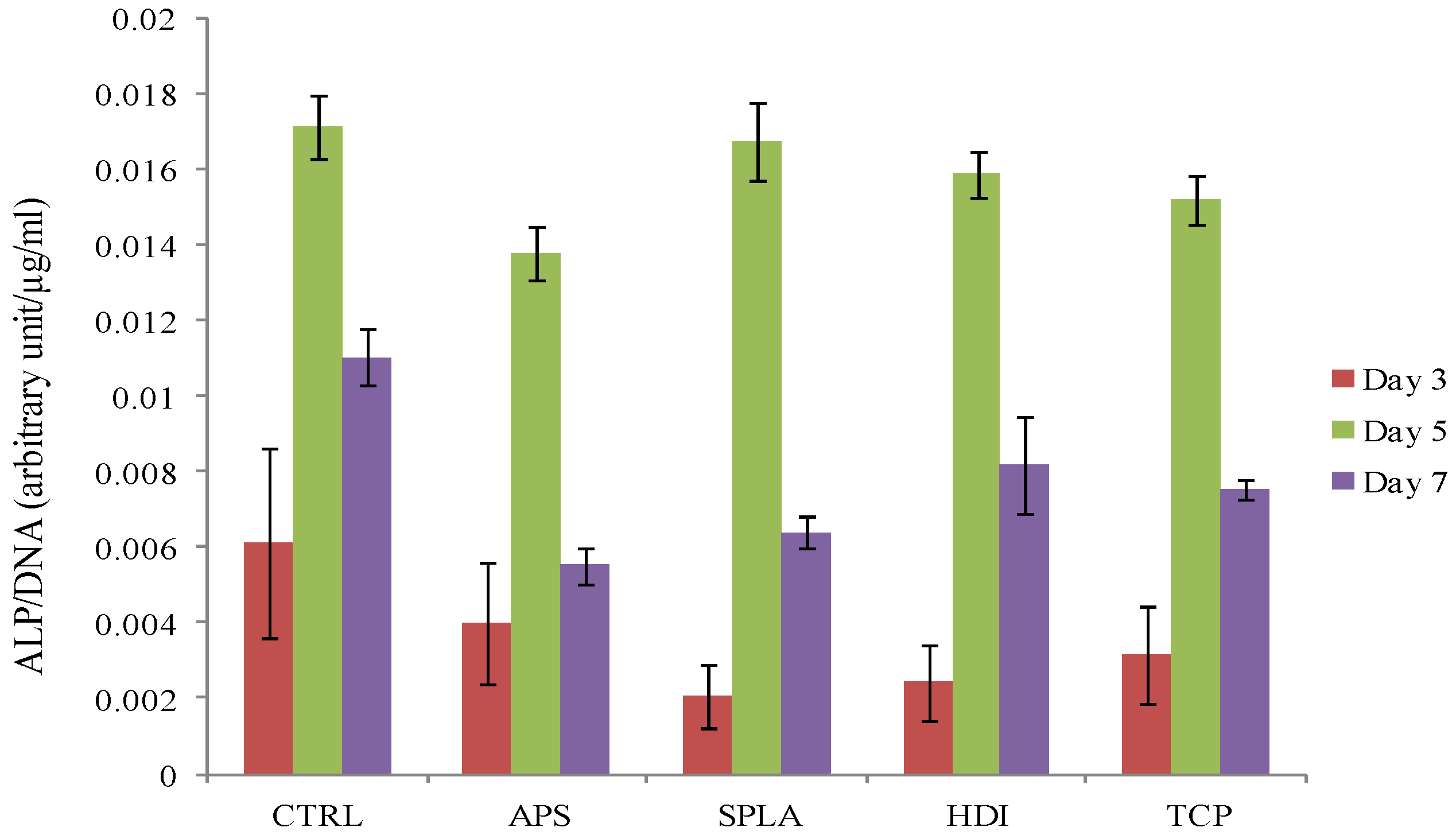

3. Experimental Section
3.1. Glass Synthesis
3.2. Fibre Production
3.3. Fibre Coupling Agent Treatment
3.4. Fibre Mat Production (Air Lay)
3.5. PLA Sheet Preparation
3.6. Composite Production
3.7. Flexural Mechanical Properties Analysis
3.8. SEM Analysis of Composite Cross-Section
3.9. Cytocompatibility Analysis
3.9.1. Cell Viability/Metabolic Activity
3.9.2. DNA Quantification
3.9.3. Alkaline Phosphatase Activity
3.9.4. Cell Morphology Analysis
3.10. Statistical Analyses
4. Conclusions
Acknowledgments
References
- Törmälä, P. Biodegradable self-reinforced composite materials; Manufacturing structure and mechanical properties. Clin. Mater. 1992, 10, 29–34. [Google Scholar] [CrossRef]
- Törmälä, P.; Pohjonen, T.; Rokkanen, P. Bioabsorbable polymers: Materials technology and surgical applications. J. Eng. Med. 1998, 212, 101–111. [Google Scholar]
- Törmälä, P.; Vasenius, J.; Vainionpää, S.; Laiho, J.; Pohjonen, T.; Rokkanen, P. Ultra-high-strength absorbable self-reinforced polyglycolide (SR-PGA) composite rods for internal fixation of bone fractures: In vitro and in vivo study. J. Biomed. Mater. Res. 1991, 25, 1–22. [Google Scholar] [CrossRef]
- Enislidis, G.; Lagogiannis, G.; Wittwer, G.; Glaser, C.; Ewers, R. Fixation of zygomatic fractures with a biodegradable copolymer osteosynthesis system: Short- and long-term results. Int. J. Oral Maxillofac. Surg. 2005, 34, 19–26. [Google Scholar]
- Wambua, P.; Ivens, J.; Verpoest, I. Natural fibres: Can they replace glass in fibre reinforced plastics? Compos. Sc. Technol. 2003, 63, 1259–1264. [Google Scholar] [CrossRef]
- Ahmed, I.; Collins, C.A.; Lewis, M.P.; Olsen, I.; Knowles, J.C. Processing, characterisation and biocompatibility of iron-phosphate glass fibres for tissue engineering. Biomaterials 2004, 25, 3223–3232. [Google Scholar]
- Felfel, R.; Ahmed, I.; Parsons, A.; Harper, L.; Rudd, C. Initial mechanical properties of phosphate-glass fibre-reinforced rods for use as resorbable intramedullary nails. J. Mater. Sci. 2012, 47, 4884–4894. [Google Scholar] [CrossRef]
- Ramsay, S.; Pilliar, R.; Yang, L.; Santerre, J. Calcium polyphosphate/polyvinyl acid-carbonate copolymer based composites for use in biodegradable load-bearing composites for orthopaedic implant fabrication. Key Eng. Mater. 2005, 284-286, 787–790. [Google Scholar] [CrossRef]
- Felfel, R.M.; Ahmed, I.; Parsons, A.J.; Haque, P.; Walker, G.S.; Rudd, C.D. Investigation of crystallinity, molecular weight change, and mechanical properties of PLA/PBG bioresorbable composites as bone fracture fixation plate. J. Biomater. Appl. 2012, 26, 765–789. [Google Scholar] [CrossRef]
- Felfel, R.M.; Ahmed, I.; Parsons, A.J.; Walker, G.S.; Rudd, C.D. In vitro degradation, flexural, compressive and shear properties of fully bioresorbable composite rods. J. Mech. Behav. Biomed. Mater. 2011, 4, 1462–1472. [Google Scholar] [CrossRef]
- Lin, T.C. Totally absorbable fiber reinforced composite for internal fracture fixation devices. Trans. Soc. Biomater. 1986, 9, 166. [Google Scholar]
- Andriano, K.P.; Daniels, A.U.; Heller, J. Biocompatibility and mechanical properties of a totally absorbable composite material for orthopaedic fixation devices. J. Appl. Biomater. 1992, 3, 197–206. [Google Scholar] [CrossRef]
- Ahmed, I.; Cronin, P.S.; Neel, E.A.A.; Parsons, A.J.; Knowles, J.C.; Rudd, C.D. Retention of mechanical properties and cytocompatibility of a phosphate-based glass fiber/polylactic acid composite. J. Biomed. Mater. Res. B Appl. Biomater. 2009, 89B, 18–27. [Google Scholar] [CrossRef]
- Brauer, D.; Rüssel, C.; Vogt, S.; Weisser, J.; Schnabelrauch, M. Degradable phosphate glass fiber reinforced polymer matrices: Mechanical properties and cell response. J. Mater. Sci. Mater. Med. 2008, 19, 121–127. [Google Scholar] [CrossRef]
- Khan, R.A.; Parsons, A.J.; Jones, I.A.; Walker, G.S.; Rudd, C.D. Surface treatment of phosphate glass fibers using 2-hydroxyethyl methacrylate: Fabrication of poly(caprolactone)-based composites. J. Appl. Polym. Sci. 2009, 111, 246–254. [Google Scholar]
- Khan, R.A.; Parsons, A.J.; Jones, I.A.; Walker, G.S.; Rudd, C.D. Preparation and characterization of phosphate glass fibers and fabrication of poly(caprolactone) matrix resorbable composites. J. Reinf. Plast. Compos. 2010, 29, 1838–1850. [Google Scholar]
- Kobayashi, H.S.; Brauer, D.S.; Rüssel, C. Mechanical properties of a degradable phosphate glass fibre reinforced polymer composite for internal fracture fixation. Mater. Sci. Eng. C 2010, 30, 1003–1007. [Google Scholar] [CrossRef]
- Ahmed, I.; Parsons, A.J.; Palmer, G.; Knowles, J.C.; Walker, G.S.; Rudd, C.D. Weight loss, ion release and initial mechanical properties of a binary calcium phosphate glass fibre/PCL composite. Acta Biomater. 2008, 4, 1307–1314. [Google Scholar]
- Parsons, A.J.; Ahmed, I.; Haque, P.; Fitzpatrick, B.; Niazi, M.I.K.; Walker, G.S.; Rudd, C.D. Phosphate glass fibre composites for bone repair. J. Bionic Eng. 2009, 6, 318–323. [Google Scholar] [CrossRef]
- Haque, P.; Barker, I.A.; Parsons, A.; Thurecht, K.J.; Ahmed, I.; Walker, G.S.; Rudd, C.D.; Irvine, D.J. Influence of compatibilizing agent molecular structure on the mechanical properties of phosphate glass fiber-reinforced PLA composites. J. Polym. Sci. A Polym. Chem. 2010, 48, 3082–3094. [Google Scholar]
- Haque, P.; Parsons, A.J.; Barker, I.A.; Ahmed, I.; Irvine, D.J.; Walker, G.S.; Rudd, C.D. Interfacial properties of phosphate glass fibres/PLA composites: Effect of the end functionalities of oligomeric PLA coupling agents. Compos. Sci. Technol. 2010, 70, 1854–1860. [Google Scholar]
- Dupraz, A.M.P.; Wijn, J.R.d.; Meer, S.; Groot, K. Characterization of silane-treated hydroxyapatite powders for use as filler in biodegradable composites. J. Biomed. Mater. Res. 1996, 30, 231–238. [Google Scholar]
- Andriano, K.P.; Daniels, A.U.; Heller, J. Effectiveness of silane treatment on absorbable microfibers. J. Appl. Biomater. 1992, 3, 191–195. [Google Scholar] [CrossRef]
- Dibenedetto, A.T.; Lex, P.J. Evaluation of surface treatments for glass fibers in composite materials. Polym. Eng. Sci. 1989, 29, 543–555. [Google Scholar] [CrossRef]
- Park, S.-J.; Jin, J.-S. Effect of silane coupling agent on mechanical interfacial properties of glass fiber-reinforced unsaturated polyester composites. J. Polym. Sci. B Polym. Phys. 2003, 41, 55–62. [Google Scholar] [CrossRef]
- DiBenedetto, A.T. Tailoring of interfaces in glass fiber reinforced polymer composites: A review. Mater. Sci. Eng. A 2001, 302, 74–82. [Google Scholar] [CrossRef]
- Yazdani, H.; Morshedian, J.; Khonakdar, H.A. Effects of silane coupling agent and maleic anhydride-grafted polypropylene on the morphology and viscoelastic properties of polypropylene-mica composites. Polym. Compos. 2006, 27, 491–496. [Google Scholar] [CrossRef]
- Haque, P. Oligomeric PLA Coupling Agents For Phosphate Glass Fibres/PLA Composites; The university of Nottingham: Nottingham, UK, 2011. [Google Scholar]
- Nowatzki, P.J.; Tirrell, D.A. Physical properties of artificial extracellular matrix protein films prepared by isocyanate crosslinking. Biomaterials 2004, 25, 1261–1267. [Google Scholar] [CrossRef]
- Dong, G.-C.; Sun, J.-S.; Yao, C.-H.; Jiang, G.J.; Huang, C.-W.; Lin, F.-H. A study on grafting and characterization of HMDI-modified calcium hydrogenphosphate. Biomaterials 2001, 22, 3179–3189. [Google Scholar] [CrossRef]
- Sun, J.-S.; Dong, G.-C.; Lin, C.-Y.; Sheu, S.-Y.; Lin, F.-H.; Chen, L.-T.; Chang, W.H.-S.; Wang, Y.-J. The effect of Gu-Sui-Bu (Drynaria fortunei J. Sm) immobilized modified calcium hydrogenphosphate on bone cell activities. Biomaterials 2003, 24, 873–882. [Google Scholar] [CrossRef]
- Liu, A.; Hong, Z.; Zhuang, X.; Chen, X.; Cui, Y.; Liu, Y.; Jing, X. Surface modification of bioactive glass nanoparticles and the mechanical and biological properties of poly(l-lactide) composites. Acta Biomater. 2008, 4, 1005–1015. [Google Scholar] [CrossRef]
- Liu, Q.; de Wijn, J.R.; van Blitterswijk, C.A. Composite biomaterials with chemical bonding between hydroxyapatite filler particles and PEG/PBT copolymer matrix. J. Biomed. Mater. Res. 1998, 40, 490–497. [Google Scholar] [CrossRef]
- Khan, R.A.; Khan, M.A.; Sultana, S.; Nuruzzaman, K.M.; Shubhra, Q.T.H.; Noor, F.G. Mechanical, degradation, and interfacial properties of synthetic degradable fiber reinforced polypropylene composites. J. Reinf. Plast. Compos. 2010, 29, 466–476. [Google Scholar]
- Khan, R.A.; Parsons, A.J.; Jones, I.A.; Walker, G.S.; Rudd, C.D. Effectiveness of 3-aminopropyl-triethoxy-silane as a coupling agent for phosphate glass fiber-reinforced poly(caprolactone)-based composites for fracture fixation devices. J. Thermoplast. Compos. Mater. 2011, 24, 517–534. [Google Scholar] [CrossRef]
- Jiang, G.; Evans, M.E.; Jones, I.A.; Rudd, C.D.; Scotchford, C.A.; Walker, G.S. Preparation of poly(ε-caprolactone)/continuous bioglass fibre composite using monomer transfer moulding for bone implant. Biomaterials 2005, 26, 2281–2288. [Google Scholar]
- Hasan, M.S. Investigation of Coupling Agents Mediated Interfacial Integrity Improvements for Phosphate Glass Fibre Reinforced Composite for Bone Repair Applications Research. PhD Diesseration, University of Nottingham, Nottingham, UK, 2012. [Google Scholar]
- Siparsky, G.L.; Voorhees, K.J.; Miao, F. Hydrolysis of polylactic acid (PLA) and polycaprolactone (PCL) in aqueous acetonitrile solutions: autocatalysis. J. Polym. Environ. 1998, 6, 31–41. [Google Scholar] [CrossRef]
- Sánchez-Vaquero, V. Characterization and cytocompatibility of hybrid aminosilane-agarose hydrogel scaffolds. Biointerphases 2010, 5, 23. [Google Scholar] [CrossRef]
- Dong, G-C.; Lin, F-H.; Sun, J-S.; Yao, C-H.; Jiang, G.; Huang, C-W. Biodegradability and cytocompatibility evaluation of surface modified calcium hydrogenphosphate. J. Med. Biol. Eng. 2001, 21, 249–256. [Google Scholar]
- Lian, J.B.; Stein, G.S. Concepts of osteoblast growth and differentiation: basis for modulation of bone cell development and tissue formation. Crit. Rev. Oral Biol. Med. 1992, 3, 269–305. [Google Scholar]
- Kim, H.-W.; Lee, H.-H.; Chun, G.-S. Bioactivity and osteoblast responses of novel biomedical nanocomposites of bioactive glass nanofiber filled poly(lactic acid). J. Biomed. Mater. Research A 2008, 85A, 651–663. [Google Scholar] [CrossRef]
- Navarro, M.; Engel, E.; Planell, J.A.; Amaral, I.; Barbosa, M.; Ginebra, M.P. Surface characterization and cell response of a PLA/CaP glass biodegradable composite material. J. Biomed. Mater. Res. A 2008, 85A, 477–486. [Google Scholar] [CrossRef]
- Ignatius, A.A.; Claes, L.E. In vitro biocompatibility of bioresorbable polymers: Poly(L,DL-lactide) and poly(L-lactide-co-glycolide). Biomaterials 1996, 17, 831–839. [Google Scholar] [CrossRef]
- Spenlehauer, G.; Vert, M.; Benoit, J.P.; Boddaert, A. In vitro and in vivo degradation of poly(D,L lactide/glycolide) type microspheres made by solvent evaporation method. Biomaterials 1989, 10, 557–563. [Google Scholar] [CrossRef]
- Daniels, A.U.; Chang, M.K.O.; Andriano, K.P.; Heller, J. Mechanical properties of biodegradable polymers and composites proposed for internal fixation of bone. J. Appl. Biomater. 1990, 1, 57–78. [Google Scholar]
- GraphPad Software, version 3.02, GraphPad Software Inc.: San Diego, CA, USA, 1992.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hasan, M.S.; Ahmed, I.; Parsons, A.; Walker, G.; Scotchford, C. Cytocompatibility and Mechanical Properties of Short Phosphate Glass Fibre Reinforced Polylactic Acid (PLA) Composites: Effect of Coupling Agent Mediated Interface. J. Funct. Biomater. 2012, 3, 706-725. https://doi.org/10.3390/jfb3040706
Hasan MS, Ahmed I, Parsons A, Walker G, Scotchford C. Cytocompatibility and Mechanical Properties of Short Phosphate Glass Fibre Reinforced Polylactic Acid (PLA) Composites: Effect of Coupling Agent Mediated Interface. Journal of Functional Biomaterials. 2012; 3(4):706-725. https://doi.org/10.3390/jfb3040706
Chicago/Turabian StyleHasan, Muhammad Sami, Ifty Ahmed, Andrew Parsons, Gavin Walker, and Colin Scotchford. 2012. "Cytocompatibility and Mechanical Properties of Short Phosphate Glass Fibre Reinforced Polylactic Acid (PLA) Composites: Effect of Coupling Agent Mediated Interface" Journal of Functional Biomaterials 3, no. 4: 706-725. https://doi.org/10.3390/jfb3040706