
The Influence of One-Electron Self-Interaction on d-Electrons

Theoretical Physics IV, University of Bayreuth, 95440 Bayreuth, Germany
*
Author to whom correspondence should be addressed.
Computation 2016, 4(3), 33; https://doi.org/10.3390/computation4030033
Submission received: 31 May 2016
/
Revised: 25 August 2016
/
Accepted: 31 August 2016
/
Published: 6 September 2016
(This article belongs to the Special Issue 50th Anniversary of the Kohn-Sham Theory—Advances in Density Functional Theory)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:We investigate four diatomic molecules containing transition metals using two variants of hybrid functionals. We compare global hybrid functionals that only partially counteract self-interaction to local hybrid functionals that are designed to be formally free from one-electron self-interaction. As d-orbitals are prone to be particularly strongly influenced by self-interaction errors, one may have expected that self-interaction-free local hybrid functionals lead to a qualitatively different Kohn–Sham density of states than global hybrid functionals. Yet, we find that both types of hybrids lead to a very similar density of states. For both global and local hybrids alike, the intrinsic amount of exact exchange plays the dominant role in counteracting electronic self-interaction, whereas being formally free from one-electron self-interaction seems to be of lesser importance.
1. Introduction
Ground-state Kohn–Sham density-functional theory (DFT) [1,2,3] grants access to the electronic structure of condensed matter in an efficient manner. While DFT is exact in principle, practical calculations require an approximation for the exchange-correlation energy as a functional of the electron ground-state density . In the last decades, numerous such approximations were developed [4], leading to a high accuracy of DFT methods for many physical applications, thus contributing to make the story of DFT one of success.
Yet, certain systems pose severe difficulties for a satisfying description with DFT due to their chemical composition. For instance, the characterization of systems containing transition metals makes high demands on the density-functional approximation that is put to task [5]. The problems of describing systems with localized d- [6,7] or f-electrons [8] are often attributed to electronic self-interaction (SI) [9,10]: Most practical density functionals, and in particular the semilocal ones, include an erroneous interaction of an electron with itself. This leads to a significant decrease in accuracy for the description of a wide range of physical processes and observables (e.g., References [11,12] and references therein). The self-interaction in particular affects systems in which some orbitals are localized and others are not [13,14], which typically is the case for systems containing d-electrons [6].
Curing the SI problem is highly non-trivial for at least two conceptual reasons. The first is that (semi)local exchange functionals—which are responsible for the largest part of the self-interaction error—implicitly model part of what in wavefunction theory is called static correlation [11,15,16]. Straightforwardly eliminating SI can therefore have detrimental effects [17]. The second is that it is straightforward to identify SI in a one-electron system—the sum of the Hartree- and the exchange-correlation energy must vanish, and if it does not, then SI is present—yet, it is much less clear what the corresponding condition is for a many-electron system.
The most often used definition [9,10] of electronic SI is based on the concept of identifying electrons with spin-orbital densities , where i counts the single-particle states and σ the electron spin. In this definition, a specific density-functional approximation is regarded as free from SI if the condition
is fulfilled [9,10]. Here, denotes the Hartree energy evaluated with the single spin-orbital density .
Equation (1) has been introduced by Perdew and Zunger [9,10] for the definition of a SI correction (SIC). The basic idea of the Perdew–Zunger SIC is subtracting the SI error as quantified by Equation (1) from an explicit expression for the xc energy [10]. However, SIC schemes can also be defined in several other ways [12,18,19,20,21,22,23,24,25,26,27,28]. There is such a wide variety of SIC schemes because not only can the expression for be defined in other ways than done by Perdew and Zunger, but also within the Perdew–Zunger definition, different variants of SIC can be defined. This is due to the fact that Equation (1) explicitly uses the single particle orbitals and is not unitarily invariant. Thus, depending on how the mapping between the density and the orbitals is defined—i.e., depending on how the potential from which the orbitals are calculated is constructed—one and the same orbital-dependent expression for can correspond to different density functionals for [11]. The Perdew–Zunger SIC energy, can, for example, be used with orbital-specific potentials [10,29,30,31,32] or as a Kohn–Sham functional using the optimized effective potential (OEP) [33,34] and approximations to it [35,36,37,38,39,40,41,42]. Additionally, with respect to how to cope with the unitary variance of Equation (1), different concepts have been developed [29,41,43,44,45,46,47,48,49]. Recently, the use of complex-valued orbitals [48,49,50,51,52,53,54,55] and the use of the Fermi orbital [56,57,58] have been introduced as new concepts for the definition of improved SIC schemes.
As an alternative way of thinking about SI, it has been advocated to focus less on one-electron conditions as in Equation (1) and to instead think about SI directly for many-electron systems. Consequently, as a conceptually different approach, “many-electron SI” has been introduced [59,60] based on the straight-line criterion [61] of the total energy as a function of fractional electron numbers. Enforcing the straight-line condition indeed leads to an improved description of the electronic structure [62,63,64,65,66,67,68]. However, the condition cannot easily be formulated in “closed form” for use in density-functional construction. Therefore, Equation (1) continues to be of considerable importance in density-functional construction (we note that this is so, even though range-separated functionals [69,70,71,72,73], and in particular tuned ones [64,74], can yield eigenvalues that closely correspond to ionization potentials without making use of Equation (1). However, tuning comes at the price of violating size-consistency [75]).
Equation (1) is relevant not only for SIC schemes, but also for other approaches that aim at reducing SI (e.g., the use of exact (Fock) exchange). Whereas global hybrids [76,77,78] that use only a fraction of Fock exchange only partially eliminate one-electron SI, local hybrid functionals [79,80,81,82,83,84,85,86,87] can be constructed to obey Equation (1). Therefore, when one-electron SI is—as is usually done—defined via Equation (1), then local hybrids can be fully one-electron SI free. In local hybrids, nonlocal and semilocal functional components are mixed together in a spatially resolved and density-dependent fashion. Detection functions are typically used to reveal spatial regions where the density is dominated by single spin-orbital densities. This information is then used in the functional design in order to reduce or eliminate SI. As a consequence, local hybrids offer the advantage of being nominally free from electronic SI in the sense of Equation (1), while providing an xc energy expression that is invariant under unitary orbital transformations [88].
Yet, it was recently demonstrated [67] that this formal attribute of local hybrids does not necessarily result in a more accurate description of observables that are expected to be substantially influenced by electronic SI. Reference [67] focuses on the description of photoemission spectra of organic molecules by interpreting the set of occupied Kohn–Sham eigenvalues as a physical density of states (DOS) [13,14,89,90,91]. Somewhat surprisingly, it became clear that global hybrid functionals [76,77,78] and local hybrids in this respect lead to rather similar results, despite the fact that local hybrids are formally SI free and global hybrids are not.
The Kohn–Sham DOS can be used in order to characterize the structural, catalytic, and magnetic properties of solid state systems. In recent years, it has particularly often been relied on in studies of clusters [92,93,94,95,96,97,98,99,100,101,102,103,104]. Many of the potentially technologically relevant clusters contain elements with d-electrons. The influence of SI on the DOS is therefore of particular interest [5] with respect to the reliable representation of d-electrons. As the investigations in Reference [67] comprised only organic molecules and no systems with d-electrons, we here close this gap and study the Kohn–Sham DOS that results from local hybrids for molecules containing transition metals.
In order to allow for a calculation of the DOS within the Kohn–Sham scheme of DFT, we evaluate local hybrids with the OEP formalism (see References [11,105,106] and references therein). Furthermore, to refrain from uncertainties arising from, for example, basis sets and pseudopotentials [67], we present results obtained on accurate numerical grids using the all-electron program package DARSEC [86,107,108]. As a consequence, we are restricted to computations of molecules with two atomic centers. We find that for the transition metal-containing molecules investigated in this work, local and global hybrids exhibit great similarities for the description of the DOS. In particular, the investigated local hybrid functionals do not show a greater sensitivity to localized d-states despite the formal fulfillment of Equation (1). Thus, the main conclusion of Reference [67] is further confirmed in this work. While transition metal atoms in diatomic molecules differ from atoms in the solid-state limit of typical d-electron metals, we see the present study as an indicator for what one can qualitatively expect from local hybrids for d-electron metals.
This paper is structured as follows: In Section 2, we briefly discuss the differences in the construction of global in contrast to local hybrid functionals, with a special focus on the treatment of SI. Section 3 provides details on the computational aspects of this work, while we show and discuss our results in Section 4. Finally, in Section 5, we summarize this work with concluding remarks.
2. Counteracting Electronic Self-Interaction with Hybrid Functionals
The exact-exchange (EXX) integral, which is defined by
provides an auspicious component for the construction of functional approximations, since EXX itself fulfills the requirement of Equation (1). In general, the xc energy can be expressed (ambiguously [85,109,110]) as , with denoting the xc energy density per particle. Here, we highlight two related, yet fundamentally different concepts of incorporating the EXX energy density per particle into practical density-functional approximations.
Global hybrid functionals combine constant amounts of EXX with the corresponding parts of semilocal exchange and correlation. In this work, we focus on the PBEh hybrid functional [111,112], which employs an xc energy density per particle defined by
Here, denotes the constant fraction of EXX, and represents the PBE exchange and correlation energy densities per particle [113,114]. It becomes evident from Equation (3) that the PBEh hybrid functional does not generally obey Equation (1), since PBE exchange and correlation are not free from SI and EXX is only partially included. Instead, one expects that SI is only effectively counteracted for large values of a (i.e., for large amounts of EXX included). This assumption is systematically confirmed by the results of Reference [63,67,115,116,117].
Local hybrid functionals, in contrast, substitute the constant hybrid parameter a by space- and density-dependent functions, resulting in a more flexible mixing of nonlocal EXX with semilocal functional components. In this work, we focus on the local hybrid functional presented in Reference [86], which is termed “ISO” functional in the following. It approximates the xc energy density by
Here, and denote the energy density of the local spin-density approximation (LSDA) [118,119,120] for exchange and correlation, respectively. The mixing functions are defined by
and
The quantity gives the von Weizsäcker and the Kohn–Sham kinetic energy density. The function denotes the reduced density gradient [113]
with the Bohr radius , , and the spin polarization . The mixing function includes an a priori undetermined parameter c in the denominator, which regulates the intrinsic amount of EXX that is involved in the local hybrid. Increasing values of c reduce the function , resulting in an increasing intrinsic amount of EXX. For more details on the construction and performance of the ISO local hybrid functional, see References [67,86,87,121].
Additionally, we investigate a modified form of the ISO local hybrid. Termed “ISOII”, it is also based on Equation (4), but uses mixing functions defined by
and
with . These mixing functions follow directly from Equations (5) and (6) by setting . Additionally, here an a priori undetermined parameter appears. It regulates the amount of EXX in analogy to the ISO functional.
Importantly, both ISO and ISOII obey Equation (1) due to their construction: When the density consists of only a single spin orbital one finds [88,122]
Consequently, the mixing functions and vanish regardless of the value of the respective functional parameter. Since in the case of a single spin orbital the spin polarization reduces to , and also vanish systematically. Thus, both ISO and ISOII can be regarded as free from SI in the sense of Equation (1) (see Reference [67] for details).
In the following, we investigate how this formal property of the functionals ISO and ISOII affects their resulting Kohn–Sham DOS for systems with d-electrons. For this, we explicitly calculate four diatomic molecules containing transition metals with the local hybrids for various values of their respective functional parameter and evaluate the resulting spectrum of occupied Kohn–Sham eigenvalues. We contrast the results of the local hybrids to the outcome of calculations using purely semilocal functionals, the PBEh global hybrid functional, and to pure EXX, which itself is free from electronic SI.
3. Computational Details
All results throughout this work were obtained with the program package DARSEC using a highly accurate real-space grid. In DARSEC, the Kohn–Sham equations are solved self-consistently with an explicit consideration of all electrons. A local, multiplicative potential for orbital-dependent functionals (i.e., EXX and the global and local hybrid functionals) was obtained by using the KLI approximation [123] to the OEP. All calculations were performed in a non-relativistic way.
We here investigate the four diatomic molecules ZnO, Cu2, CuCl, and Pd2. The former three molecules were evaluated based on their experimental ground-state bond lengths , , and (see Reference [124]). The bond length of Pd2 was determined as based on a geometry optimization using the LSDA, since no experimental value is available.
All molecules were evaluated in the state with no unpaired electrons, and consequently, a spin polarization of . In this context, a fundamental difference between ISO and ISOII becomes evident: for such systems, the detection function effectively only contributes to the mixing functions in the case of the ISOII functional, since for ISO it is multiplied by zero. Yet, the argument that formally both local hybrids are free from SI still holds (see Reference [67] for more detailed arguments).
When plotting the Kohn–Sham DOS, we broaden the Kohn–Sham eigenvalue spectrum by convolution with Gaussians using a standard deviation of 0.08 eV. Furthermore, in order to allow for a better comparison of the different spectra, the eigenvalue spectra were aligned to match , where denotes the highest occupied (ho) Kohn–Sham eigenvalue. We present and discuss the Kohn–Sham DOS of the four diatomic molecules obtained with PBEh, ISO, ISOII, and EXX in the next section.
4. Results and Discussion
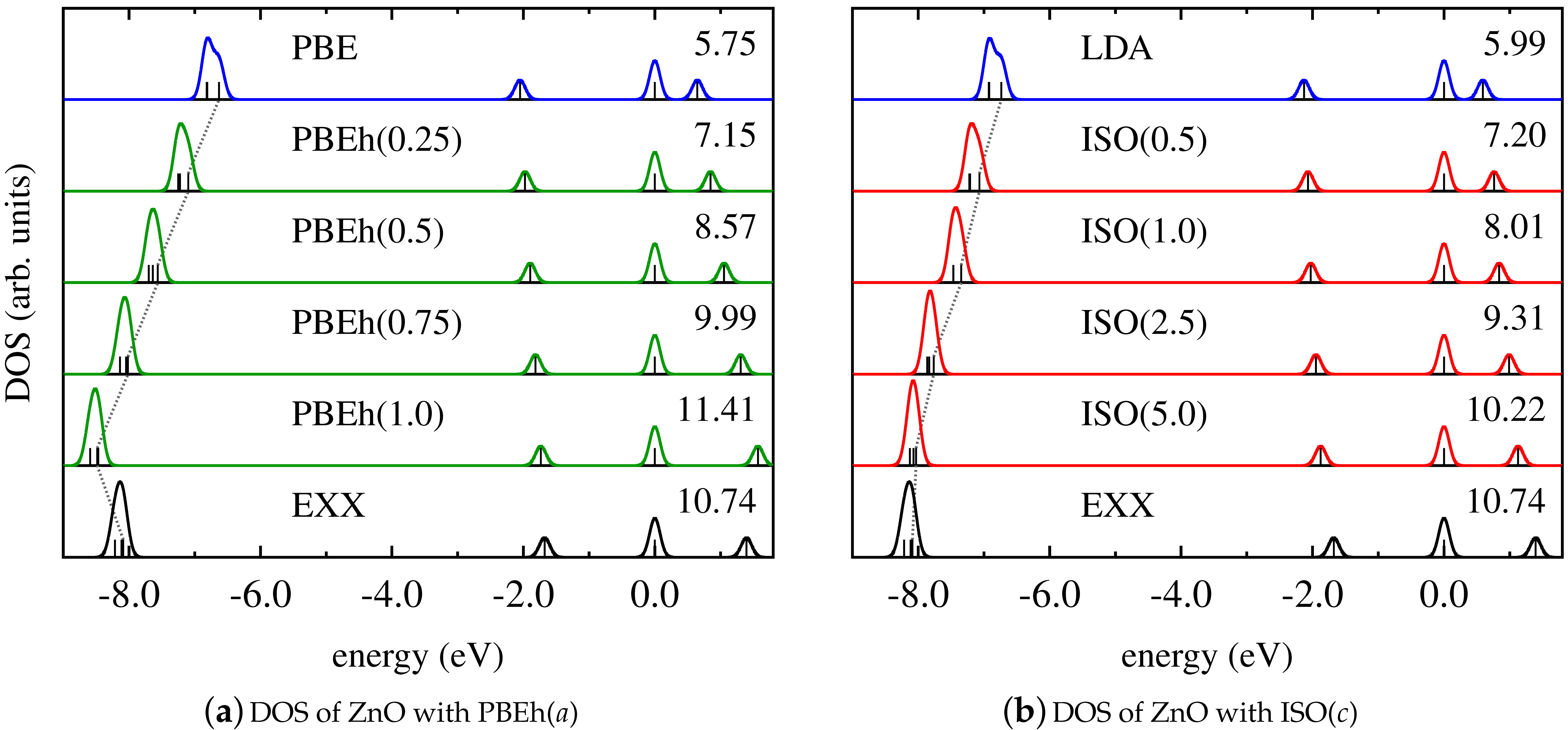
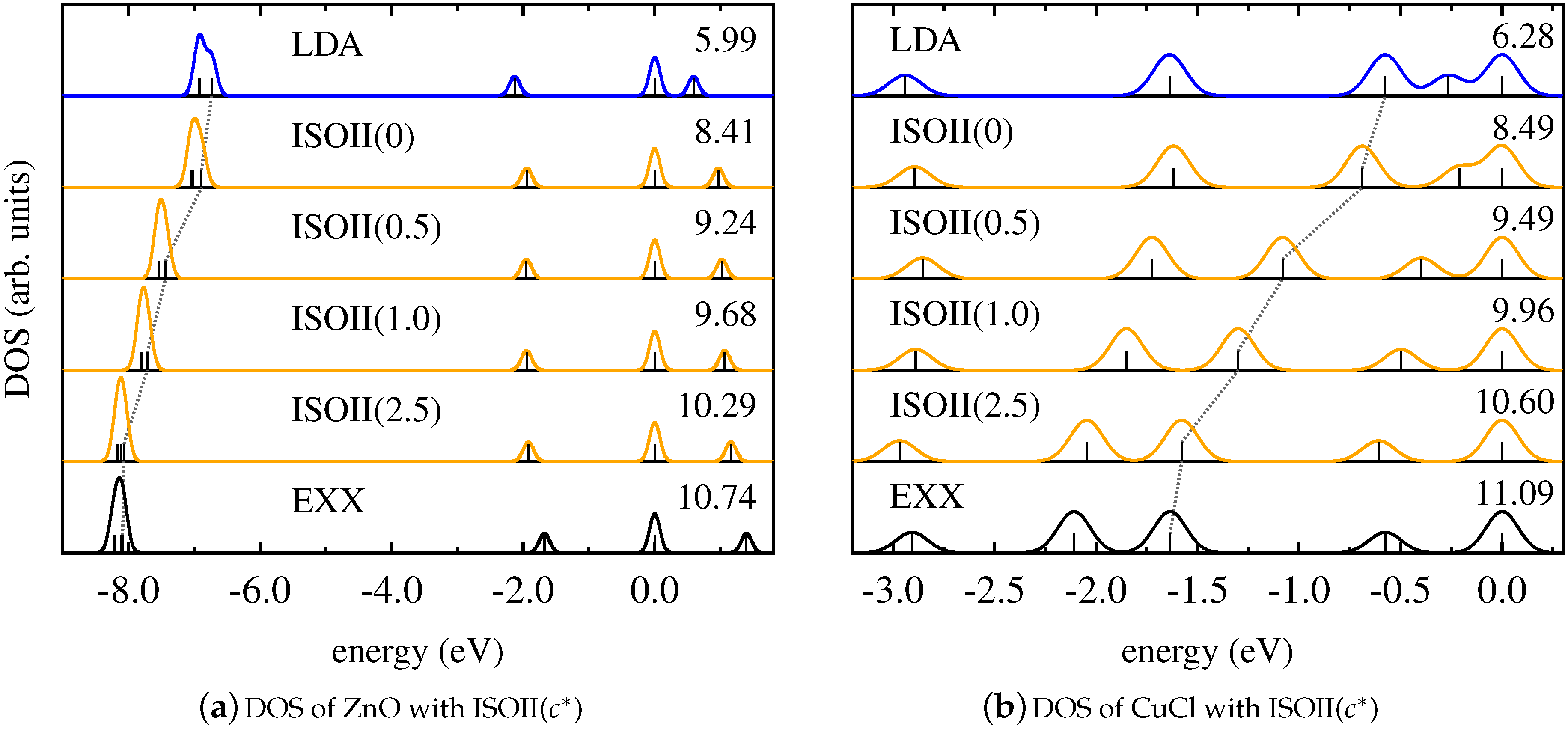
We begin our discussion by examining the Kohn–Sham DOS of ZnO as presented in Figure 1a. For this system, the group of eigenvalues that can be found around below if evaluated with pure PBE can be identified as d-electrons originating from the Zn atom. When comparing the DOS of PBE to the one of pure EXX, it becomes evident that the position of these eigenvalues is significantly influenced by SI, as their positions are moved by towards lower energies. In contrast, the eigenstate directly below the ho Kohn–Sham state remains nearly constant in its position relative to the ho state when going from PBE to EXX.
The DOS obtained with PBEh for various values of the parameter a (as shown in Figure 1a) reveals how the global hybrid functional copes with the electronic SI: the positions of the eigenvalues that are affected by SI are systematically shifted towards lower energies with increasing values of a. This behavior can be understood from the consideration that PBEh in general only partially counteracts SI, and the deviation from fulfilling Equation (1) can be reduced by increasing the value of a (see Reference [67] for a more detailed argument). Furthermore, the observed behavior is consistent with the fact that PBE and EXX can be considered as limiting cases of PBEh with and , respectively (note that PBEh with does not exactly correspond to pure EXX due to the semilocal correlation term in Equation (3)).
In contrast, the local hybrid functional ISO formally obeys Equation (1) for any value of the parameter c, as argued in Section 2. Yet, this feature does not directly translate into an improved DOS. Instead, as shown in Figure 1b, the DOS of the ISO functional for various values of c behaves similar to the one of PBEh: with increasing values of c, the group of eigenvalues that is most affected by SI moves from the position given by the LSDA to the one of EXX. These functionals provide the limiting cases for ISO in the case of spin-unpolarized systems with and , respectively, as can be seen from Equations (4)–(6) [86]. In any case, the formal freedom from SI for ISO does not necessarily result in a DOS that is comparable to EXX. In fact, the intrinsic amount of EXX that is involved in the local hybrid plays the dominant role in determining the Kohn–Sham DOS.
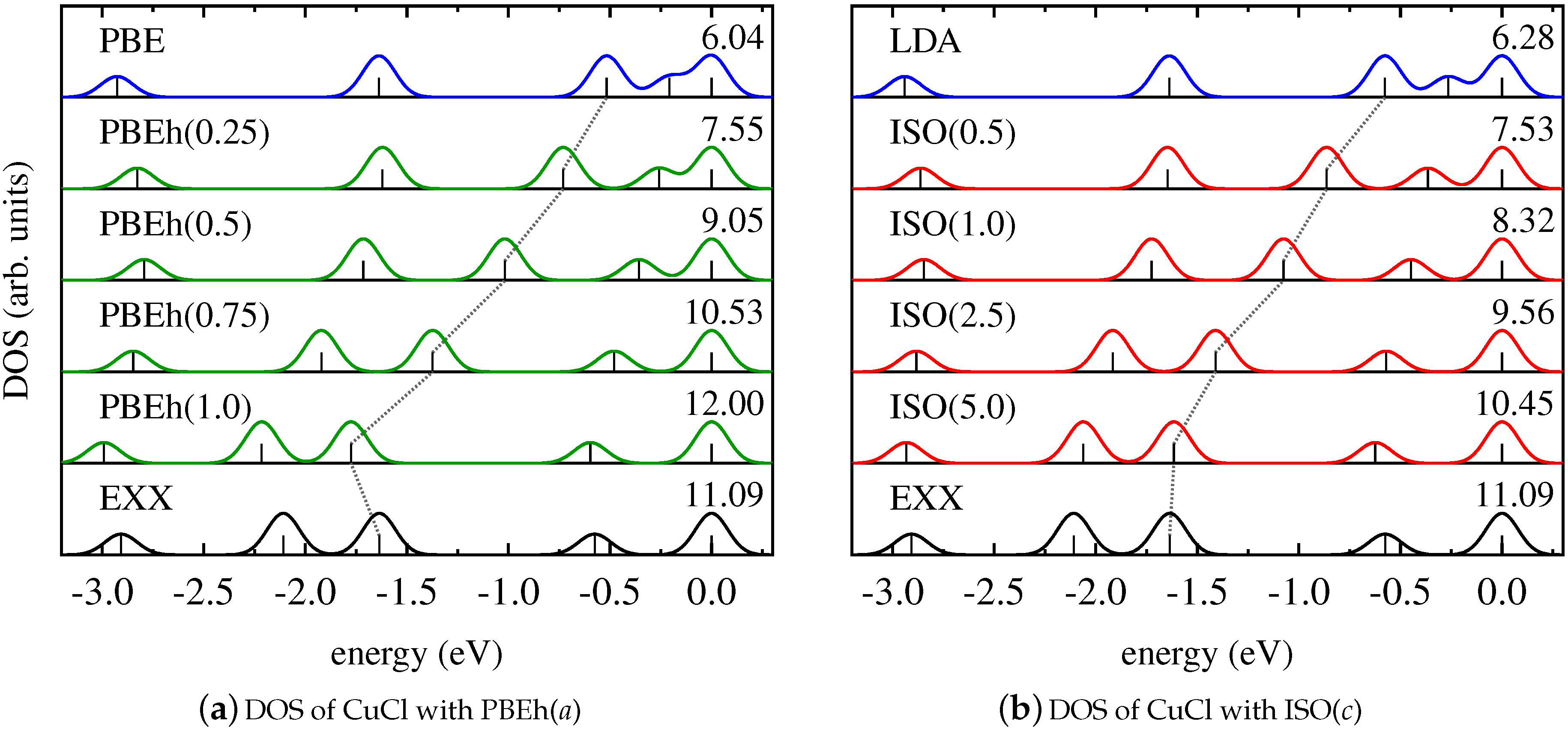
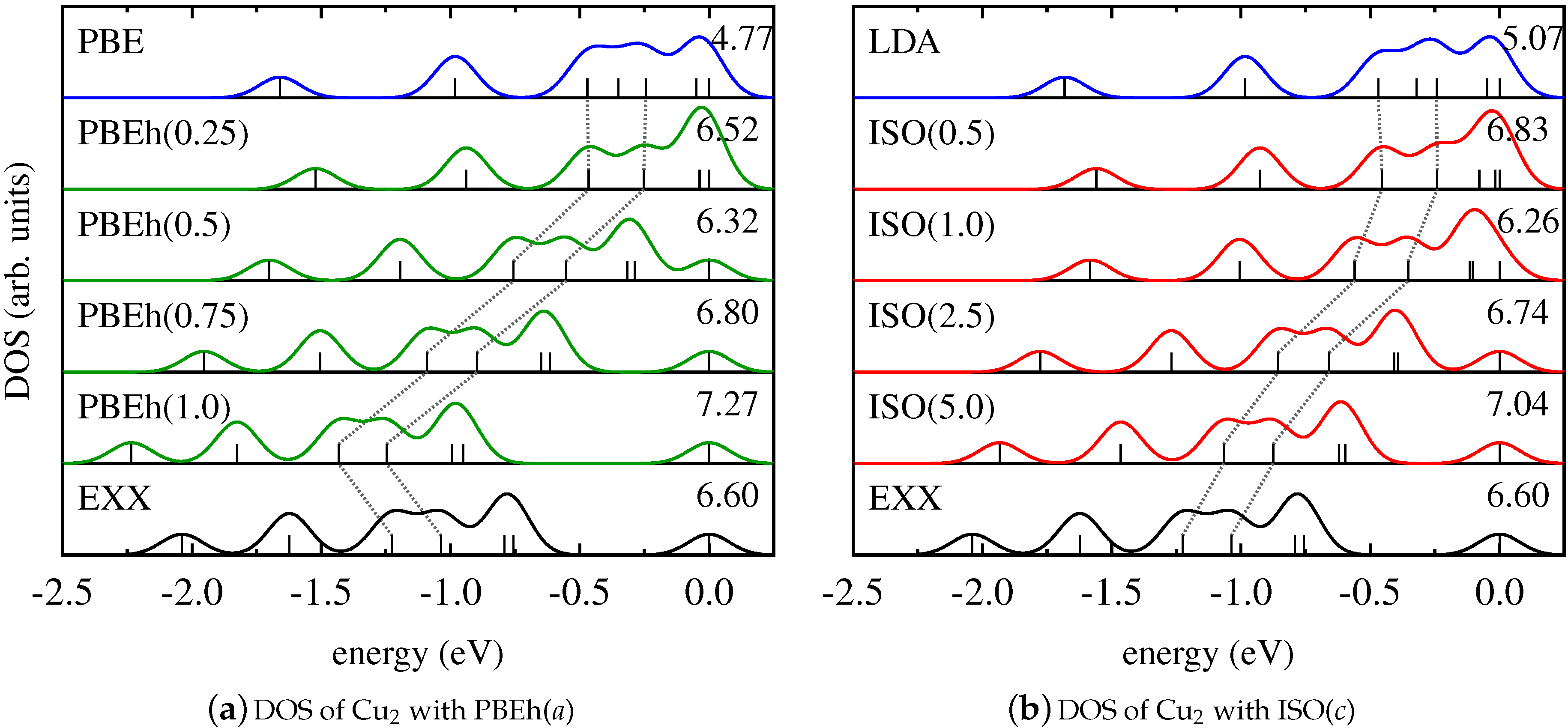
This finding is further supported by the results for the Kohn–Sham DOS of CuCl and Cu2, as shown in Figure 2 and Figure 3. It becomes evident that both PBEh and ISO with varying values of their parameters gradually change the energetic position of Kohn–Sham states that are affected by SI between the limiting cases of the semilocal PBE/LSDA and nonlocal, SI-free EXX. Interestingly, for some states—such as, for instance, the lowest state shown for Cu2—inclusion of small parts of EXX results in an upshift on the energy scale, while larger amounts of EXX reverse the direction of the shift. However, the local hybrid functional does not exhibit a special sensitivity to states affected by SI, as can be seen be comparing the DOS obtained with PBEh() and ISO(). These functionals can be regarded as comparable in the sense that both use the parametrization that was determined as ideal for the description of thermochemical properties such as binding energies [86,111]. Yet, despite the fact that ISO in contrast to PBEh is formally free from SI, the spectra of PBEh() and ISO() are rather similar for the systems investigated here.
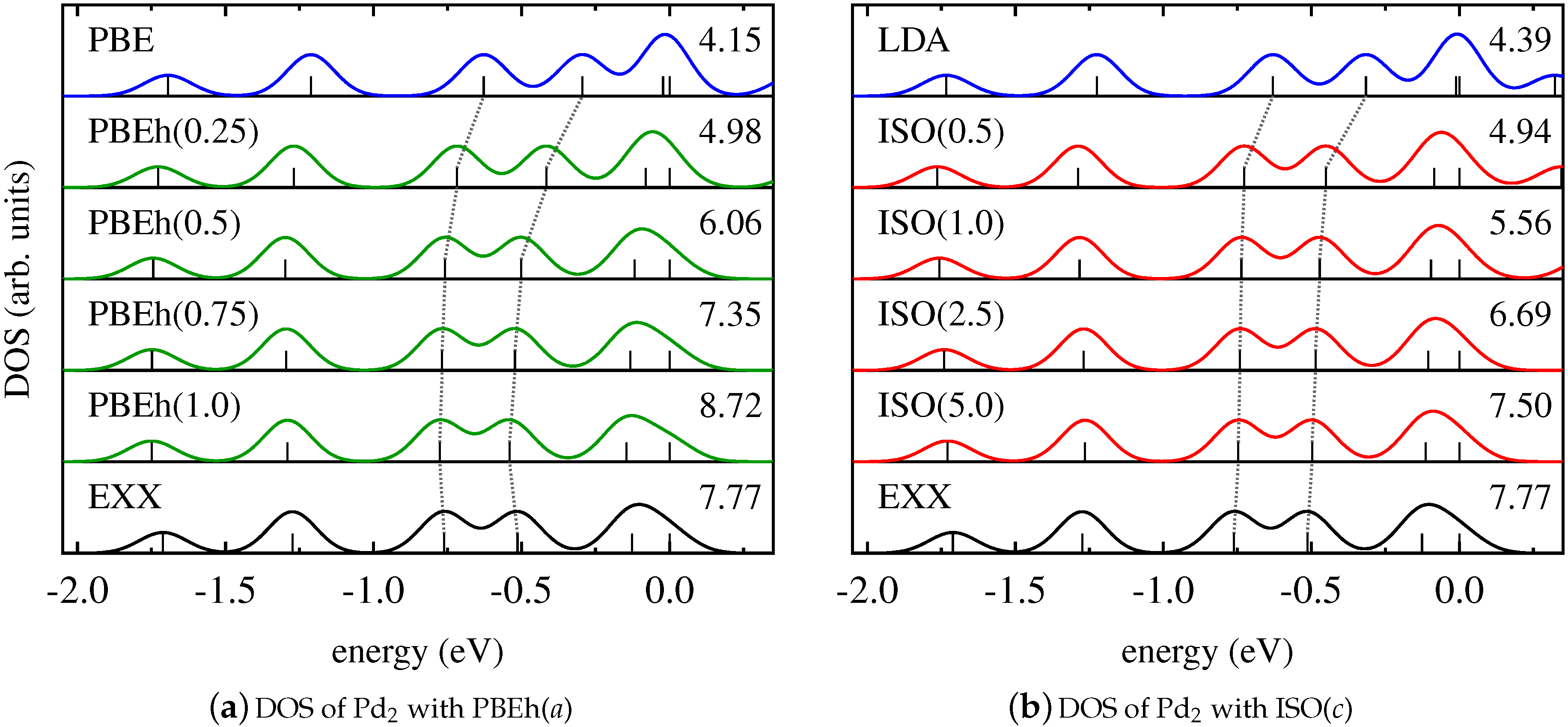
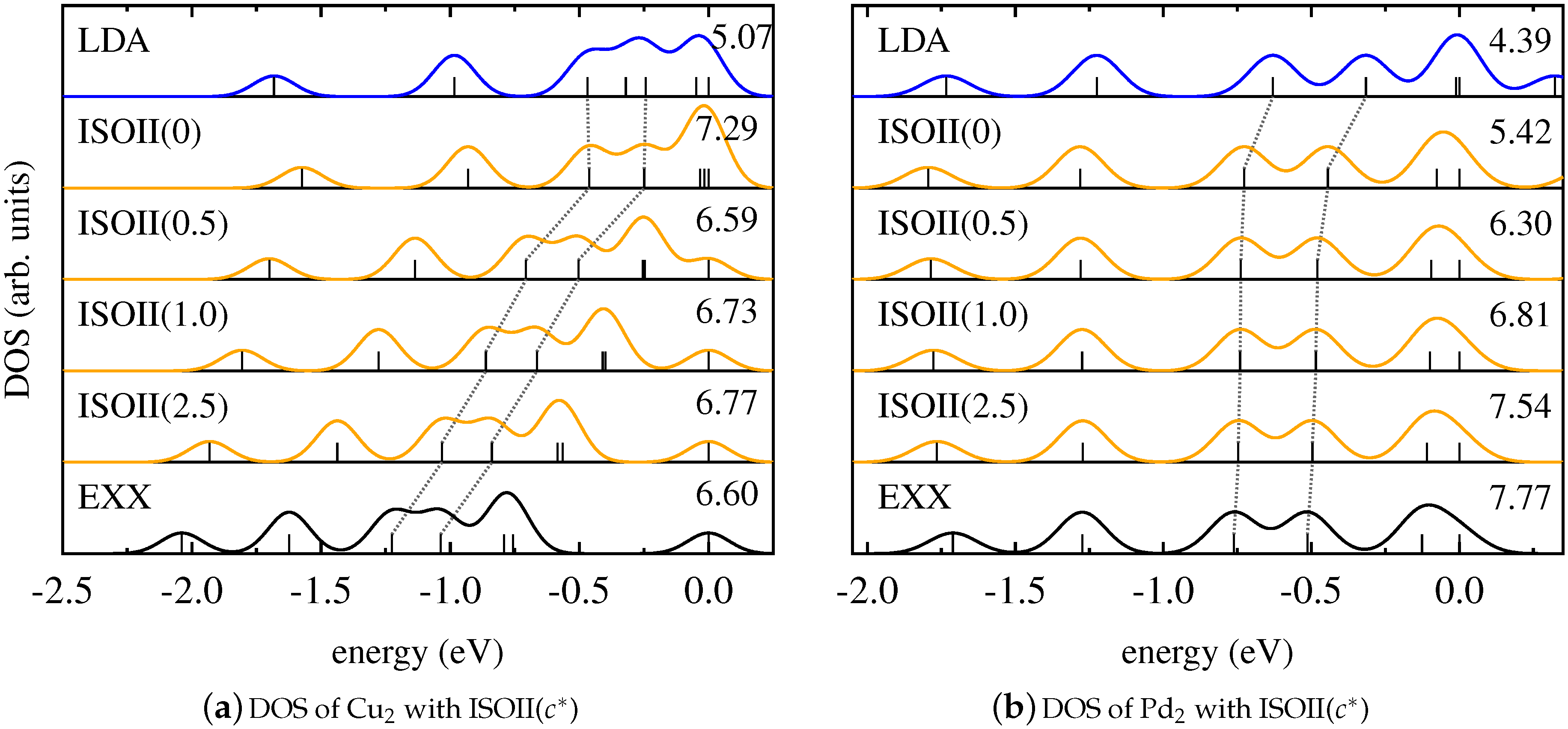
For Pd2, a different scenario occurs. Figure 4 demonstrates that the DOS changes insignificantly when using EXX instead of semilocal functionals. This indicates that all states are affected by SI to the same extent [13,115]. In this case, counteracting SI by using a global hybrid functional with increasing amounts of EXX does not change the relative energetic positions of the Kohn–Sham states, as can be seen from Figure 4a. Similarly, the results obtained with ISO in Figure 4b exhibit a DOS that is nearly independent of the value of the functional parameter. Consequently, also for systems whose electronic structure is less influenced by SI, the local and global hybrid perform very similarly.
We note that both the DOS of the homonuclear dimer Cu2 and the DOS of the heteronuclear dimer CuCl are greatly affected by SI. The relative position of the Kohn–Sham eigenvalues of the homonuclear diatomic molecule Pd2, on the other hand, is hardly influenced by SI. We conclude from this finding that it is difficult to predict trends in the DOS based on general arguments about formally being free from SI. For this reason, the results for the Pd2 dimer also cannot be generalized to bulk Pd.
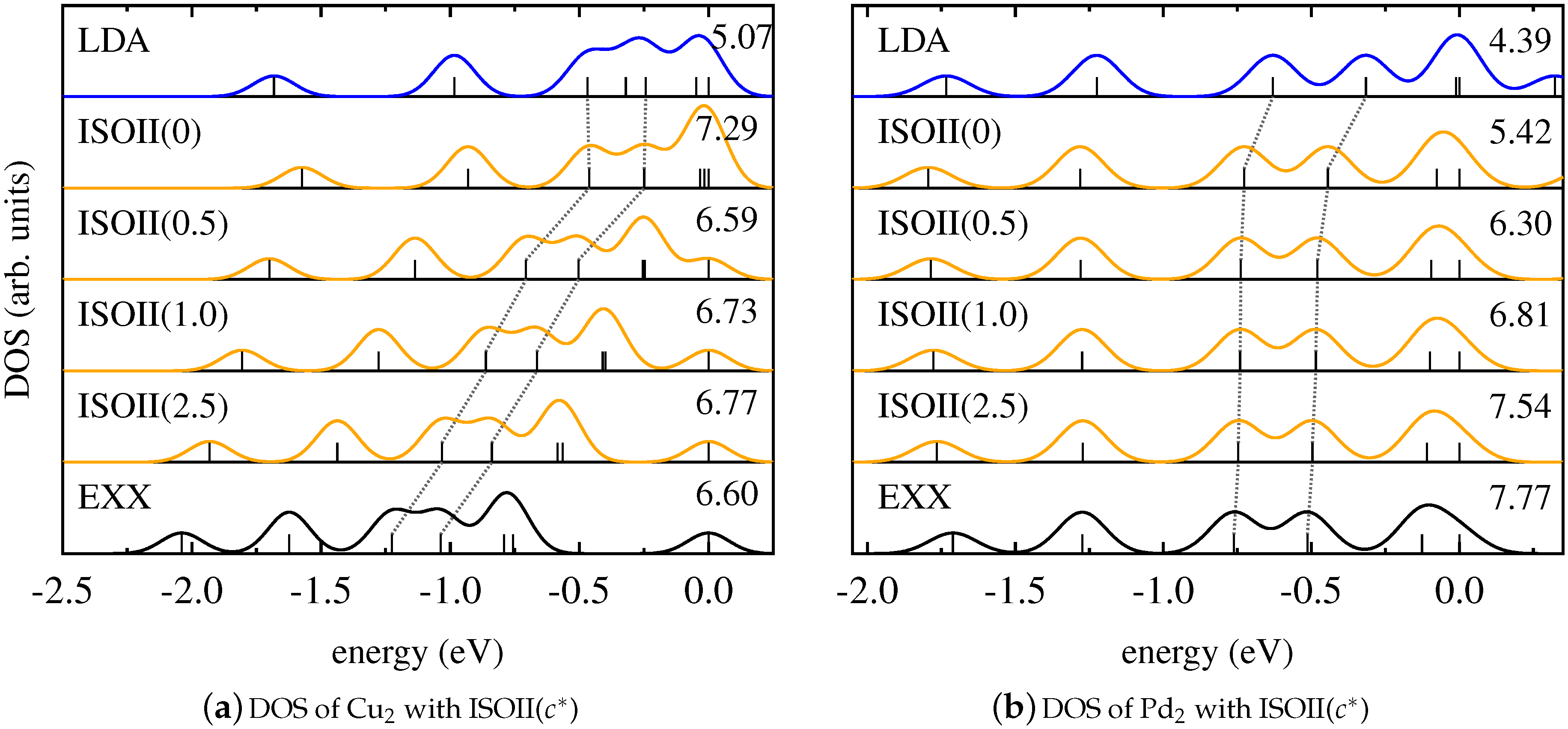
As discussed in Section 3, the detection function is canceled in the mixing functions of ISO if , as it is the case for the four molecules considered in this work. Thus, the detection function effectively does not contribute in the mixing process. Since this function (according to Equation (10)) is crucial for the local hybrid to be free from SI, one might argue that the similar performance of PBEh and ISO is a consequence of the appearance of the spin polarization in Equations (5) and (6). In order to contradict this conclusion, and to emphasize that the similarity between global and local hybrids is a fundamental feature rather than a specific result observed for one particular functional, we evaluate the Kohn–Sham DOS of the four molecules obtained with the modified local hybrid ISOII.
The corresponding results are shown in Figure 5 for ZnO and CuCl, and in Figure 6 for Cu2 and Pd2. These graphs demonstrate that ISOII, even though it explicitly uses the detection function also for spin-unpolarized systems, provides a Kohn–Sham DOS that is similar to the results of PBEh and ISO. For ZnO, CuCl, and Cu2, the energetic positions of the states affected by SI change gradually from the result of the LSDA to the one of pure EXX with increasing values of the functional parameter , and good agreement with SI-free calculations is only achieved for large values of . In the case of Pd2, again, changing the parametrization of ISOII has virtually no effect on the relative Kohn–Sham eigenvalue spectrum. In general, also for ISOII, the intrinsic amount of EXX appears to be the decisive factor for the outcome of the Kohn–Sham DOS.
5. Conclusions
In this article, we investigated diatomic molecules containing transition metals under the aspect of electronic SI. Based on the Kohn–Sham eigenvalue spectrum as an indicator for the influence of SI, we compared local hybrid functionals (which are designed to be free from SI in the sense of Equation (1)) to global hybrids (which only partially counteract SI). We found that both types of hybrid functionals show striking similarities in their Kohn–Sham DOS. More specifically, the intrinsic amount of EXX included in the hybrid construction appears as the important criterion, independent of the formal reduction of SI via an iso-orbital detection function. Good agreement with the result of EXX calculations was only obtained for hybrid functionals with large EXX admixture. Thus, correcting SI via detection functions such as does not generally result in a Kohn–Sham DOS comparable to pure EXX.
The observed behavior of the DOS with variation of the functional parameter marks another manifestation of the so-called parameter dilemma which, as discussed in the conclusion of Reference [86], affects local hybrids, global hybrids, range-separated hybrids, and SIC schemes alike: Properties determined by the total energy require a different parametrization of the functional than properties determined by the potential; e.g., the KS eigenvalues. Our investigation regarding transition metal dimers underlines the finding that an equally satisfying description of both domains remains challenging. Our finding that a functional’s property of being formally one-electron SI free is not necessarily important is in line with other recent reports [125] and extends the conclusions of Reference [67] to systems containing transition elements.
Acknowledgments
We acknowledge financial support by the German–Israeli Foundation and by the German Science Foundation via SFB840.
Author Contributions
Both authors discussed the conceptual aspects of the work and the research plan. All presented calculations were carried out by Tobias Schmidt. Both authors contributed to the writing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Becke, A.D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [CrossRef] [PubMed]
- Szotek, Z.; Temmerman, W.M.; Winter, H. Application of the self-interaction correction to transition-metal oxides. Phys. Rev. B 1993, 47, 4029–4032. [Google Scholar] [CrossRef]
- Schulthess, T.C.; Temmerman, W.M.; Szotek, Z.; Butler, W.H.; Stocks, G.M. Electronic structure and exchange coupling of Mn impurities in III–V semiconductors. Nat. Mater. 2005, 4, 838–844. [Google Scholar] [CrossRef]
- Strange, P.; Svane, A.; Temmerman, W.M.; Szotek, Z.; Winter, H. Understanding the valency of rare earths from first-principles theory. Nature 1999, 399, 756–758. [Google Scholar] [CrossRef]
- Perdew, J.P. Orbital functional for exchange and correlation: Self-interaction correction to the local density approximation. Chem. Phys. Lett. 1979, 64, 127–130. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Kümmel, S.; Kronik, L. Orbital-dependent density functionals: Theory and applications. Rev. Mod. Phys. 2008, 80, 3–60. [Google Scholar] [CrossRef]
- Tsuneda, T.; Hirao, K. Self-interaction corrections in density functional theory. J. Chem. Phys. 2014, 140, 18A513. [Google Scholar] [CrossRef] [PubMed]
- Körzdörfer, T.; Kümmel, S.; Marom, N.; Kronik, L. When to trust photoelectron spectra from Kohn–Sham eigenvalues: The case of organic semiconductors. Phys. Rev. B 2009, 79, 201205(R). [Google Scholar] [CrossRef]
- Körzdörfer, T.; Kümmel, S.; Marom, N.; Kronik, L. Erratum: When to trust photoelectron spectra from Kohn–Sham eigenvalues: The case of organic semiconductors [Phys. Rev. B 79, 201205 (2009)]. Phys. Rev. B 2010, 82, 129903. [Google Scholar] [CrossRef]
- Handy, N.C.; Cohen, A.J. Left-right correlation energy. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Cremer, D. Density functional theory: Coverage of dynamic and non-dynamic electron correlation effects. Mol. Phys. 2001, 99, 1899–1940. [Google Scholar] [CrossRef]
- Polo, V.; Kraka, E.; Cremer, D. Electron correlation and the self-interaction error of density functional theory. Mol. Phys. 2002, 100, 1771–1790. [Google Scholar] [CrossRef]
- Fermi, E.; Amaldi, E. Le Orbite [infinito] s Ddegli Elementi; R. Accademia d’Italia: Roma, Italy, 1934; Volume 6, p. 119. [Google Scholar]
- Cortona, P. New self-interaction-corrected local-density approximation to tzhe density-functional theory. Phys. Rev. A 1986, 34, 769–776. [Google Scholar] [CrossRef]
- Guo, Y.; Whitehead, M.A. An alternative self-interaction correction in the generalized exchange local-density functional theory. J. Comput. Chem. 1991, 12, 803–810. [Google Scholar] [CrossRef]
- Lundin, U.; Eriksson, O. Novel method of self-interaction corrections in density functional calculations. Int. J. Quantum Chem. 2001, 81, 247–252. [Google Scholar] [CrossRef]
- Unger, H.J. Self-interaction correction with an explicitly density-dependent functional. Phys. Lett. A 2001, 284, 124–129. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. A simple method to selectively scale down the self-interaction correction. J. Chem. Phys. 2006, 124, 191101. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.; Capelle, K. Investigation of self-interaction corrections for an exactly solvable model system: Orbital dependence and electron localization. J. Chem. Theory Comput. 2010, 6, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Constantin, L.A.; Fabiano, E.; Della Sala, F. Improving atomization energies of molecules and solids with a spin-dependent gradient correction from one-electron density analysis. Phys. Rev. B 2011, 84, 233103. [Google Scholar] [CrossRef]
- Dinh, P.M.; Reinhard, P.G.; Suraud, E.; Vincendon, M. The two-set and average-density self-interaction corrections applied to small electronic systems. In Advances in Atomic, Molecules, and Optical Physics; Elsevier: Amsterdam, The Netherlands, 2015; Volume 64, pp. 87–103. [Google Scholar]
- Borghi, G.; Ferretti, A.; Nguyen, N.L.; Dabo, I.; Marzari, N. Koopmans-compliant functionals and their performance against reference molecular data. Phys. Rev. B 2014, 90, 075135. [Google Scholar] [CrossRef]
- Nguyen, N.L.; Borghi, G.; Ferretti, A.; Dabo, I.; Marzari, N. First-principles photoemission spectroscopy and orbital tomography in molecules from koopmans-compliant functionals. Phys. Rev. Lett. 2015, 114, 166405. [Google Scholar] [CrossRef] [PubMed]
- Pederson, M.R.; Heaton, R.A.; Lin, C.C. Local-density Hartree-Fock theory of electronic states of molecules with self-interaction correction. J. Chem. Phys. 1984, 80, 1972. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Effect of the Perdew-Zunger self-interaction correction on the thermochemical performance of approximate density functionals. J. Chem. Phys. 2004, 121, 8187–8193. [Google Scholar] [CrossRef] [PubMed]
- Messud, J.; Dinh, P.M.; Reinhard, P.G.; Suraud, E. Time-dependent density-functional theory with a self-interaction correction. Phys. Rev. Lett. 2008, 101, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ruzsinszky, A.; Perdew, J.P.; Csonka, G.I.; Scuseria, G.E.; Vydrov, O.A. Understanding and correcting the self-interaction error in the electrical response of hydrogen chains. Phys. Rev. A 2008, 77, 060502. [Google Scholar] [CrossRef]
- Körzdörfer, T.; Mundt, M.; Kümmel, S. Electrical Response of Molecular Systems: The Power of Self-Interaction Corrected Kohn–Sham Theory. Phys. Rev. Lett. 2008, 100, 133004. [Google Scholar] [CrossRef] [PubMed]
- Kümmel, S. Self-interaction correction as a Kohn–Sham scheme in ground-state and time-dependent density functional theory. In Advances in Atomic, Molecular, and Optical Physics; Elsevier: Amsterdam, The Netherlands, 2015; Volume 64, pp. 143–151. [Google Scholar]
- Chen, J.; Krieger, J.B.; Li, Y.; Iafrate, G.J. Kohn–Sham calculations with self-interaction-corrected local-spin-density exchange-correlation energy functional for atomic systems. Phys. Rev. A 1996, 54, 3939–3947. [Google Scholar] [CrossRef] [PubMed]
- Garza, J.; Nichols, J.A.; Dixon, D.A. The optimized effective potential and the self-interaction correction in density functional theory: Application to molecules. J. Chem. Phys. 2000, 112, 7880. [Google Scholar] [CrossRef]
- Patchkovskii, S.; Autschbach, J.; Ziegler, T. Curing difficult cases in magnetic properties prediction with self-interaction corrected density functional theory. J. Chem. Phys. 2001, 115, 26–42. [Google Scholar] [CrossRef]
- Legrand, C.; Suraud, E.; Reinhard, P.G. Comparison of self-interaction-corrections for metal clusters. J. Phys. B Atomic Mol. Opt. Phys. 2002, 35, 1115–1128. [Google Scholar] [CrossRef]
- Vieira, D.; Capelle, K.; Ullrich, C.A. Physical signatures of discontinuities of the time-dependent exchange-correlation potential. Phys. Chem. Chem. Phys. 2009, 11, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, C.D.; Sanvito, S.; Burke, K. Polarizability of molecular chains: A self-interaction correction approach. Phys. Rev. B 2008, 77, 121204(R). [Google Scholar] [CrossRef]
- Körzdörfer, T.; Kümmel, S.; Mundt, M. Self-interaction correction and the optimized effective potential. J. Chem. Phys. 2008, 129, 014110. [Google Scholar] [CrossRef] [PubMed]
- Körzdörfer, T.; Kümmel, S. Self-interaction correction in the Kohn–Sham framework. In Theoretical Computational Developments in Modern Density Functional Theory; Roy, A.K., Ed.; Nova Science Publishers: New York, NY, USA, 2012. [Google Scholar]
- Svane, A.; Gunnarsson, O. Transition-metal oxides in the self-interaction-corrected density-functional formalism. Phys. Rev. Lett. 1990, 65, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Temmerman, W.M.; Szotek, Z.; Winter, H. Self-interaction-corrected electronic strucutre of La2CuO4. Phys. Rev. B 1993, 47, 11533–11536. [Google Scholar] [CrossRef]
- Goedecker, S.; Umrigar, C.J. Critical assessment of the self-interaction-corrected-local-density-functional method and its algorithmic implementation. Phys. Rev. A 1997, 55, 1765–1771. [Google Scholar] [CrossRef]
- Pederson, M.R.; Heaton, R.A.; Lin, C.C. Density-functional theory with self-interaction correction: Application to the lithium molecule. J. Chem. Phys. 1985, 82, 2688. [Google Scholar] [CrossRef]
- Pederson, M.R.; Lin, C.C. Localized and canonical atomic orbitals in self-interaction corrected local density functional approximation. J. Chem. Phys. 1988, 88, 1807–1817. [Google Scholar] [CrossRef]
- Klüpfel, S.; Klüpfel, P.; Jónsson, H. Importance of complex orbitals in calculating the self-interaction- corrected ground state of atoms. Phys. Rev. A 2011, 84, 050501. [Google Scholar] [CrossRef]
- Hofmann, D.; Klüpfel, S.; Klüpfel, P.; Kümmel, S. Using complex degrees of freedom in the Kohn–Sham self-interaction correction. Phys. Rev. A 2012, 85, 062514. [Google Scholar] [CrossRef]
- Klüpfel, S.; Klüpfel, P.; Jónsson, H. The effect of the Perdew-Zunger self-interaction correction to density functionals on the energetics of small molecules. J. Chem. Phys. 2012, 137, 124102. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, D.; Körzdörfer, T.; Kümmel, S. Kohn–Sham Self-Interaction Correction in Real Time. Phys. Rev. Lett. 2012, 108, 146401. [Google Scholar] [CrossRef] [PubMed]
- Lehtola, S.; Jónsson, H. Variational, self-consistent implementation of the Perdew-Zunger self-interaction correction with complex optimal orbitals. J. Chem. Theory Comput. 2014, 10, 5324–5337. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdóttir, H.; Jónsson, E.Ö.; Jónsson, H. Calculations of Al dopant in α-quartz using a variational implementation of the Perdew-Zunger self-interaction correction. New J. Phys. 2015, 17, 83006. [Google Scholar] [CrossRef]
- Lehtola, S.; Jónsson, E.Ö.; Jónsson, H. Effect of complex-valued optimal orbitals on atomization energies with the Perdew–Zunger self-interaction correction to density functional theory. J. Chem. Theory Comput. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lehtola, S.; Head-Gordon, M.; Jónsson, H. Complex orbitals, multiple local minima and symmetry breaking in Perdew-Zunger self-interaction corrected density-functional theory calculations. J. Chem. Theory Comput. 2016, 12, 3195–3207. [Google Scholar] [CrossRef] [PubMed]
- Pederson, M.R.; Ruzsinszky, A.; Perdew, J.P. Communication: Self-interaction correction with unitary invariance in density functional theory. J. Chem. Phys. 2014, 140, 121103. [Google Scholar] [CrossRef] [PubMed]
- Pederson, M.R.; Baruah, T. Self-interaction corrections within the fermi-orbital-based formalism. In Advance in Atomic, Molecular, and Optical Physics; Elsevier: Amsterdam, The Netherlands, 2015; Volume 64, pp. 153–180. [Google Scholar]
- Hahn, T.; Liebing, S.; Kortus, J.; Pederson, M.R. Fermi orbital self-interaction corrected electronic structure of molecules beyond local density approximation. J. Chem. Phys. 2015, 143, 224104. [Google Scholar] [CrossRef] [PubMed]
- Ruzsinszky, A.; Perdew, J.P.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E. Spurious fractional charge on dissociated atoms: Pervasive and resilient self-interaction error of common density functionals. J. Chem. Phys. 2006, 125, 194112. [Google Scholar] [CrossRef] [PubMed]
- Mori-Sánchez, P.; Cohen, A.J.; Yang, W. Many-electron self-interaction error in approximate density functionals. J. Chem. Phys. 2006, 125, 201102. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Parr, R.G.; Levy, M.; Balduz, J.L. Density-Functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy. Phys. Rev. Lett. 1982, 49, 1691–1694. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Sai, N.; Barbara, P.F.; Leung, K. Hole localization in molecular crystals from hybrid density functional theory. Phys. Rev. Lett. 2011, 106, 226403. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Autschbach, J.; Govind, N.; Kronik, L.; Baer, R. Curvature and frontier orbital energies in density functional theory. J. Phys. Chem. Lett. 2012, 3, 3740–3744. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, X.; Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Local scaling correction for reducing delocalization error in density functional approximations. Phys. Rev. Lett. 2015, 114, 053001. [Google Scholar] [CrossRef] [PubMed]
- Dauth, M.; Caruso, F.; Kümmel, S.; Rinke, P. Piecewise linearity in the GW approximation for accurate quasiparticle energy predictions. Phys. Rev. B 2016, 93, 121115. [Google Scholar] [CrossRef]
- Schmidt, T.; Kümmel, S. One- and many-electron self-interaction error in local and global hybrid functionals. Phys. Rev. B 2016, 93, 165120. [Google Scholar] [CrossRef]
- Atalla, V.; Zhang, I.Y.; Hofmann, O.T.; Ren, X.; Rinke, P.; Scheffler, M. Enforcing the linear behavior of the total energy with hybrid functionals: Implications for charge transfer, interaction energies, and the random-phase approximation. Phys. Rev. B 2016, 94, 035140. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Peach, M.J.G.; Helgaker, T.; Sałek, P.; Keal, T.W.; Lutnæs, O.B.; Tozer, D.J.; Handy, N.C. Assessment of a Coulomb-attenuated exchange-correlation energy functional. Phys. Chem. Chem. Phys. 2006, 8, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient- approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, T.B.; Kümmel, S. Tuned range separated hybrid functionals for solvated low bandgap oligomers. J. Chem. Phys. 2015, 143, 034101. [Google Scholar] [CrossRef] [PubMed]
- Karolewski, A.; Kronik, L.; Kümmel, S. Using optimally tuned range separated hybrid functionals in ground-state calculations: Consequences and caveats. J. Chem. Phys. 2013, 138, 204115. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Cruz, F.G.; Lam, K.C.; Burke, K. Exchange-correlation energy density from virial theorem. J. Phys. Chem. A 1998, 102, 4911–4917. [Google Scholar] [CrossRef]
- Jaramillo, J.; Scuseria, G.E.; Ernzerhof, M. Local hybrid functionals. J. Chem. Phys. 2003, 118, 1068–1073. [Google Scholar] [CrossRef]
- Arbuznikov, A.V.; Kaupp, M.; Bahmann, H. From local hybrid functionals to “localized local hybrid” potentials: Formalism and thermochemical tests. J. Chem. Phys. 2006, 124, 204102. [Google Scholar] [CrossRef] [PubMed]
- Janesko, B.G.; Scuseria, G.E. Local hybrid functionals based on density matrix products. J. Chem. Phys. 2007, 127, 164117. [Google Scholar] [CrossRef] [PubMed]
- Bahmann, H.; Rodenberg, A.; Arbuznikov, A.V.; Kaupp, M. A thermochemically competitive local hybrid functional without gradient corrections. J. Chem. Phys. 2007, 126, 011103. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, M.; Bahmann, H.; Arbuznikov, A.V. Local hybrid functionals: An assessment for thermochemical kinetics. J. Chem. Phys. 2007, 127, 194102. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Staroverov, V.N.; Tao, J.; Scuseria, G.E. Density functional with full exact exchange, balanced nonlocality of correlation, and constraint satisfaction. Phys. Rev. A 2008, 78, 052513. [Google Scholar] [CrossRef]
- Schmidt, T.; Kraisler, E.; Makmal, A.; Kronik, L.; Kümmel, S. A self-interaction-free local hybrid functional: Accurate binding energies vis-à-vis accurate ionization potentials from Kohn–Sham eigenvalues. J. Chem. Phys. 2014, 140, 18A510. [Google Scholar] [CrossRef] [PubMed]
- de Silva, P.; Corminboeuf, C. Local hybrid functionals with orbital-free mixing functions and balanced elimination of self-interaction error. J. Chem. Phys. 2015, 142, 074112. [Google Scholar] [CrossRef] [PubMed]
- Kümmel, S.; Perdew, J.P. Two avenues to self-interaction correction within Kohn–Sham theory: Unitary invariance is the shortcut. Mol. Phys. 2003, 101, 1363–1368. [Google Scholar] [CrossRef]
- Duffy, P.; Chong, D.P.; Casida, M.E.; Salahub, D.R. Kohn–Sham density-functional orbitals as approximate Dyson orbitals scattering for the calculation. Phys. Rev. A 1994, 50, 4707–4728. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.P.; Gritsenko, O.V.; Baerends, E.J. Interpretation of the Kohn–Sham orbital energies as approximate vertical ionization potentials. J. Chem. Phys. 2002, 116, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Kronik, L.; Kümmel, S. Gas-phase valence-electron photoemission spectroscopy using density functional theory. In First Principles Approaches to Spectroscopic Properties of Complex Materials; Topics in Current Chemistry; di Valentin, C., Botti, S., Coccoccioni, M., Eds.; Springer: Berlin, Germany, 2014. [Google Scholar]
- Akola, J.; Manninen, M.; Häkkinen, H.; Landman, U.; Li, X.; Wang, L.S. Aluminum cluster anions: Photoelectron spectroscopy and ab initio simulations. Phys. Rev. B 2000, 62, 13216. [Google Scholar] [CrossRef]
- Khanna, S.N.; Beltran, M.; Jena, P. Relationship between photoelectron spectroscopy and the magnetic moment of Ni7 clusters. Phys. Rev. B 2001, 64, 235419. [Google Scholar] [CrossRef]
- Kronik, L.; Fromherz, R.; Ko, E.; Ganteför, G.; Chelikowsky, J.R. Highest electron affinity as a predictor of cluster anion structures. Nat. Mater. 2002, 1, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Moseler, M.; Huber, B.; Häkkinen, H.; Landman, U.; Wrigge, G.; Hoffmann, M.A.; Issendorff, B.V. Thermal effects in the photoelectron spectra of Na-N clustres (N = 4–19). Phys. Rev. B 2003, 68, 165413. [Google Scholar] [CrossRef]
- Häkkinen, H.; Moseler, M.; Kostko, O.; Morgner, N.; Hoffmann, M.A.; Issendorff, B.V. Symmetry and electronic structure of noble-metal nanoparticles and the role of relativity. Phys. Rev. Lett. 2004, 93, 093401. [Google Scholar] [CrossRef] [PubMed]
- Mundt, M.; Kümmel, S.; Huber, B.; Moseler, M. Photoelectron spectra of sodium clusters: The problem of interpreting Kohn–Sham eigenvalues. Phys. Rev. B 2006, 73, 205407. [Google Scholar] [CrossRef]
- Leppert, L.; Kümmel, S. The electronic structure of gold-platinum nanoparticles: Collecting clues for why they are special. J. Phys. Chem. C 2011, 115, 6694–6702. [Google Scholar] [CrossRef]
- Leppert, L.; Albuquerque, R.Q.; Foster, A.S.; Kümmel, S. Interplay of electronic structure and atomic mobility in nanoalloys of Au and Pt. J. Phys. Chem. C 2013, 117, 17268–17273. [Google Scholar] [CrossRef]
- Zamudio-Bayer, V.; Leppert, L.; Hirsch, K.; Langenberg, A.; Rittmann, J.; Kossick, M.; Vogel, M.; Richter, R.; Terasaki, A.; Möller, T.; et al. Coordination-driven magnetic-to-nonmagnetic transition in manganese-doped silicon clusters. Phys. Rev. B 2013, 88, 115425. [Google Scholar] [CrossRef]
- Capelo, R.G.; Leppert, L.; Albuquerque, R.Q. The concept of localized atomic mobility: Unraveling properties of nanoparticles. J. Phys. Chem. C 2014, 118, 21647–21654. [Google Scholar] [CrossRef]
- Leppert, L.; Kempe, R.; Kümmel, S. Hydrogen binding energies and electronic structure of Ni–Pd particles: A clue to their special catalytic properties. Phys. Chem. Chem. Phys. 2015, 17, 26140–26148. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.V.; Melnyk, I.; Skorb, E.V.; Fratzl, P.; Zolotoyabko, E.; Dubrovinskaia, N.; Dubrovinsky, L.; Avadhut, Y.S.; Senker, J.; Leppert, L.; et al. The use of ultrasonic cavitation for near-surface structuring of robust and low-cost AlNi catalysts for hydrogen production. Green Chem. 2015, 17, 2745. [Google Scholar] [CrossRef]
- Aslan, M.; Davis, J.B.A.; Johnston, R.L. Global optimization of small bimetallic Pd–Co binary nanoalloy clusters: A genetic algorithm approach at the DFT level. Phys. Chem. Chem. Phys. 2016, 18, 6676. [Google Scholar] [CrossRef] [PubMed]
- Grabo, T.; Kreibich, T.; Gross, E.K.U. Optimized effective potential for atoms and molecules. Mol. Eng. 1997, 7, 27–50. [Google Scholar] [CrossRef]
- Kümmel, S.; Perdew, J.P. Simple Iterative Construction of the Optimized Effective Potential for Orbital Functionals, Including Exact Exchange. Phys. Rev. Lett. 2003, 90, 043004. [Google Scholar] [CrossRef] [PubMed]
- Makmal, A.; Kümmel, S.; Kronik, L. Fully Numerical All-Electron Solutions of the Optimized Effective Potential Equation for Diatomic Molecules. J. Chem. Theory Comput. 2009, 5, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Makmal, A.; Kümmel, S.; Kronik, L. Dissociation of diatomic molecules and the exact-exchange Kohn–Sham potential: The case of LiF. Phys. Rev. A 2011, 83, 062512. [Google Scholar] [CrossRef]
- Burke, K.; Cruz, F.G.; Lam, K.C. Unambiguous exchange-correlation energy density. J. Chem. Phys. 1998, 109, 8161–8167. [Google Scholar] [CrossRef]
- Arbuznikov, A.V.; Kaupp, M. Towards improved local hybrid functionals by calibration of exchange-energy densities. J. Chem. Phys. 2014, 141, 204101. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Erratum: Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Körzdörfer, T.; Kümmel, S. Single-particle and quasiparticle interpretation of Kohn–Sham and generalized Kohn–Sham eigenvalues for hybrid functionals. Phys. Rev. B 2010, 82, 155206. [Google Scholar] [CrossRef]
- Imamura, Y.; Kobayashi, R.; Nakai, H. Linearity condition for orbital energies in density functional theory (II): Application to global hybrid functionals. Chem. Phys. Lett. 2011, 513, 130–135. [Google Scholar] [CrossRef]
- Atalla, V.; Yoon, M.; Caruso, F.; Rinke, P.; Scheffler, M. Hybrid density functional theory meets quasiparticle calculations: A consistent electronic structure approach. Phys. Rev. B 2013, 88, 165122. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Wang, Y.; Perdew, J.P. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar]
- Schmidt, T.; Kraisler, E.; Kronik, L.; Kümmel, S. One-electron self-interaction and the asymptotics of the Kohn–Sham potential: An impaired relation. Phys. Chem. Chem. Phys. 2014, 16, 14357–14367. [Google Scholar] [CrossRef] [PubMed]
- Kurth, S.; Perdew, J.P.; Blaha, P. Molecular and solid-state tests of density functional approximations: LSD, GGAs, and meta-GGAs. Int. J. Quantum Chem. 1999, 75, 889–909. [Google Scholar] [CrossRef]
- Krieger, J.B.; Li, Y.; Iafrate, G.J. Construction and application of an accurate local spin-polarized Kohn–Sham potential with integer discontinuity: Exchange-only theory. Phys. Rev. A 1992, 45, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 92nd ed.; CRC: London, UK, 2011.
- Gritsenko, O.V.; Mentel, L.M.; Baerends, E.J. On the errors of local density (LDA) and generalized gradient (GGA) approximations to the Kohn–Sham potential and orbital energies. J. Chem. Phys. 2016, 144, 204114. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Kohn–Sham density of states (DOS) of ZnO obtained with (a) PBE (blue), exact-exchange (EXX, black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red). In this and all following figures, each panel contains the value of in eV in order to indicate the absolute position of the DOS on the energy scale. Further, the relative shift of the d-states—which were identified in DARSEC as the Kohn–Sham states with angular momentum quantum number (cf. Reference [107])—is highlighted by the dashed grey line.
Figure 1.
Kohn–Sham density of states (DOS) of ZnO obtained with (a) PBE (blue), exact-exchange (EXX, black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red). In this and all following figures, each panel contains the value of in eV in order to indicate the absolute position of the DOS on the energy scale. Further, the relative shift of the d-states—which were identified in DARSEC as the Kohn–Sham states with angular momentum quantum number (cf. Reference [107])—is highlighted by the dashed grey line.
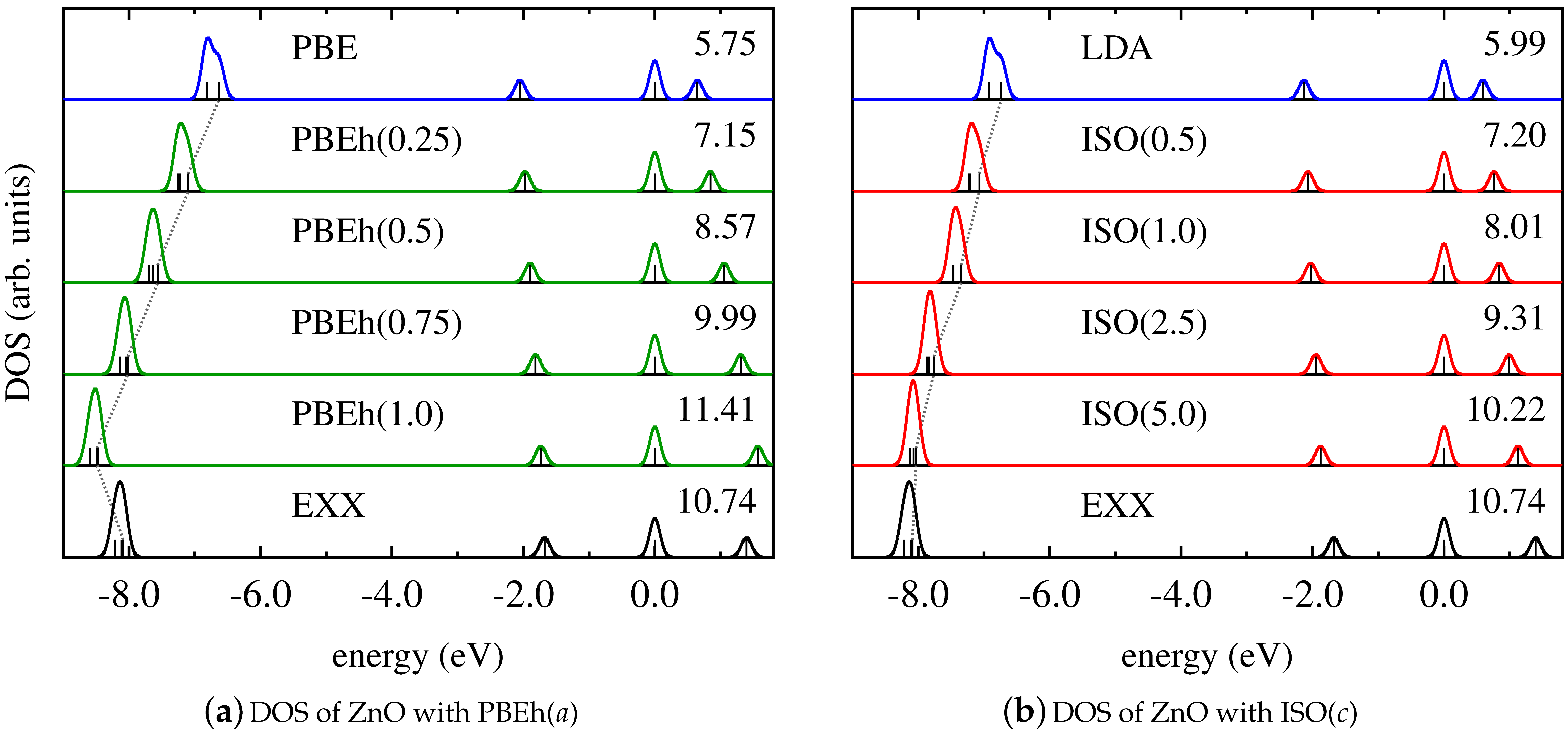
Figure 2.
Kohn–Sham DOS of CuCl obtained with (a) PBE (blue), EXX (black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red).
Figure 2.
Kohn–Sham DOS of CuCl obtained with (a) PBE (blue), EXX (black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red).
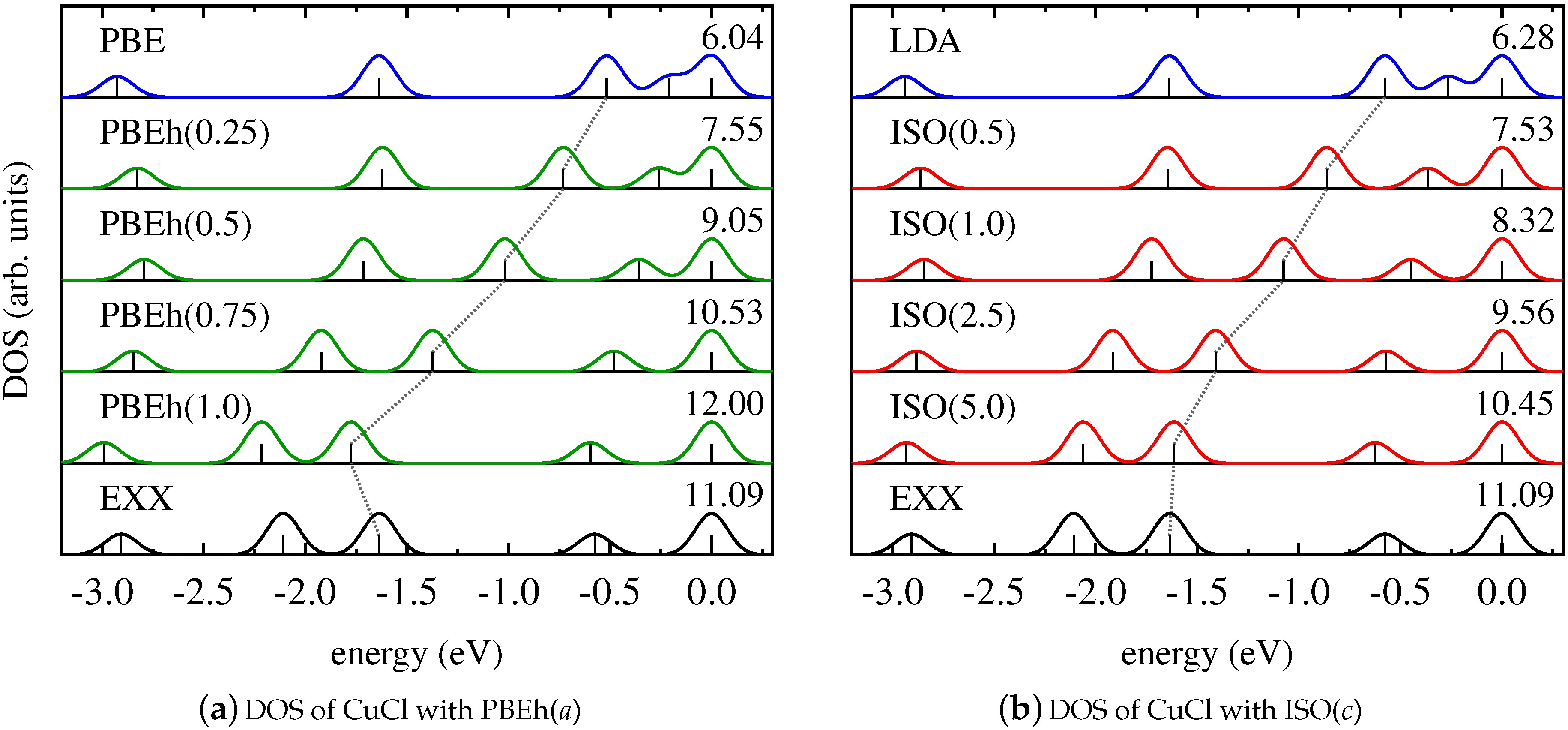
Figure 3.
Kohn–Sham DOS of Cu2 obtained with (a) PBE (blue), EXX (black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red).
Figure 3.
Kohn–Sham DOS of Cu2 obtained with (a) PBE (blue), EXX (black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red).
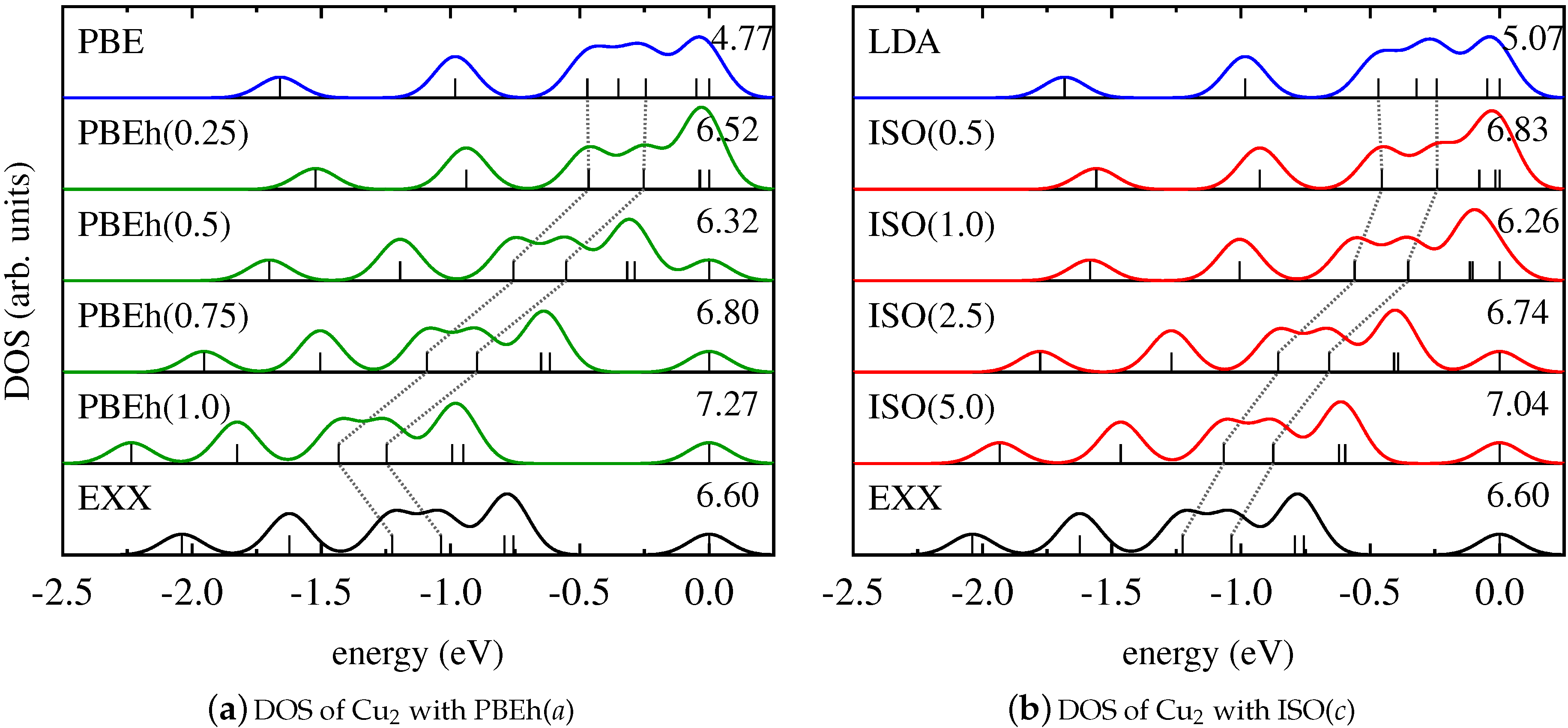
Figure 4.
Kohn–Sham DOS of Pd2 obtained with (a) PBE (blue), EXX (black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red).
Figure 4.
Kohn–Sham DOS of Pd2 obtained with (a) PBE (blue), EXX (black), and PBEh in dependence on a (green); (b) LDA (blue), EXX (black), and ISO in dependence on c (red).
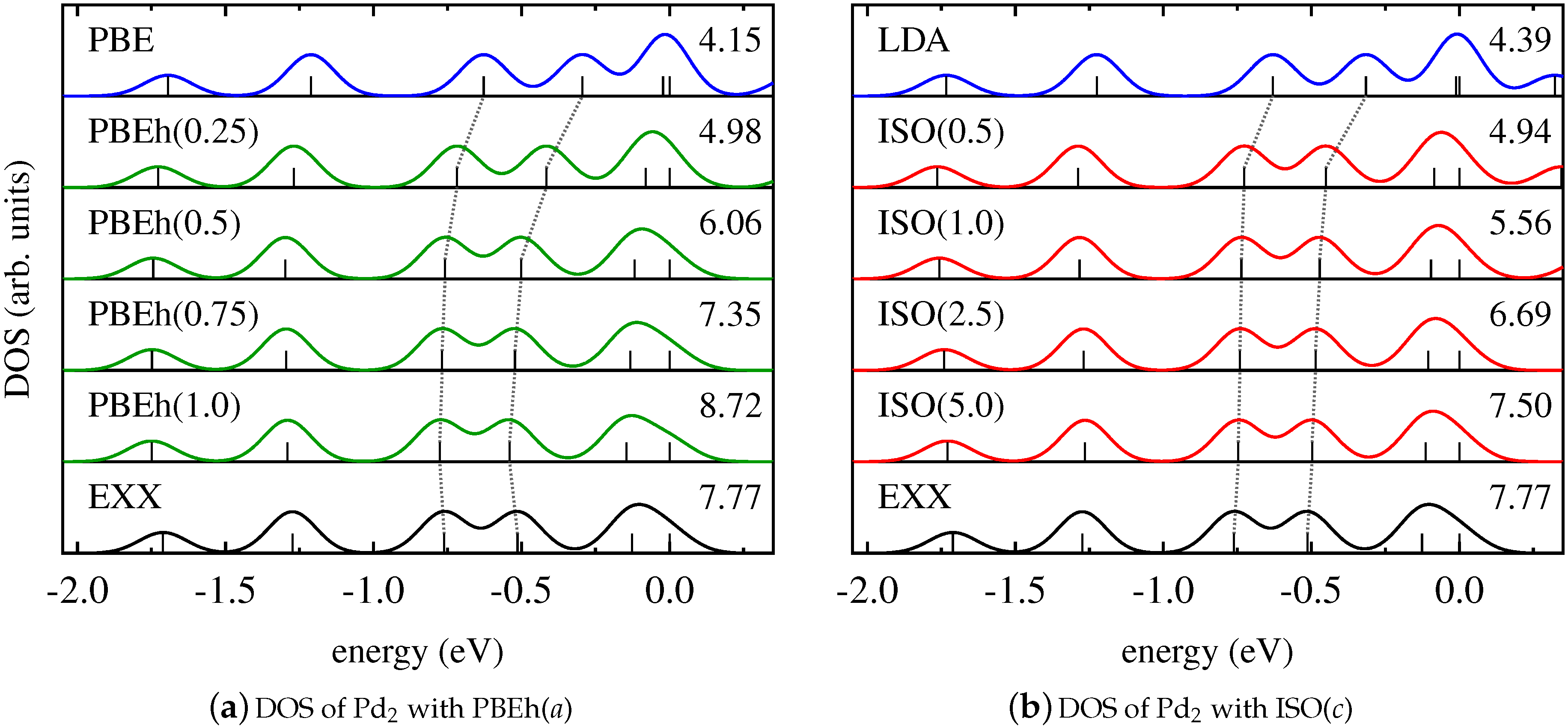
Figure 5.
Kohn–Sham DOS obtained with LDA (blue), pure EXX (black), and ISOII in dependence on (orange) for (a) ZnO and (b) CuCl.
Figure 5.
Kohn–Sham DOS obtained with LDA (blue), pure EXX (black), and ISOII in dependence on (orange) for (a) ZnO and (b) CuCl.
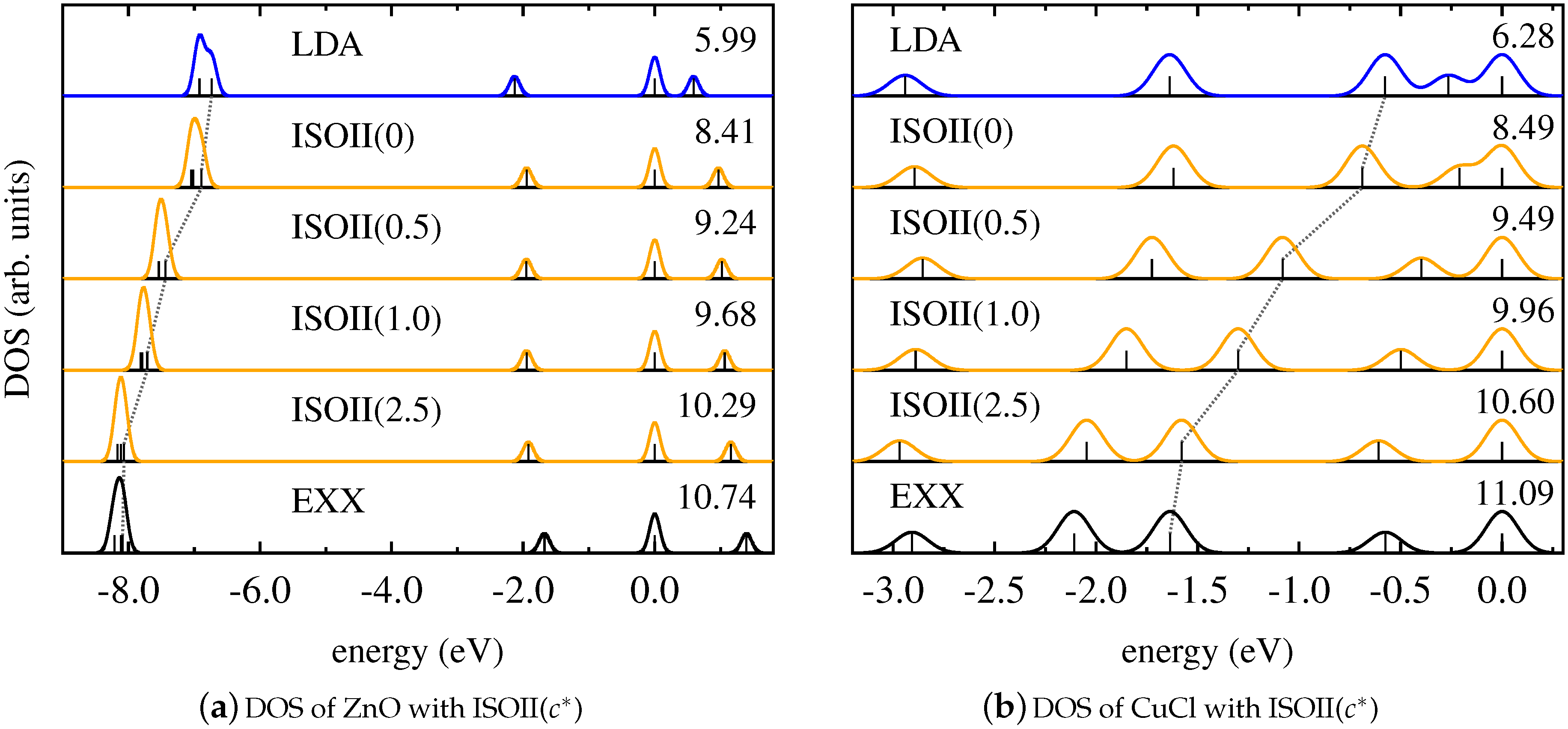
Figure 6.
Kohn–Sham DOS obtained with LDA (blue), pure EXX (black), and ISOII in dependence on (orange) for (a) Cu2 and (b) Pd2.
Figure 6.
Kohn–Sham DOS obtained with LDA (blue), pure EXX (black), and ISOII in dependence on (orange) for (a) Cu2 and (b) Pd2.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schmidt, T.; Kümmel, S. The Influence of One-Electron Self-Interaction on d-Electrons. Computation 2016, 4, 33. https://doi.org/10.3390/computation4030033
AMA Style
Schmidt T, Kümmel S. The Influence of One-Electron Self-Interaction on d-Electrons. Computation. 2016; 4(3):33. https://doi.org/10.3390/computation4030033
Chicago/Turabian StyleSchmidt, Tobias, and Stephan Kümmel. 2016. "The Influence of One-Electron Self-Interaction on d-Electrons" Computation 4, no. 3: 33. https://doi.org/10.3390/computation4030033
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.