Structural Features That Stabilize ZnO Clusters: An Electronic Structure Approach



Abstract
:
1. Introduction
2. Computational Methods
3. Results and Discussion
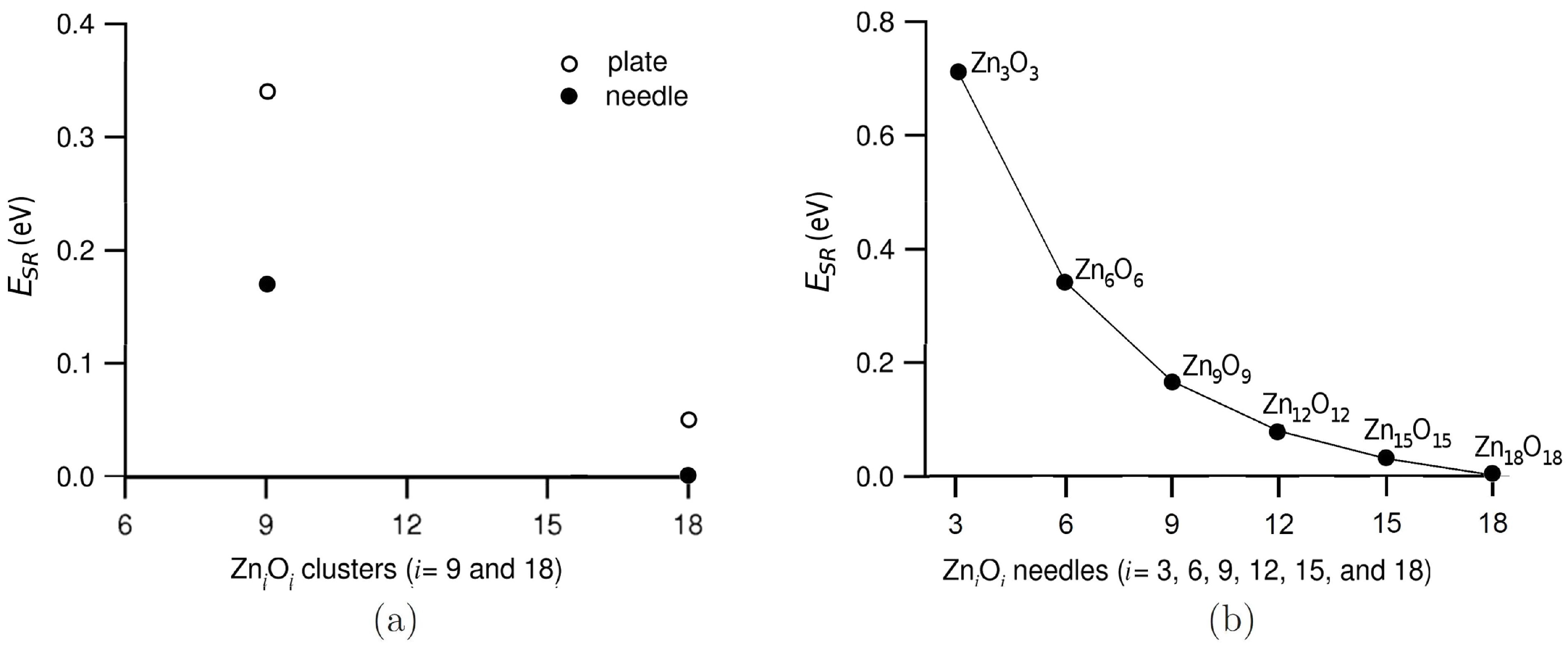
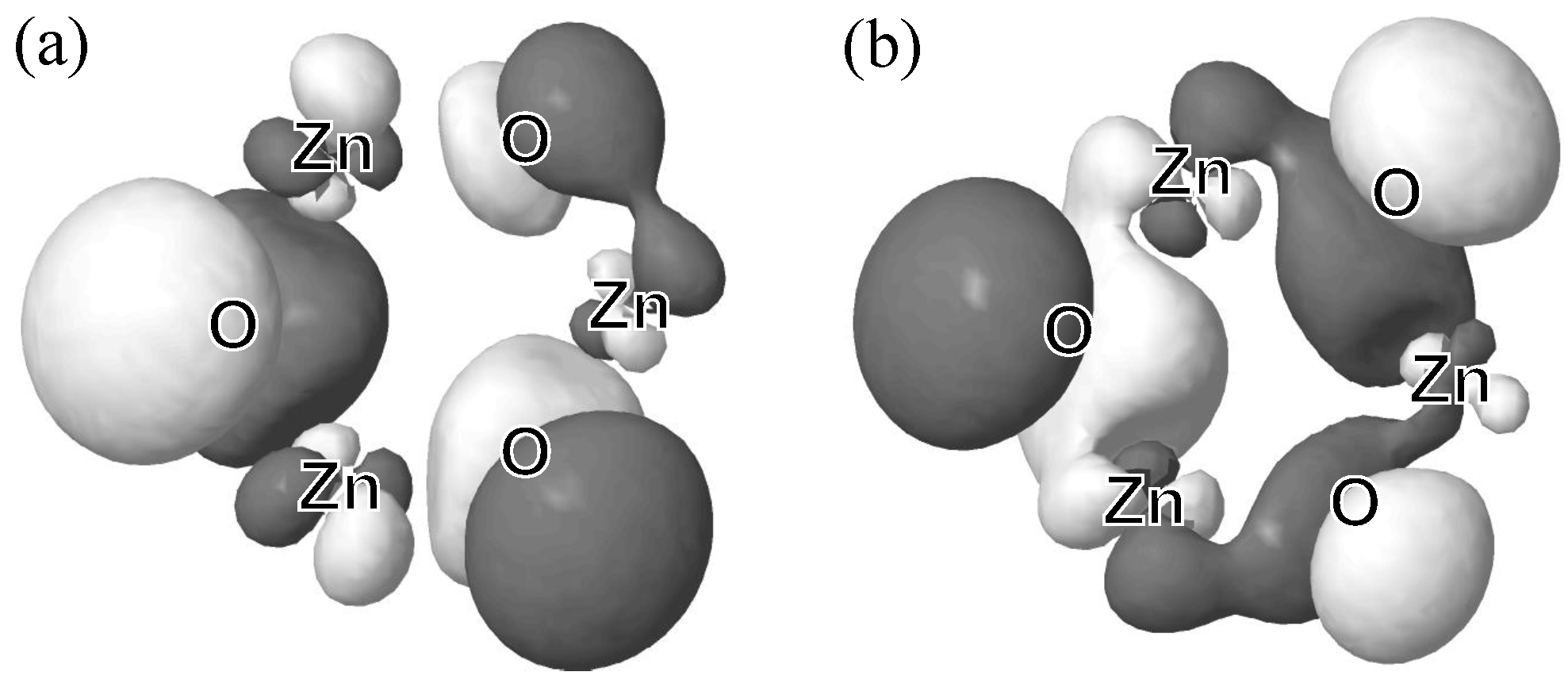
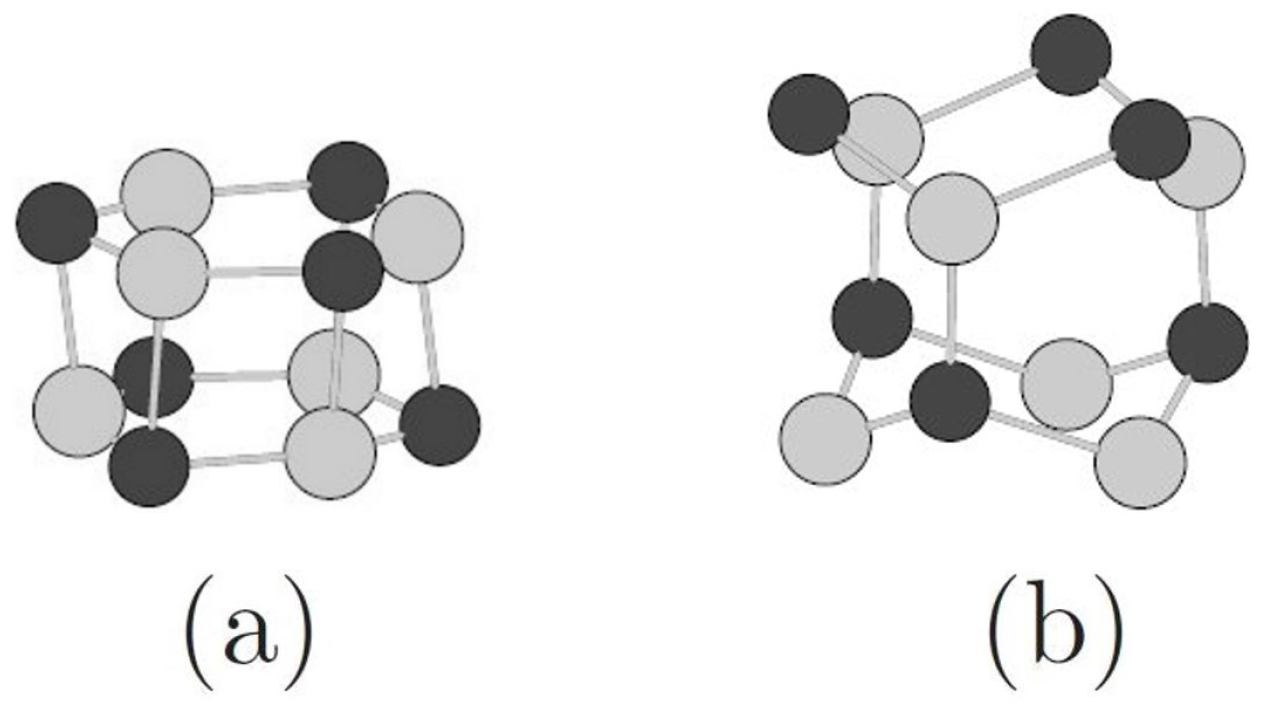
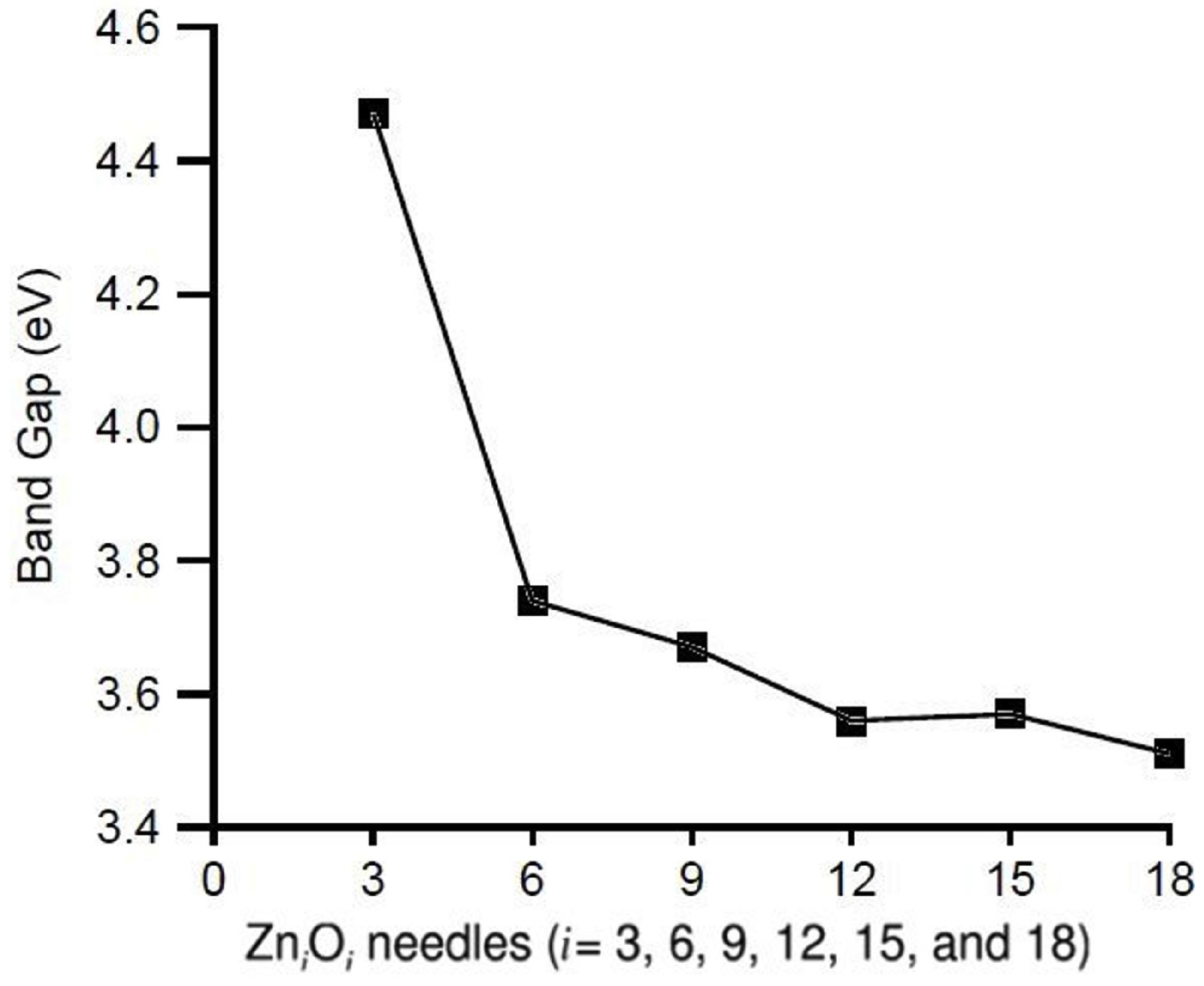
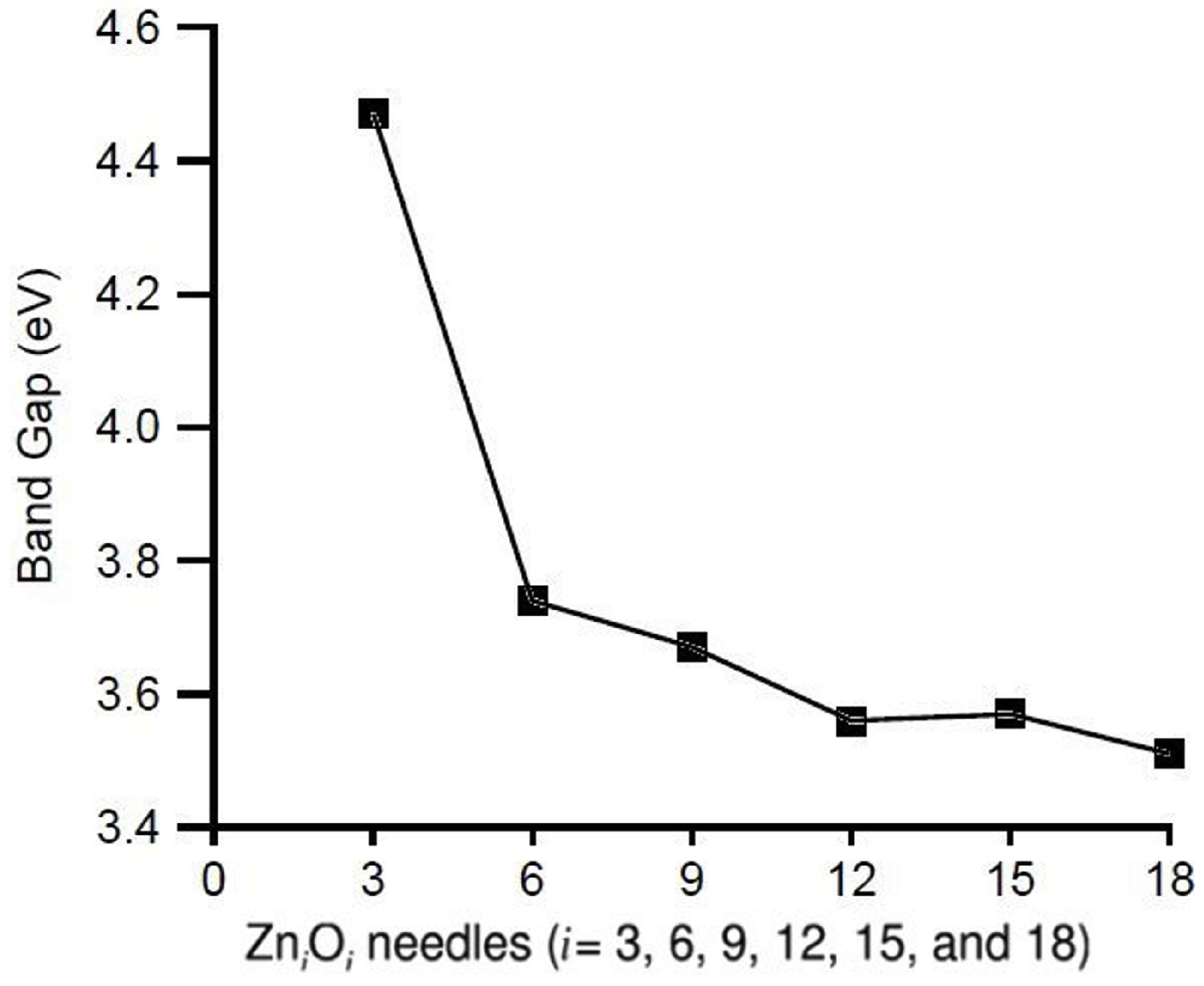
3.1. Relative Crystal Shape Stabilities
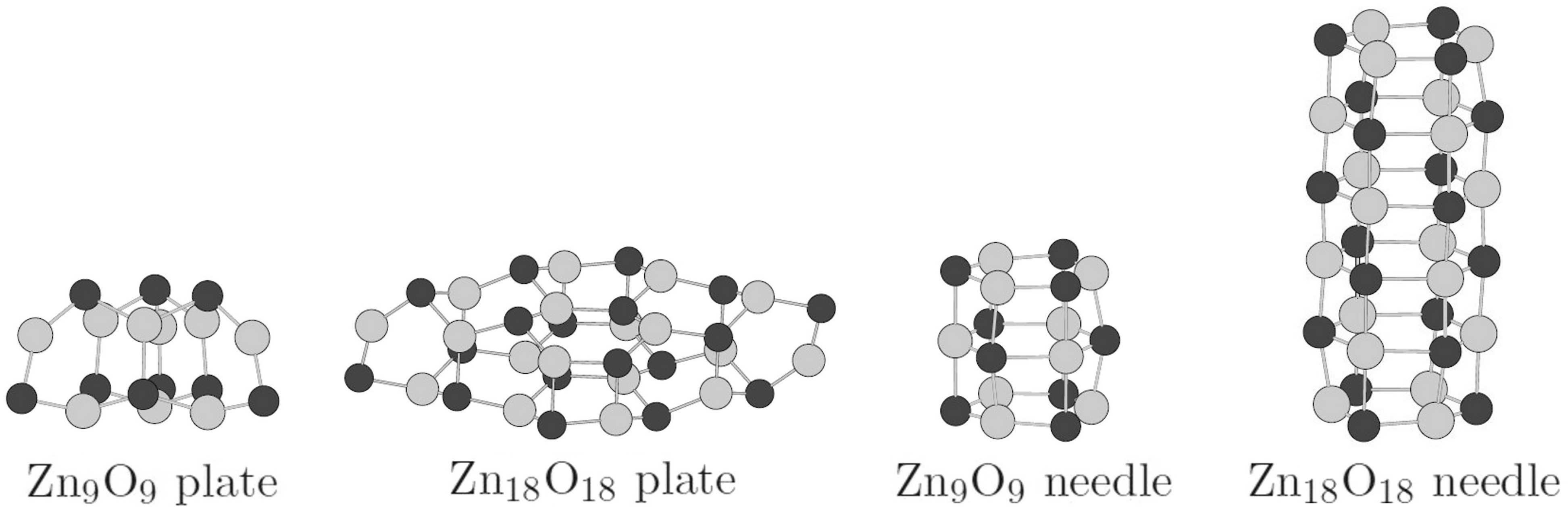
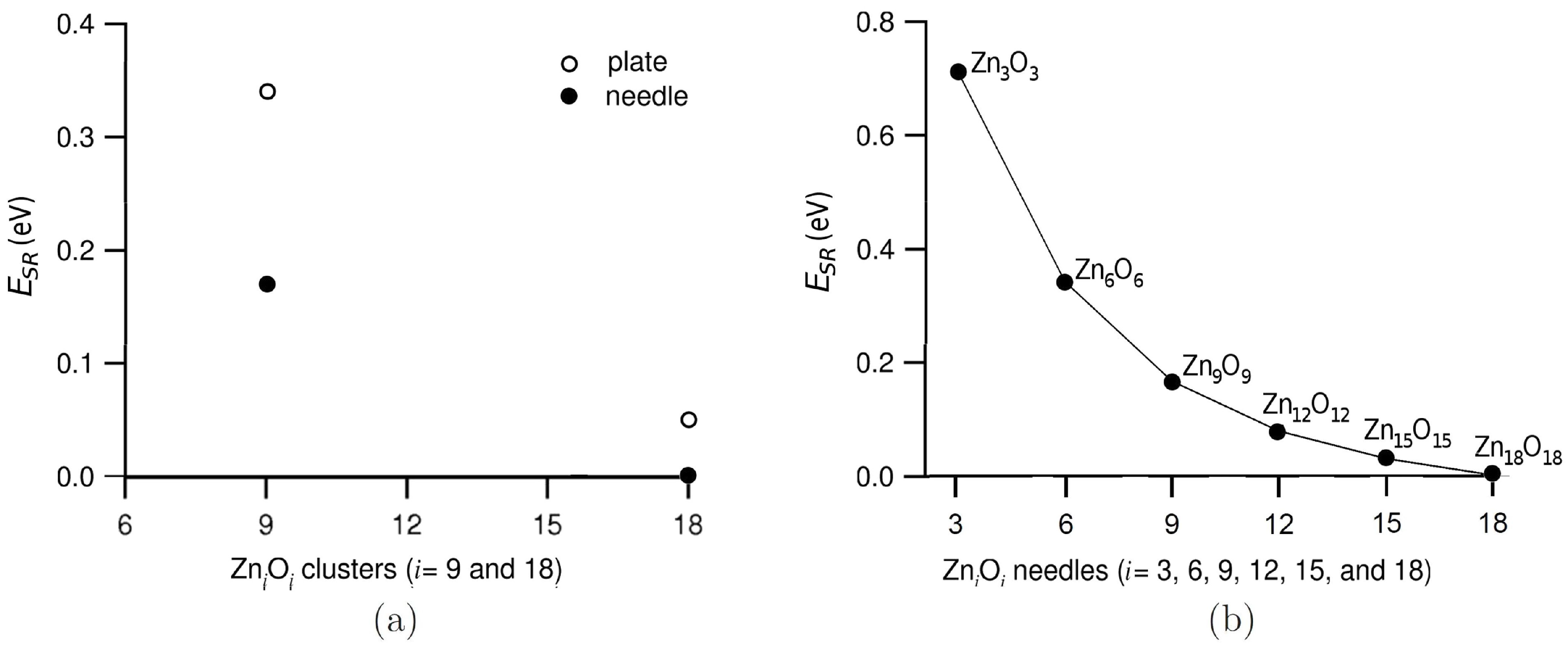


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Needles | OP | OP | |
|---|---|---|---|
| Terminal rings | Middle rings | ||
| ZnO | 0.236 | - | - |
| ZnO | 0.167 | - | 0.170 |
| ZnO | 0.192 | 0.077 | 0.156 |
| ZnO | 0.188 | 0.110 | 0.152 |
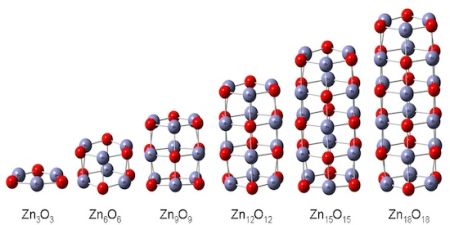
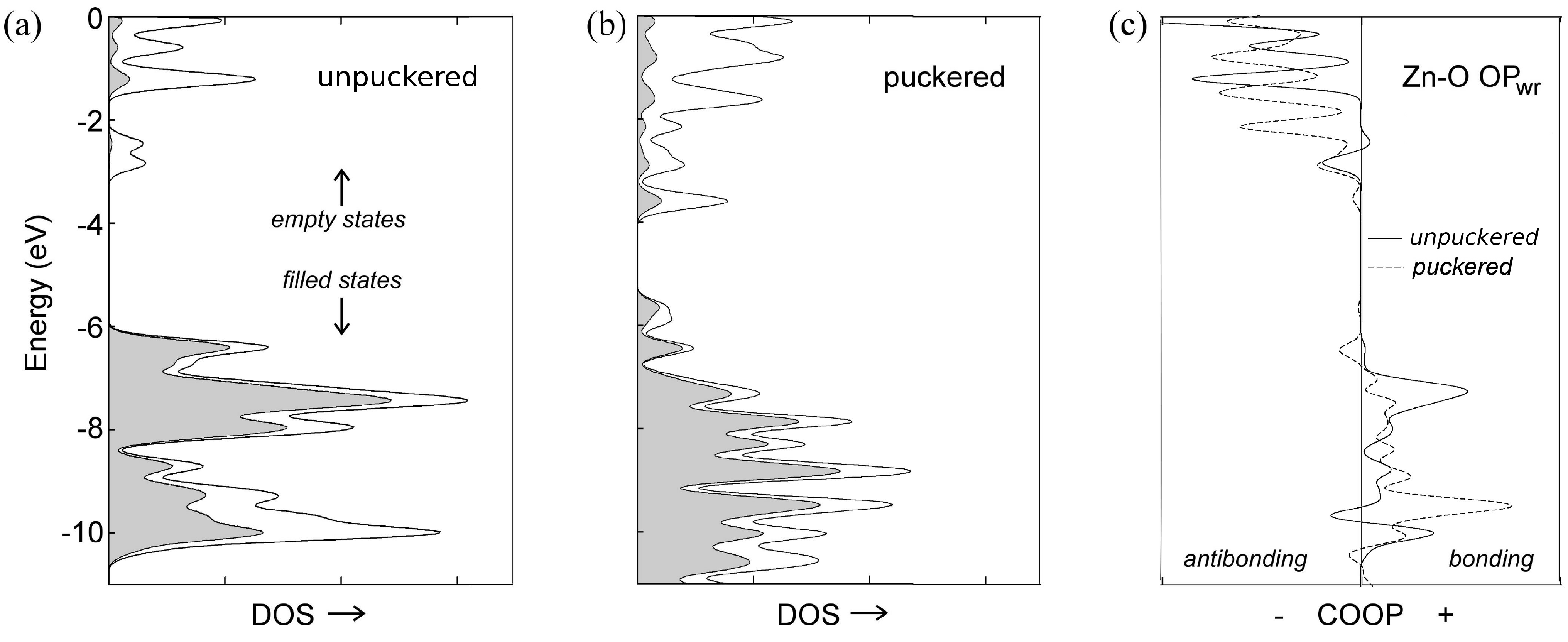
3.2. Toward Wurtzite: Puckering and Extending the Needles
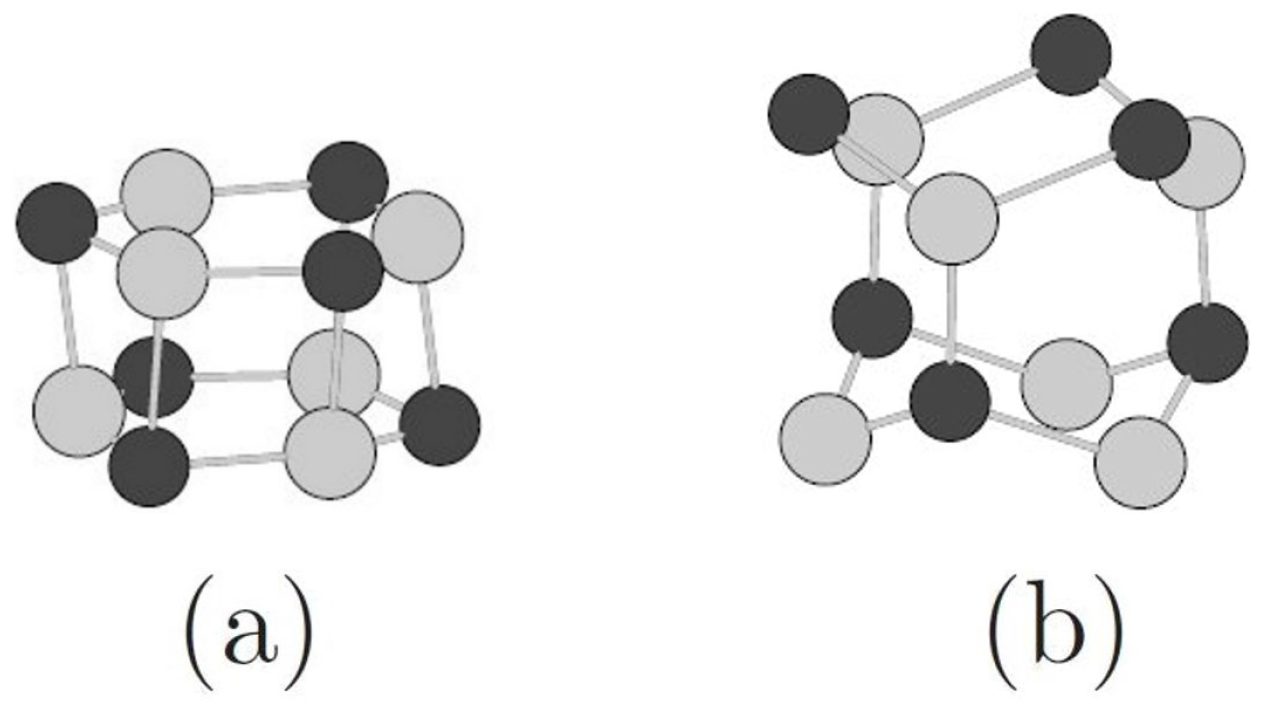
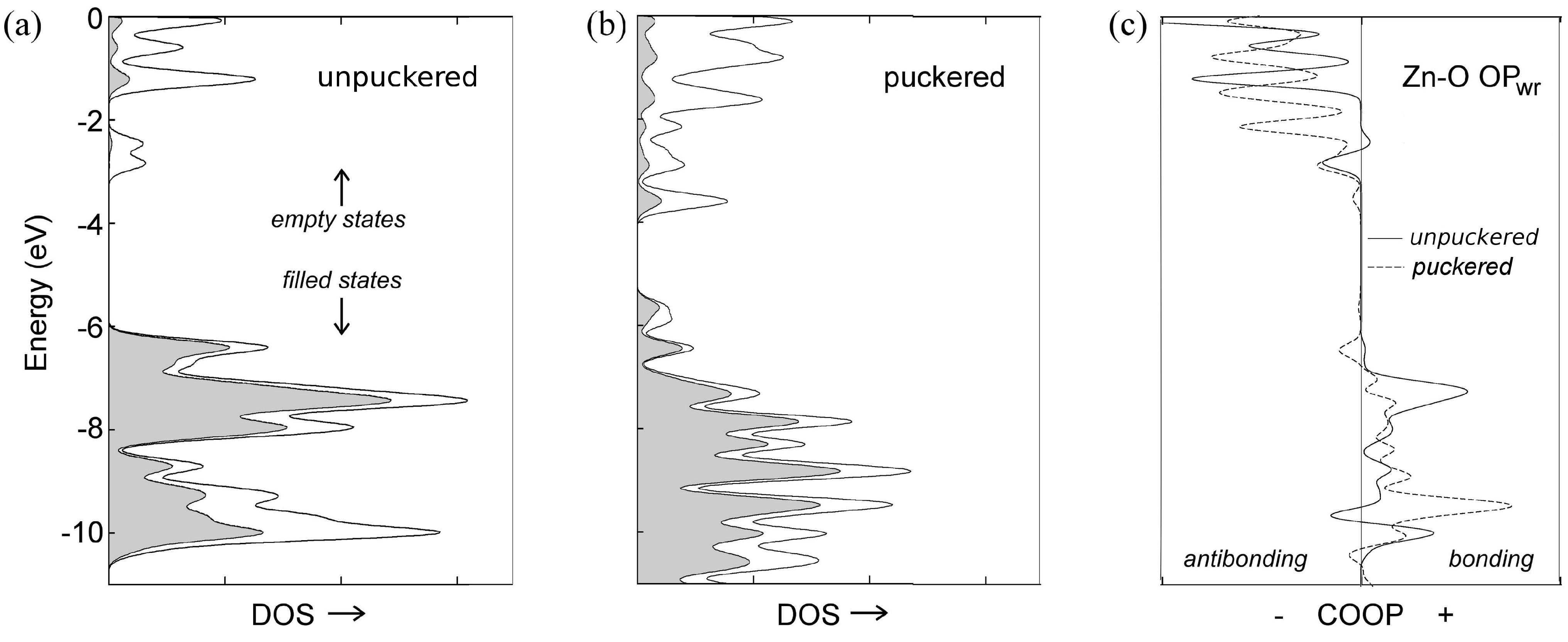
| Needles | ||
|---|---|---|
| unpuckered ZnO | –25224.683 | 0 |
| puckered ZnO | –25224.178 | 0.505 |
| unpuckered [ZnO] | –25224.732 | 0 |
| puckered [ZnO] | –25224.727 | 0.004 |
| Needles | Band Gap (eV) |
|---|---|
| unpuckered ZnO | 1.70 |
| puckered ZnO | 1.67 |
| unpuckered [ZnO] | 1.60 |
| puckered [ZnO] | 1.84 |
4. Conclusions
Acknowledgements
References
- Özgür, U.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Doǧan, S.; Avrutin, V.; Cho, S.J.; Morkoç, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 2005, 98, 041301–041404. [Google Scholar] [CrossRef]
- Brewster, M.M.; Zhou, X.; Lu, M.Y.; Gradec̆ak, S. The interplay of structural and optical properties in individual ZnO nanostructures. Nanoscale 2012, 4, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Cui, J. Zinc oxide nanowires. Mater. Charact. 2012, 64, 43–52. [Google Scholar] [CrossRef]
- Spencer, M.J. Gas sensing applications of 1D-nanostructured zinc oxide: Insights from density functional theory calculations. Prog. Mater. Sci. 2012, 57, 437–486. [Google Scholar] [CrossRef]
- Mowbray, D.J.; Martínez, J.I.; Calle-Vallejo, F.; Rossmeisl, J.; Thygesen, K.S.; Jacobsen, K.W.; Nørskov, J.K. Trends in metal oxide stability for nanorods, nanotubes, and surfaces. J. Phys. Chem. C 2011, 115, 2244–2252. [Google Scholar] [CrossRef]
- Oliva, J.M.; Llunll, M.; Alemany, P.; Canadell, E. Quantitative vs. qualitative approaches to the electronic structure of solids. J. Solid State Chem. 2003, 176, 375–389. [Google Scholar] [CrossRef]
- Lü, X.; Xu, X.; Wang, N.; Zhang, Q.; Ehara, M.; Nakatsuji, H. Cluster modeling of metal oxides: How to cut out a cluster? Chem. Phys. Lett. 1998, 291, 445–452. [Google Scholar] [CrossRef]
- Gunaratne, K.D.D.; Berkdemir, C.; Harmon, C.L.; Castleman, A.W., Jr. Investigating the relative stabilities and electronic properties of small zinc oxide clusters. J. Phys. Chem. A 2012, 116, 12429–12437. [Google Scholar] [CrossRef] [PubMed]
- Caddeo, C.; Malloci, G.; de Angelis, F.; Colombo, L.; Mattoni, A. Optoelectronic properties of (ZnO)60 isomers. Phys. Chem. Chem. Phys. 2012, 14, 14293–14298. [Google Scholar] [CrossRef]
- Malloci, G.; Chiodo, L.; Rubio, A.; Mattoni, A. Structural and optoelectronic properties of unsaturated ZnO and ZnS nanoclusters. J. Phys. Chem. C 2012, 116, 8741–8746. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Huang, S.; Liu, H.; Wang, P.; Tian, H. Theoretical investigation of growth, stability, and electronic properties of beaded ZnO nanoclusters. J. Mater. Chem. 2011, 21, 16905–16910. [Google Scholar] [CrossRef]
- Wang, X.; Wang, B.; Tang, L.; Sai, L.; Zhao, J. What is atomic structures of (ZnO)34 magic cluster. Phys. Lett. A 2010, 374, 850–853. [Google Scholar] [CrossRef]
- Sanyal, B.; Mookerjee, A. Study of the electronic and structural properties of ZnO clusters. Int. J. Mod. Phys. B 2010, 24, 3297–3309. [Google Scholar] [CrossRef]
- Zhao, M.; Xia, Y.; Tan, Z.; Liu, X.; Mei, L. Design and energetic characterization of ZnO clusters from first-principles calculations. Phys. Lett. A 2007, 372, 39–43. [Google Scholar] [CrossRef]
- Azpiroz, J.M.; Mosconi, E.; de Angelis, F. Modeling ZnS and ZnO nanostructures: Structural, electronic, and optical properties. J. Phys. Chem. C 2011, 115, 25219–25226. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Slavetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Matxain, J.M.; Fowler, J.E.; Ugalde, J.M. Small clusters of II-VI materials: ZniOi, i = 1–9. Phys. Rev. A 2000, 62, 053201–053211. [Google Scholar] [CrossRef]
- Muscat, J.; Wander, A.; Harrison, N.M. On the prediction of band gaps from hybrid functional theory. Chem. Phys. Lett. 2001, 342, 397–401. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.J.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jmol: An open-source Java viewer for chemical structures in 3D. Available online: http://www.jmol.org/ (accessed on 29 May 2013).
- Tenderholt, A.L. QMForge, Version 2.1. Available online: http://qmforge.sourceforge.net (accessed on 29 May 2013).
- O’Boyle, N.M.; Tenderholt, A.L.; Lagner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Illy, B.; Shollock, B.A.; MacManus-Driscoll, J.L.; Ryan, M.P. Electrochemical growth of ZnO nanoplates. Nanotechnology 2005, 16, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S.; Raj, A.D.; Mangalaraj, D.; Nataraj, D.; Ponpandian, N.; Li, L.; Chabrol, G. Growth of hierarchical based ZnO micro/nanostructured films and their tunable wettability behavior. Appl. Surf. Sci. 2011, 257, 6678–6686. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Liu, L.; He, C.; Tan, J.; Liang, Y. Morphology-controlled synthesis and growth mechanism of ZnO nanostructures via the NaCl nonaqueous ionic liquid route. CrystEngComm 2012, 14, 4997–5004. [Google Scholar] [CrossRef]
- Gao, S.Y.; Li, H.D.; Yuan, J.J.; Yang, X.X.; Liu, J.W. ZnO nanorods/plates on Si substrate grown by low-temperature hydrothermal reaction. Appl. Surf. Sci. 2010, 256, 2781–2785. [Google Scholar] [CrossRef]
- Wang, J.S.; Yang, C.S.; Chen, P.I.; Su, C.F.; Chen, W.J.; Chiu, K.C.; Chou, W.C. Catalyst-free highly vertically aligned ZnO nanoneedle arrays grown by plasma assisted molecular beam epitaxy. Appl. Phys. A 2009, 97, 553–557. [Google Scholar] [CrossRef]
- Ahn, M.W.; Park, K.S.; Heo, J.H.; Kim, D.W.; Choi, K.J.; Park, J.G. On-chip fabrication of ZnO-nanowire gas sensor with high gas sensitivity. Sens. Actuators B 2009, 138, 168–173. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Chen, Y.; Xiang, Q.; Pan, Q.; Shi, L. Uniform ZnO nanorods can be used to improve the response of ZnO gas sensor. Mater. Sci. Eng. B 2008, 150, 55–60. [Google Scholar] [CrossRef]
- Azpiroz, J.M.; Infante, I.; Lopez, X.; Ugalde, J.M.; de Angelis, F. A first-principles study of II-VI (II = Zn; VI = O, S, Se, Te) semiconductor nanostructures. J. Mater. Chem. 2012, 22, 21453–21465. [Google Scholar] [CrossRef]
- Komsa, H.P.; Broqvist, P.; Pasquarello, A. Alignment of defect levels and band edges through hybrid functionals: Effect of screening in the exchange term. Phys. Rev. B 2010, 81, 205118. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Calculation of semiconductor band gaps with the M06-L density functional. J. Chem. Phys. 2009, 130, 074103. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, Y.H.; Zheng, J.C.; Zhu, Z.Z. Direct to indirect band gap transition in ultrathin ZnO nanowires under uniaxial compression. Appl. Phys. Lett. 2009, 94, 113114:1–113114:3. [Google Scholar] [CrossRef]
- Kumar, S.; Sahare, P. Observation of band gap and surface defects of ZnO nanoparticles synthesized via hydrothermal route at different reaction temperature. Opt. Commun. 2012, 285, 5210–5216. [Google Scholar] [CrossRef]
- Mohanta, K.; Pal, A.J. Diode junctions between two ZnO nanoparticles: Current rectification and the role of particle size (and bandgap). Nanotechnology 2009, 20, 185203. [Google Scholar] [CrossRef] [PubMed]
- Sans, J.A.; Sánchez-Royo, J.F.; Segura, A.; Tobias, G.; Canadell, E. Chemical effects on the optical band-gap of heavily doped ZnO: MIII(M = Al,Ga,In): An investigation by means of photoelectron spectroscopy, optical measurements under pressure, and band structure calculations. Phys. Rev. B 2009, 79, 195105:1–195105:9. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Szakacs, C.E.; Merschrod S., E.F.; Poduska, K.M. Structural Features That Stabilize ZnO Clusters: An Electronic Structure Approach. Computation 2013, 1, 16-26. https://doi.org/10.3390/computation1010016
Szakacs CE, Merschrod S. EF, Poduska KM. Structural Features That Stabilize ZnO Clusters: An Electronic Structure Approach. Computation. 2013; 1(1):16-26. https://doi.org/10.3390/computation1010016
Chicago/Turabian StyleSzakacs, Csaba E., Erika F. Merschrod S., and Kristin M. Poduska. 2013. "Structural Features That Stabilize ZnO Clusters: An Electronic Structure Approach" Computation 1, no. 1: 16-26. https://doi.org/10.3390/computation1010016