Shannon Entropy in Atoms: A Test for the Assessment of Density Functionals in Kohn-Sham Theory

Dipartimento di Chimica e Chimica Industriale, University of Pisa, Via Giuseppe Moruzzi 13, 56124 Pisa, Italy
*
Author to whom correspondence should be addressed.
Computation 2018, 6(2), 36; https://doi.org/10.3390/computation6020036
Submission received: 10 March 2018
/
Revised: 7 April 2018
/
Accepted: 22 April 2018
/
Published: 3 May 2018
(This article belongs to the Special Issue In Memory of Walter Kohn—Advances in Density Functional Theory)
Abstract
:Electron density is used to compute Shannon entropy. The deviation from the Hartree–Fock (HF) of this quantity has been observed to be related to correlation energy. Thus, Shannon entropy is here proposed as a valid quantity to assess the quality of an energy density functional developed within Kohn–Sham theory. To this purpose, results from eight different functionals, representative of Jacob’s ladder, are compared with accurate results obtained from diffusion quantum Monte Carlo (DMC) computations. For three series of atomic ions, our results show that the revTPSS and the PBE0 functionals are the best, whereas those based on local density approximation give the largest discrepancy from DMC Shannon entropy.
1. Introduction
In a recent work [1], we proposed an alternative and more practical way to relate an entropy measure to the correlation energy of atomic systems.
Correlation energy has been related to the entropy of entanglement [2] by Collins [3], and this conjecture has been assessed on a series of light atomic ions by Esquivel et al. [4]. Relations with the correlation strength have been also established by Ziesche [5], while many other works have been focused on the link between the nonidempotency of the one-particle density matrix and complexity measures (see, for example, Nagy and Romera [6] and other references therein). In a slightly different context, Romera and Dehesa [7] have introduced a rather sophisticated measure of the electron correlation based on the combination of Shannon and Fischer information. Instead, in the framework of density functional theory (DFT), more recently, Grimme and Hansen [8] have proposed a method of particular interest to recover information about static correlation from the entanglement entropy.
In our approach, we used densities, tested on atomic model systems, obtained from diffusion Monte Carlo (DMC) and Hartree–Fock (HF) calculations, and we observed that the difference between the two sets of entropy (correlation Shannon entropy) shows a significant positive deviation in approaching the conditions at which Coulomb correlation is stronger, namely at critical nuclear binding condition. This study seems to confirm the original idea of Amovilli and March [9] that there might be a relation between such difference and the entanglement entropy. Thus, we reached the conclusion that the correlation Shannon entropy should also be related to the correlation energy [1]. An interesting discussion about the connection of Shannon entropy and many electron correlation has been given recently by Delle Site [10] (see also Ghiringhelli et al. [11]). Trickey et al. [12] criticized this view because of the problems connected to the logarithmic density dependence of Shannon entropy. In their work, they suggest a renormalization of the Shannon term in order to limit this kind of problem. However, this could not be the case of correlation Shannon entropy, that is a difference and is not an absolute quantity.
In this paper, we propose Shannon entropy as a possible indicator to assess the quality of an energy density functional. Typically, such functionals are written in terms of a set of parameters that are calibrated on some large training set made of a variety of molecular properties. Although the fit of the training set is sufficiently good, one cannot be certain that such a functional works properly in all cases. In this regard, Wasserman et al. [13] analyzed errors on both energy and density in order to review the role of self-consistency in DFT, giving simple pedagogical examples. The variational validity of density functionals has been investigated in a previous work by one of us [14]. In that paper, the authors classified some largely used functionals into heuristic and possibly variationally valid on the basis of a comparison with accurate ab initio calculations.
We remark that Shannon entropy depends explicitly on electron density and can be compared directly with the corresponding accurate DMC value. Thus, the purpose of this work is to compare electron density from various functionals with the one obtained from a DMC calculation through the computation of Shannon entropy.
2. Calculations and Discussion of Results
Let us begin with an overview of the calculations. We computed the electron density for three series of atomic ions in the ground state, namely He-like, N-like, and Ne-like ions. In order to have a fairly general view of the results, we have chosen at least a representative functional for any rung of the so-called Jacob’s ladder [15]. A standard Kohn–Sham (KS) DFT calculation was performed on each system by using the GAMESS-US package [16]. A universal basis set of even tempered (16s,16p,3d,2f) Gaussian atomic orbitals was used, with 0.007 458.752, 0.7 2.8 and 1 2. For N-like ions, we used an unrestricted approach. For N- and Ne-like series, we considered only valence electrons and we used Burkatzki, Filippi, and Dolg (BFD) pseudopotentials [17]. Quantum Monte Carlo (QMC) computations were performed by means of the program CHAMP [18]. In this case, the atomic wavefunction is optimized at the variational Monte Carlo (VMC) level in the form of a one-determinant Slater–Jastrow wavefunction [19,20] and subsequently used at the DMC level. In DMC, we used a time step of 0.025 a.u. and the method of Casula [21] to go beyond the locality approximation. At DMC level, the electron density was recovered from the fractional number of electrons contained in a sphere of arbitrary radius centered on the nucleus. This quantity can be easily obtained from the configurations generated from fixed node DMC mixed distribution during the simulation thanks to the spherical symmetry of the test cases studied in this work. Electron densities are finally utilized to compute the Shannon entropy according to the definition
as performed in our recent work [1].
Table 1, Table 2 and Table 3 record our resulting entropies. In these tables, we show also total energy and kinetic energy for comparison between the various approaches. For KS-DFT, the kinetic energy is the single particle kinetic energy that deviates significantly from correlated kinetic energy computed at the QMC level. In all three series of data, the most interesting cases are those of the anions H, C, and F, being closer to the limit of weak binding. We remind the reader that the critical nuclear charges are, respectively, 0.911028 [22], 5.85 [23], and 8.74 [23] for the three series of atomic ions. In all cases, the DMC value of entropy is larger than the HF one due to the effect of electronic Coulomb correlation on this property. The highest difference occurs for the three anions considered in this work. In this regard, a good energy density functional should be in agreement with the DMC corresponding value. In order to verify if this is a valid criterion to assess the quality of a given functional, we have tested eight functionals representative of the Jacob’s ladder.
In Figure 1, we show the deviation from the DMC reference of Shannon entropy computed for the three anions and with different approaches. The HF and the functional based on local density approximation (SVWN) show the highest discrepancy from the DMC results. All the other functionals considered in this work fall in between these two sets of results. While the deviation for HF is negative because of Coulomb correlation, an approximate functional could lead to both positive or negative deviations. For the three anions, the best functionals appear to be the revTPSS and the PBE0, the first one belonging to the meta-GGA category and the second to the hybrid-GGA one. Moreover, it is interesting to note that the quantity plotted in Figure 1 is strictly connected to the Kullback–Leibler divergence [24] of the various calculated densities with respect to the DMC one.
In Table 4, we report the mean absolute relative errors (MARE) computed by considering all the entropy data of this work for each method with respect to the DMC set of results. In this Table, HF and VMC values are also reported in order to fix two reference values, namely, non-correlated and correlated ab initio calculations. Again, revTPSS and PBE0 appear to be the best ones. None of the eight functionals achieve the quality of VMC, whereas SVWN and PW91 lead to worse results than HF.
3. Conclusions
In this work, we have presented results of Shannon entropy for a list of selected atomic systems by means of electron density obtained from various functionals in KS theory and from QMC. The purpose of this test study is to verify the sensitivity of Shannon entropy with respect to the variation of density functionals in order to use this property in the design of new approximate energy density functionals. The main results of the present work are displayed in Figure 1 and Table 4. The density functionals considered in this study show clearly a different behavior in the comparison with DMC. None of them reach the accuracy of, for example, VMC, namely a good ab initio case. SVWN and PW91 present an MARE greater than HF and the best functionals of this study are PBE0 and revTPSS. PBE0 energies lie above the DMC energies and can also be classified, following Amovilli et al. [14], as possibly variationally valid, while revTPSS is more heuristic, the energies in this case being lower than those from DMC.
Although the number of illustrative examples considered in this work is limited, this suggests that different functionals give different Shannon entropies and that such differences are not negligible, being a significant fraction of the difference between HF and DMC Shannon entropy. We remark that the correlation Shannon entropy is strongly related to Coulomb correlation [1]. DMC is a good reference for testing Shannon entropy computed at the KS-DFT level. The KS electron density is easily achievable from standard packages of quantum chemistry calculations, while DMC electron density needs more attention if one requires a very accurate density function. For the future, if the electron density cannot be obtained from any convenient method, like, for example, a full CI calculation, it is very important to improve the techniques for the reconstruction of highly accurate electron densities for molecular systems that can be studied at a DMC level. We believe that the approach presented in this note could be a valid tool for the development of new energy density functionals.
Author Contributions
Claudio Amovilli conceived and supervised the work and wrote the paper. Franca Maria Floris performed calculations and contributed to the analysis of data and to the preparation of the manuscript.
Funding
This research was funded by Università di Pisa grant number FA 2016.
Acknowledgments
We thank Claudia Filippi for providing the version of CHAMP code under development at the University of Twente (NL).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| SWVN | Slater, Vosko, Wilk, and Nusair functional [25] |
| PW91 | Perdew and Wang 1991 functional [26] |
| PBE0 | Perdew–Burke–Ernzerhof with 0.25 Hartree–Fock exchange functional [27,28] |
| B3LYP | Becke, three-parameter, Lee–Yang–Parr hybrid functional [29,30] |
| revTPSS | revised Tao, Perdew, Staroverov, and Scuseria meta-GGA functional [31,32] |
| CAMB3LYP | Cambridge B3LYP functional [33] |
| B97X | long-range corrected Becke 97 hybrid functional [34] |
| B2PLYP | double hybrid BLYP functional [35] |
References and Note
- Amovilli, C.; Floris, F.M. Shannon entropy and correlation energy for electrons in atoms. In Many-Body Approaches at Different Scales; Angilella, G.G.N., Amovilli, C., Eds.; Springer International Publishing: Heidelberg, Germany, 2018; pp. 187–198. ISBN 978-3-319-72373-0. [Google Scholar]
- Janzing, D. Entropy of Entanglement. In Compendium of Quantum Physics; Greenberger, D., Hentschel, K., Weinert, F., Eds.; Springer International Publishing: Heidelberg, Germany, 2009; pp. 205–209. ISBN 978-3-540-70622-9. [Google Scholar]
- Collins, D.M. Entropy Maximizations on Electron-Density. Z. Naturforsc. A 1993, 48, 68–74. [Google Scholar] [CrossRef]
- Esquivel, R.O.; Rodriguez, A.L.; Sagar, R.P.; Ho, M.; Smith, V.H., Jr. Physical interpretation of information entropy: Numerical evidence of the Collins conjecture. Phys. Rev. A 1996, 54, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Ziesche, P. Correlation strength and information entropy. Int. J. Quantum Chem. 1995, 56, 363–369. [Google Scholar] [CrossRef]
- Nagy, A.; Romera, E. Link between generalized nonidempotency and complexity measures. J. Mol. Model. 2017, 23, 159. [Google Scholar] [CrossRef] [PubMed]
- Romera, E.; Dehesa, J.S. The Fisher-Shannon information plane, an electron correlation tool. J. Chem. Phys. 2004, 120, 8906–8912. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A. Practicable Real-Space Measure and Visualization of Static Electron-Correlation Effects. Angew. Chem. Int. Ed. 2015, 54, 12308–12313. [Google Scholar] [CrossRef] [PubMed]
- Amovilli, C.; March, N.H. Use of ab initio methods to classify four existing energy density functionals according to their possible variational validity. Phys. Rev. A 2004, 69, 054302. [Google Scholar] [CrossRef]
- Delle Site, L. Shannon entropy and many-electron correlations: Theoretical concepts, numerical results, and Collins conjecture. Int. J. Quantum Chem. 2015, 115, 1396–1404. [Google Scholar] [CrossRef]
- Ghiringhelli, L.M.; Hamilton, I.P.; Delle Site, L. Interacting electrons, spin statistics, and information theory. J. Chem. Phys. 2010, 132, 014106. [Google Scholar] [CrossRef] [PubMed]
- Trickey, S.B.; Karasiev, V.V.; Vela, A. Positivity constraints and information-theoretical kinetic energy functionals. Phys. Rev. B 2011, 84, 075146. [Google Scholar] [CrossRef]
- Wasserman, A.; Nafziger, J.; Jiang, K.L.; Kim, M.C.; Sim, E.; Burke, K. The Importance of Being Inconsistent. Ann. Rev. Phys. Chem. 2017, 68, 555–581. [Google Scholar] [CrossRef] [PubMed]
- Amovilli, C.; March, N.H.; Bogar, F.; Gál, T. Use of ab initio methods to classify four existing energy density functionals according to their possible variational validity. Phys. Lett. A 2009, 373, 3158–3160. [Google Scholar] [CrossRef]
- Perdew, J.P.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, N.K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Burkatzki, M.; Filippi, C.; Dolg, M. Energy-consistent pseudopotentials for quantum Monte Carlo calculations. J. Chem. Phys. 2007, 126, 234105. [Google Scholar] [CrossRef] [PubMed]
- The Cornell-Holland Ab-initio Materials Package (CHAMP) is a quantum Monte Carlo suite of programs for electronic structure calculations of atomic and molecular systems. See, for more information: http://www.utwente.nl/tnw/ccp/research/CHAMP.html (28 April 2018).
- Filippi, C.; Umrigar, C.J. Multiconfiguration wave functions for quantum Monte Carlo calculations of first row diatomic molecules. J. Chem. Phys. 1996, 105, 213–226. [Google Scholar] [CrossRef]
- Umrigar, C.J.; Toulouse, J.; Filippi, C.; Sorella, S.; Hennig, R.G. Alleviation of the Fermion-Sign Problem by Optimization of Many-Body Wave Functions. Phys. Rev. Lett. 2007, 98, 110201. [Google Scholar] [CrossRef] [PubMed]
- Casula, M. Beyond the locality approximation in the standard diffusion Monte Carlo method. Phys. Rev. B 2006, 74, 161102. [Google Scholar] [CrossRef]
- Drake, G.W.F.; Swainson, R.A. Bethe logarithms for hydrogen up to n = 20, and approximations for two-electron atoms. Phys. Rev. A 1990, 41, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Hogreve, H. On the maximal electronic charge bound by atomic nuclei. J. Phys. B At. Mol. Phys. 1998, 31, L439–L446. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Burke, K.; Perdew, J.P.; Wang, Y. Derivation of a Generalized Gradient Approximation: The PW91 Density Functional. In Electronic Density Functional Theory; Dobson, J.F., Vignale, G., Das, M.P., Eds.; Springer: Boston, MA, USA, 1988; pp. 81–111. ISBN 978-1-4899-0316-7. [Google Scholar]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. TPSS: Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Marsman, M.; Csonka, G.; Ruzsinszky, A.; Hao, P.; Kim, Y.-S.; Kresse, G.; Perdew, J.P. Self-consistent meta-generalized gradient approximation within the projector-augmented-wave method. Phys. Rev. B 2011, 84, 035117. [Google Scholar] [CrossRef]
- Yanaia, T.; Handy, D.N. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Da Chaia, J.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, T.; Grimme, S. Double-hybrid density functionals with long-range dispersion corrections: Higher accuracy and extended applicability. Phys. Chem. Chem. Phys. 2007, 9, 3397–3406. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Deviation from DMC of Shannon entropy computed for the three anions, H, C, and F, using eight functionals representative of the Jacob’s ladder and HF.
Figure 1.
Deviation from DMC of Shannon entropy computed for the three anions, H, C, and F, using eight functionals representative of the Jacob’s ladder and HF.
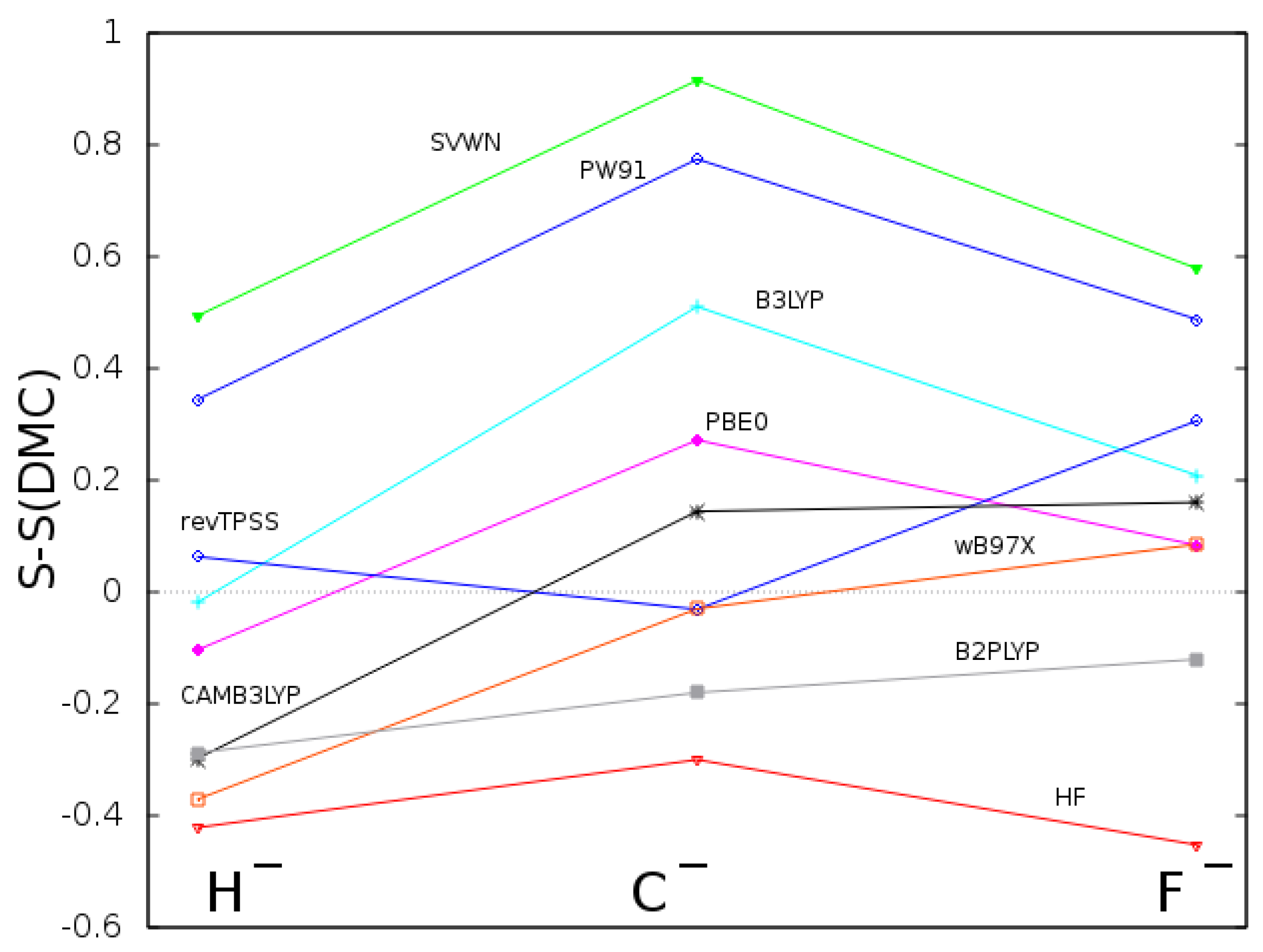

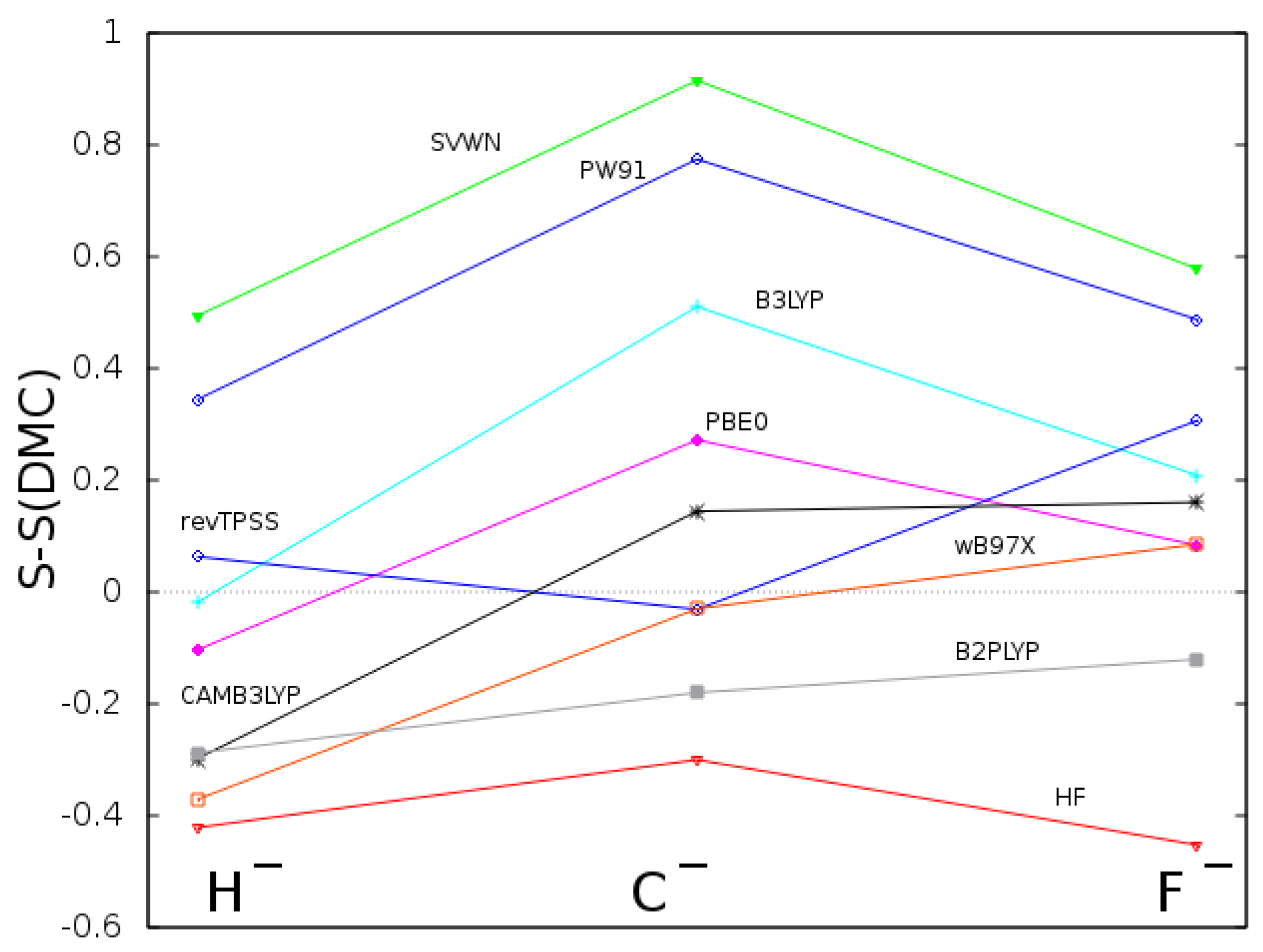{kind=link}
Table 1.
Total energy, kinetic energy, and Shannon entropy for some atomic systems of the He-like series calculated at different level of computation. Data are in atomic units (a.u.).
Table 1.
Total energy, kinetic energy, and Shannon entropy for some atomic systems of the He-like series calculated at different level of computation. Data are in atomic units (a.u.).
| Method | Total Energy | Kinetic Energy | Entropy | Total Energy | Kinetic Energy | Entropy | Total Energy | Kinetic Energy | Entropy |
|---|---|---|---|---|---|---|---|---|---|
| H− | He | Li+ | |||||||
| RHF | −0.48793 | 0.48793 | 9.8889 | −2.86163 | 2.86148 | 4.0100 | −7.23599 | 7.23474 | 1.1203 |
| SVWN | −0.51425 | 0.48054 | 10.8040 | −2.83479 | 2.76771 | 4.2066 | −7.14240 | 7.05513 | 1.2467 |
| PW91 | −0.53052 | 0.50957 | 10.6541 | −2.89995 | 2.86266 | 4.1155 | −7.26720 | 7.22341 | 1.1834 |
| PBE0 | −0.52450 | 0.50023 | 10.2064 | −2.89513 | 2.85773 | 4.0816 | −7.26205 | 7.21907 | 1.1659 |
| B3LYP | −0.53065 | 0.51090 | 10.2912 | −2.90806 | 2.86808 | 4.0927 | −7.27821 | 7.22723 | 1.1733 |
| revTPSS | −0.53325 | 0.50611 | 10.3734 | −2.91200 | 2.87003 | 4.0424 | −7.28824 | 7.24107 | 1.1396 |
| CAMB3LYP | −0.53100 | 0.50802 | 10.0120 | −2.90138 | 2.85372 | 4.1173 | −7.26518 | 7.21801 | 1.1816 |
| B97X | −0.52898 | 0.50860 | 9.9390 | −2.90437 | 2.87235 | 4.0709 | −7.28655 | 7.27475 | 1.1302 |
| B2PLYP | −0.52488 | 0.50280 | 10.0224 | −2.90463 | 2.86924 | 4.0547 | −7.26813 | 7.23682 | 1.1480 |
| VMC | −0.52755(4) | 0.5248(6) | 10.3775 | −2.90359(7) | 2.912(4) | 4.0106 | −7.27991(4) | 7.292(6) | 1.1204 |
| DMC | −0.52779(2) | 0.5237(9) | 10.3119 | −2.90386(7) | 2.908(3) | 4.0256 | −7.27997(4) | 7.298(6) | 1.1143 |
Table 2.
Total energy, kinetic energy and Shannon entropy for some atomic systems of the N-like series calculated at different level of computation. Data are in a.u.
Table 2.
Total energy, kinetic energy and Shannon entropy for some atomic systems of the N-like series calculated at different level of computation. Data are in a.u.
| Method | Total Energy | Kinetic Energy | Entropy | Total Energy | Kinetic Energy | Entropy | Total Energy | Kinetic Energy | Entropy |
|---|---|---|---|---|---|---|---|---|---|
| C− | N | O+ | |||||||
| UHF | −5.35111 | 3.59151 | 16.5669 | −9.67304 | 6.80209 | 11.2305 | −15.27536 | 10.78233 | 7.5464 |
| USVWN | −5.44029 | 3.52699 | 17.7825 | −9.72520 | 6.75214 | 11.5299 | −15.29526 | 10.73464 | 7.7365 |
| UPW91 | −5.48765 | 3.58086 | 17.6413 | −9.79981 | 6.83133 | 11.4282 | −15.39928 | 10.84028 | 7.6524 |
| UPBE0 | −5.47450 | 3.58184 | 17.1388 | −9.79225 | 6.81302 | 11.3493 | −15.39264 | 10.81020 | 7.6112 |
| UB3LYP | −5.46483 | 3.59524 | 17.3775 | −9.78493 | 6.83758 | 11.3852 | −15.38900 | 10.83889 | 7.6330 |
| UrevTPSS | −5.49420 | 3.61922 | 16.8359 | −9.81463 | 6.82378 | 11.3659 | −15.41653 | 10.82372 | 7.6172 |
| UCAMB3LYP | −5.46771 | 3.62211 | 17.0112 | −9.78632 | 6.83303 | 11.4052 | −15.38512 | 10.82775 | 7.6555 |
| UB97X | −5.46535 | 3.66699 | 16.8368 | −9.78760 | 6.86984 | 11.3268 | −15.39130 | 10.87310 | 7.5819 |
| UB2PLYP | −5.43696 | 3.61515 | 16.6868 | −9.74356 | 6.79620 | 11.3129 | −15.38589 | 10.81562 | 7.5928 |
| VMC | −5.46486(4) | 3.6547(5) | 16.8599 | −9.78662(5) | 6.8672(9) | 11.2796 | −15.39153(8) | 10.855(2) | 7.5655 |
| DMC | −5.47112(2) | 3.6689(3) | 16.8686 | −9.79314(2) | 6.9054(5) | 11.2699 | −15.39865(2) | 10.9094(6) | 7.5608 |
Table 3.
Total energy, kinetic energy, and Shannon entropy for some atomic systems of the Ne-like series calculated at different level of computation. Data are in a.u.
Table 3.
Total energy, kinetic energy, and Shannon entropy for some atomic systems of the Ne-like series calculated at different level of computation. Data are in a.u.
| Method | Total Energy | Kinetic Energy | Entropy | Total Energy | Kinetic Energy | Entropy |
|---|---|---|---|---|---|---|
| F− | Ne | |||||
| RHF | −23.98621 | 18.82316 | 10.994 | −34.70905 | 27.73954 | 5.272 |
| SVWN | −24.21194 | 19.00417 | 12.025 | −34.87089 | 27.72792 | 5.722 |
| PW91 | −24.35071 | 18.97835 | 11.934 | −35.05853 | 27.97595 | 5.601 |
| PBE0 | −24.30801 | 18.93859 | 11.530 | −35.02224 | 27.90528 | 5.481 |
| B3LYP | −24.32192 | 18.96881 | 11.655 | −35.03774 | 27.94395 | 5.545 |
| revTPSS | −24.34359 | 18.95492 | 11.753 | −35.05088 | 27.93728 | 5.553 |
| CAMB3LYP | −24.32105 | 18.97777 | 11.607 | −35.03248 | 27.93379 | 5.571 |
| B97X | −24.32782 | 19.00417 | 11.532 | −35.04599 | 27.98958 | 5.475 |
| B2PLYP | −24.20896 | 18.92158 | 11.326 | −34.93208 | 27.87075 | 5.422 |
| VMC | −24.3011(1) | 19.075(3) | 11.4523 | −35.01487(8) | 27.998(3) | 5.441 |
| DMC | −24.31239(3) | 19.201(1) | 11.4481 | −35.02504(3) | 28.166(1) | 5.440 |
Table 4.
Mean absolute relative error (MARE) on Shannon entropy computed at different levels with respect to DMC.
Table 4.
Mean absolute relative error (MARE) on Shannon entropy computed at different levels with respect to DMC.
| Method | MARE |
|---|---|
| RHF | 0.0329 |
| SVWN | 0.0508 |
| PW91 | 0.0405 |
| PBE0 | 0.0111 |
| B3LYP | 0.0167 |
| revTPSS | 0.0115 |
| CAMB3LYP | 0.0171 |
| B97X | 0.0151 |
| B2PLYP | 0.0165 |
| VMC | 0.0024 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Amovilli, C.; Floris, F.M. Shannon Entropy in Atoms: A Test for the Assessment of Density Functionals in Kohn-Sham Theory. Computation 2018, 6, 36. https://doi.org/10.3390/computation6020036
AMA Style
Amovilli C, Floris FM. Shannon Entropy in Atoms: A Test for the Assessment of Density Functionals in Kohn-Sham Theory. Computation. 2018; 6(2):36. https://doi.org/10.3390/computation6020036
Chicago/Turabian StyleAmovilli, Claudio, and Franca Maria Floris. 2018. "Shannon Entropy in Atoms: A Test for the Assessment of Density Functionals in Kohn-Sham Theory" Computation 6, no. 2: 36. https://doi.org/10.3390/computation6020036
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.