
Variations in Microbial Community Structure through the Stratified Water Column in the Tyrrhenian Sea (Central Mediterranean)


 ,
,
Abstract
:1. Introduction
2. Experimental Section
2.1. Sampling Site and Sampling Procedure
2.2. Nucleic Acids Extraction and RT-PCR of Total RNA Templates
2.3. CARD-FISH Analysis

{kind=link}
{kind=link}
| Probe | Sequence (5′ to 3′) of Probe | Target Organisms | % Formamide | References |
|---|---|---|---|---|
| Eub338 | GCTGCCTCCCGTAGGAGT | Bacteria | 55 | (Amann et al., 1990) [29] |
| Cren537 | TGACCACTTGAGGTGCTG | Crenarchaea | 20 | (Teira et al., 2004) [31] |
| Eury806 | CACAGCGTTTACACCTAG | Euryarchaea | 20 | (Teira et al., 2004) [31] |
| Non338 | ACTCCTACGGGAGGCAGC | For non-specific probe binding | 55 | (Wallner et al., 1993) [33] |
2.4. Sequencing and Phylogenetic Analysis
2.5. Statistical Analysis
2.6. Nucleotide Sequence Accession Numbers
3. Results
3.1. Hydrological and Hydrochemical Conditions of the Sampling Site
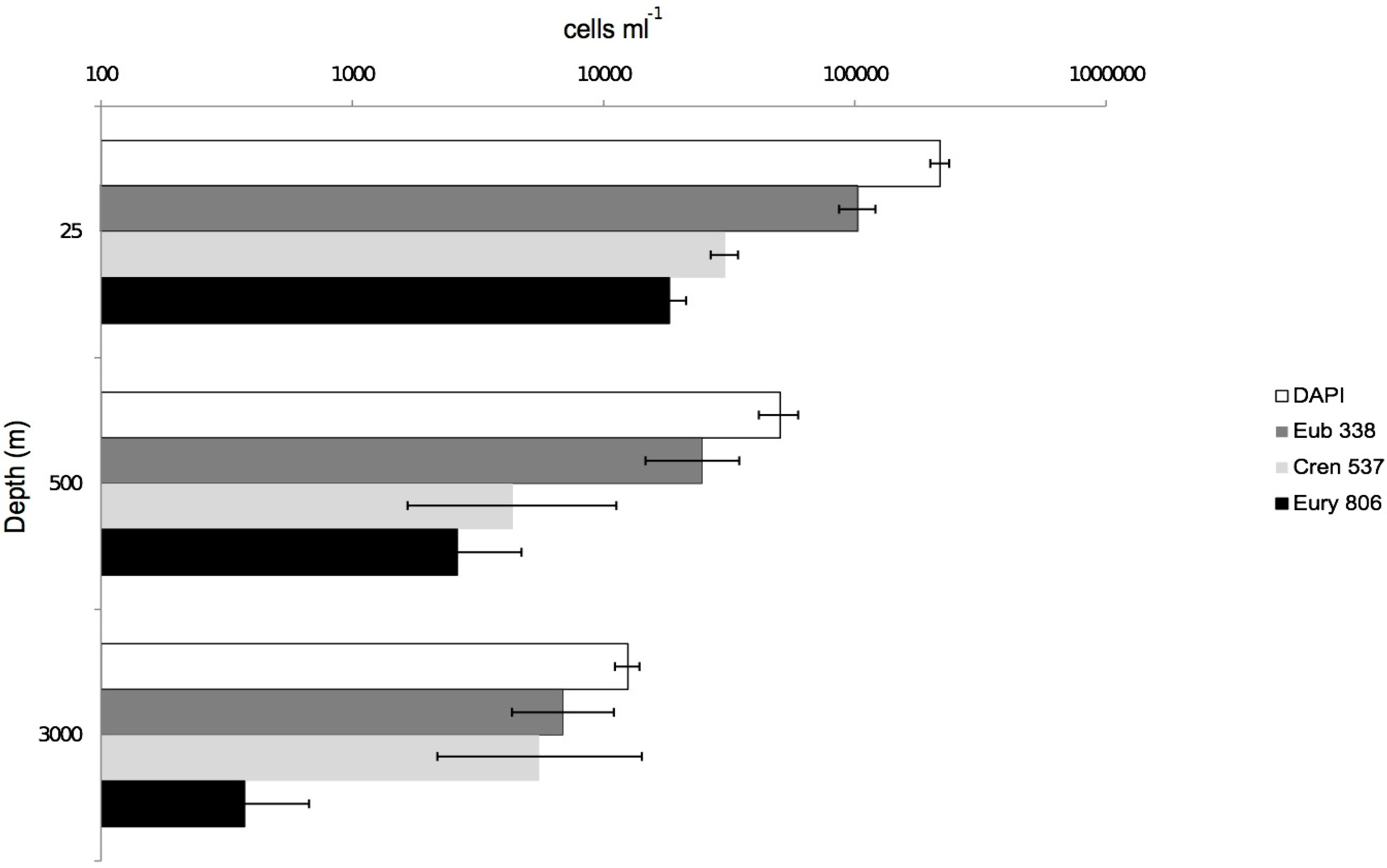
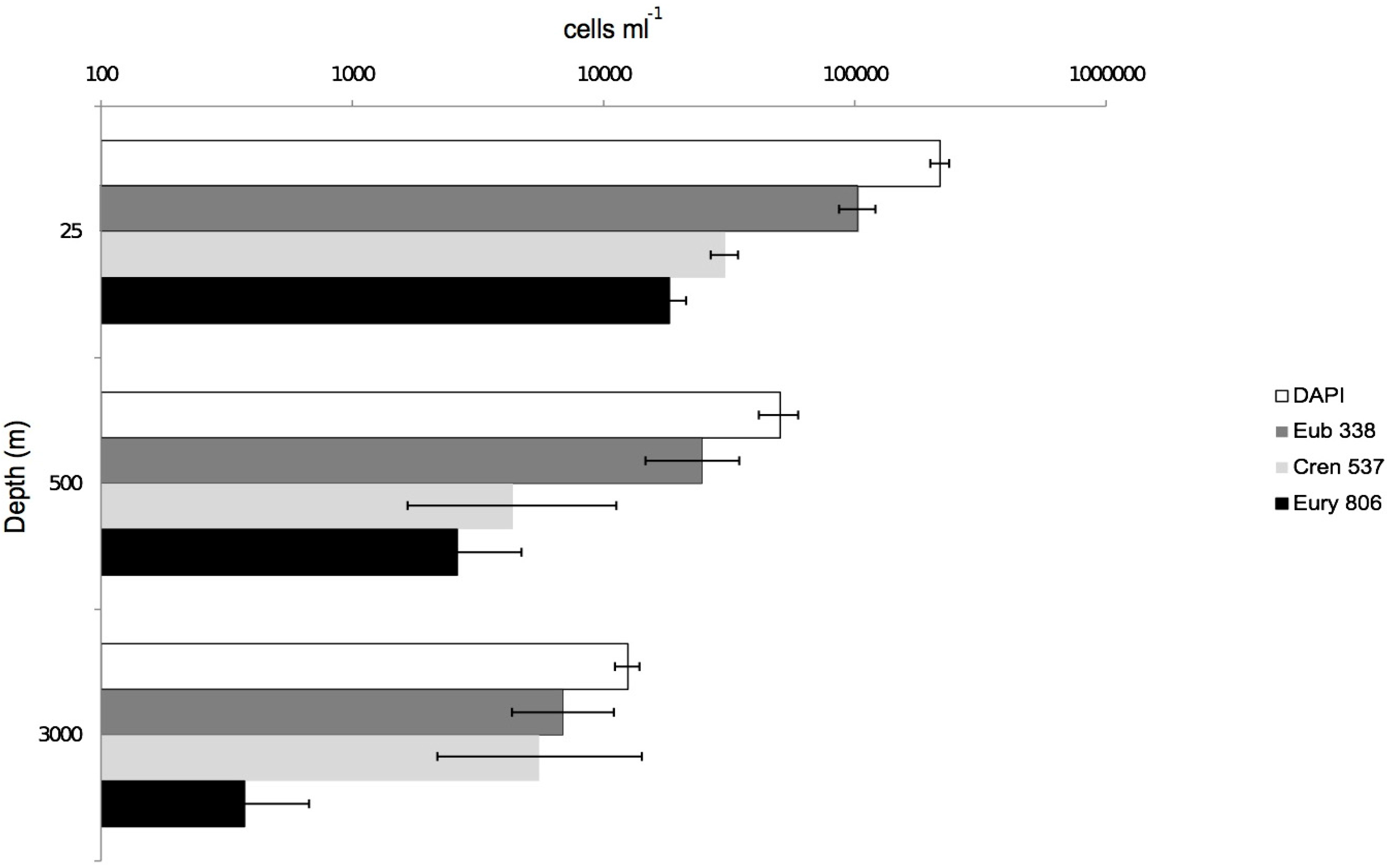
3.2. CARD-FISH Distribution
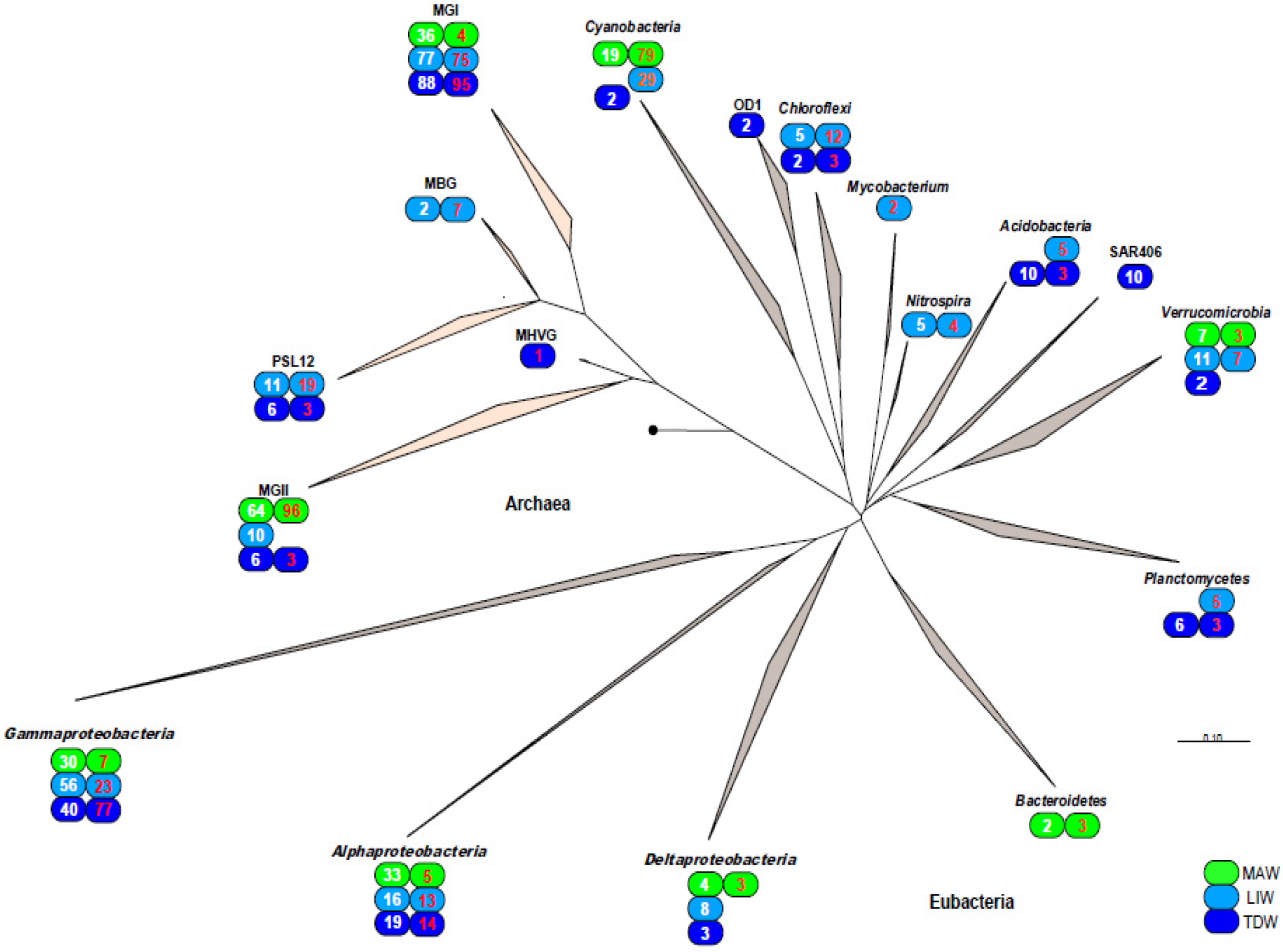
3.3. Phylogenetic Analysis of 16S rDNA and 16S rRNA Bacterial Clones Retrieved from Different Depths
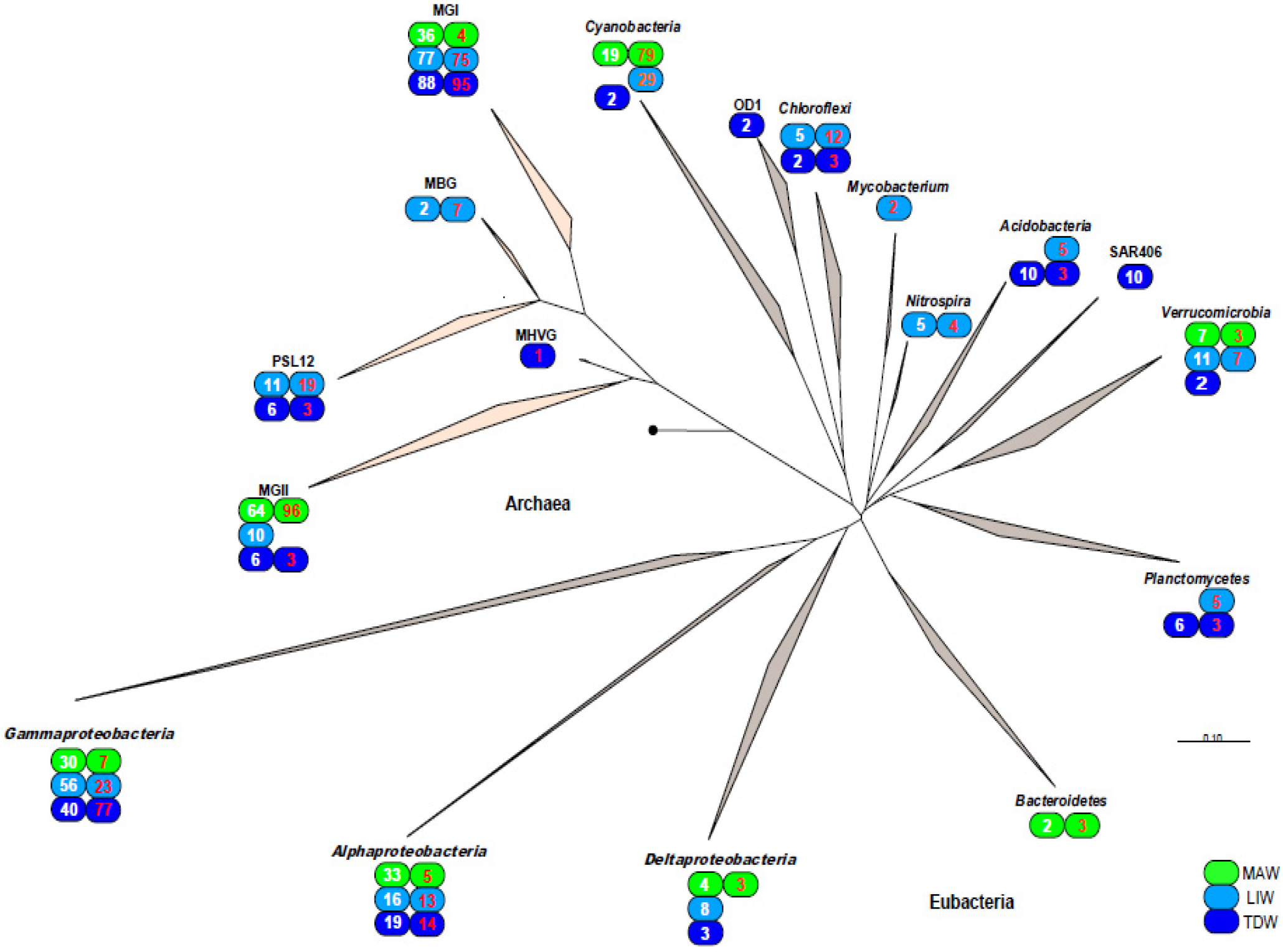
| Bacterial or Archaeal Division | Genus/Clade | Closest Relative (EMBL Accession No.) | % Identity | MAW | LIW | TDW |
|---|---|---|---|---|---|---|
| Gammaproteobacteria | SAR 86 | Clone S23_686 (EF572587) | 99 | 8D-2R | ||
| JL-ENTP-VE | Clone HOT157_350m7 (JN166339.1) | 100 | 2D-2R | |||
| Alteromonadaceae | Alteromonas macleodii str. “Deep ecotype” (CP001103) | 99 | 10D | 3D-17R | ||
| OM60 clade | Clone SHTA403 (GU235460) | 97 | 2D | |||
| Umboniibacter | Umboniibacter marinipuniceus strain: KMM 3891 (AB467279) | 99 | 3D | |||
| Pseudospirillum | Clone HOT157_350m9 (JN166361) | 99 | 2D-2R | 2D | ||
| E01-9C-26 | Clone ASTS_SIM_500m_310 (KJ589919) | 99 | 2D | 5D-1R | 1D | |
| E01-9C-26 | Clone PRTBB84 (HM798879) | 98 | 2D-1R | 1D | ||
| Alphaproteobacteria | SAR 11 | Clone C146Chl358 (JX530113) | 99 | 5D | 3D | |
| SAR 11 | Clone C146300253 (JX530857) | 99 | 2D | 1D | ||
| SAR 11 | Clone PRTAB7655 (HM798613) | 98 | 3D | 3D | 1D | |
| DB1-14 | Clone HglFeb5D7m (JX017019) | 98 | 5D-1R | |||
| AEGEAN-169 | Clone SPOTSFEB02_70m27 (DQ009450) | 97 | 2D | 2D | ||
| AEGEAN-169 | Cone 20162U78 (EU237459) | 99 | 2R | |||
| S25-593 | Clone CTD005-32B-02 (DQ513053) | 99 | 4D-1R | |||
| SAR 116 | Clone 20154U16 (EU237278) | 99 | 5D-1R | |||
| SAR 116 | Clone SPOTSAUG01_5m101 (DQ009267) | 99 | 3D | |||
| Deltaproteobacteria | SAR 324 | Clone SHAB608 (GQ348695) | 99 | 2D-2R | ||
| OM27 | Clone 52-3-11 (JN018836) | 99 | 2D | |||
| Planctomycetes | Clone OS2BR2-03 (JN233146) | 99 | 2R | |||
| Verrucomicrobia | Clone SPOTSMAY03_150m14 (DQ009456) | 98 | 2D | |||
| Acidobacteria | Clone J8P41000_1H01 (GQ351195) | 97 | 2R | 2D-2R | ||
| Nitrospira | Clone HMMVPog-53 (AJ704710) | 95 | 2R | |||
| Chloroflexi | Clone SAR250 (AY534097) | 99 | 2R | |||
| Clone CTD005-79B-02 (AY704386) | 100 | 1R | 1D-2R | |||
| Cyanobacteria | Prochlorococcus | Prochlorococcus marinus str. NATL2A (CP000095) | 100 | 10D-32R | ||
| Synechococcus | Synechococcus sp. RS9920 (AY172830) | 99 | 6D-6R | |||
| Geitlerinema | Geitlerinema sp. A28DM (FJ410907) | 100 | 18R | |||
| Thaumarchaeota | MGI | Clone HF770_015N17 (DQ300532) | 99 | 9D-1R | 2D-2R | |
| MGI | Clone TS_HP_RNAA_69 (AM937105) | 99 | 9D-5R | 8R | ||
| MGI | Clone KM3_193_A03 (KF900772) | 99 | 10D-4R | |||
| MGI | Clone MT793-NA08 (AB193963) | 99 | 2D-6R | |||
| MGI | Clone SC5_2222_T2_E3 (KJ814656) | 99 | 33D-19R | 14D-69R | ||
| MGI | Nitrosopumilus maritimus SCM1 (NR_102913) | 99 | 13D-8R | 1D | ||
| Crenarchaeota | MBG | Clone TS_NB_DNAA_125 (AM937086) | 99 | 1D-4R | ||
| PSL12 | Clone TS_HP_RNAA_67 (AM937090) | 100 | 2R | |||
| PSL12 | Clone TS_NB_RNAA_25 (AM937096) | 100 | 3R | 1D | ||
| PSL12 | Clone TS_NB_DNAA_66 (AM937087) | 99 | 4D-3R | |||
| Euryarchaeota | MGII | Clone HF70_25A12 (DQ156469) | 99 | 4D-10R | ||
| MGII | Clone HF200_89A11 (DQ156453) | 99 | 6D | 1D | ||
| MGII | Clone HF770_037J18 (DQ300558) | 99 | 2D-5R | |||
| MGII | Clone KM3-136-D10 (EF597686) | 99 | 1D-1R | 1D | ||
| MGII | Clone HF4000_48H06 (DQ300561) | 99 | 4D | 2D-2R | ||
| MGII | Clone PD_18 (JX103589) | 98 | 8D-2R |
3.4. Phylogenetic Analysis of 16S rDNA and 16S rRNA Archaeal Clones Retrieved from Different Depths
3.5. Rarefaction Analysis, Diversity Index and Coverage Values of Analysed Clone Libraries
3.6. Cluster Analyses of Clone Libraries
| LIBSHUFF results between bacterial libraries | delta-C | p-value | LIBSHUFF results between archaeal libraries | delta-C | p-value |
|---|---|---|---|---|---|
| MAW 25 m (DNA vs. RNA) | 6.198 | 0.001 | MAW 25 m (DNA vs. RNA) | 0.019 | 0.824 |
| LIW 500 m (DNA vs. RNA) | 3.528 | 0.001 | LIW 500 m (DNA vs. RNA) | 0.511 | 0.002 |
| TDW 3000 m (DNA vs. RNA) | 7.647 | 0.001 | TDW 3000 m (DNA vs. RNA) | 0.090 | 0.037 |
| Bacteria | R statistic | Significance level of sample statistic (%) | Archaea | R statistic | Significance level of sample statistic (%) |
|---|---|---|---|---|---|
| DNA-derived libraries | 0.048 | 0.8 | DNA-derived libraries | 0.378 | 0.1 |
| MAW, LIW(D) | 0.053 | 0.3 | MAW, LIW (D) | 0.446 | 0.1 |
| MAW, TDW(D) | 0.077 | 0.5 | MAW, TDW (D) | 0.470 | 0.1 |
| LIW, TDW (D) | 0.000 | 46.4 | LIW, TDW (D) | −0.007 | 57.2 |
| RNA-derived libraries | 0.302 | 0.1 | RNA-derived libraries | 0.476 | 0.1 |
| MAW, LIW (R) | 0.311 | 0.1 | MAW, LIW (R) | 0.684 | 0.1 |
| MAW, TDW (R) | 0.409 | 0.1 | MAW, TDW (R) | 0.842 | 0.1 |
| LIW, TDW (R) | 0.037 | 15.2 | LIW, TDW (R) | 0.039 | 0.9 |
4. Discussion
5. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gasol, J.M.; Del Giorgio, P.A.; Duarte, C. Biomass distribution in marine planktonic communities. Limnol. Oceanogr. 1997, 42, 1353–1363. [Google Scholar] [CrossRef]
- Azam, F. Microbial control of oceanic carbon flux: The plot thickens. Science 1998, 280, 694–696. [Google Scholar] [CrossRef]
- Pedros-Alio, C. Marine microbial diversity: Can it be determined? Trends Microbiol. 2006, 14, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, M.T.; Kirchman, D.L. Contribution of major bacterial groups to bacterial biomass production (thymidine and leucine incorporation) in the Delaware estuary. Limnol. Oceanogr. 2003, 48, 168–178. [Google Scholar] [CrossRef]
- Venter, J.; Remington, K.; Heidelberg, J.; Halpern, A.; Rusch, D.; Eisen J Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; Fouts, D.E.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Quaiser, A.; Zivanovic, Y.; Moreira, D.; López-García, P. Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J. 2011, 5, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Smedile, F.; Messina, E.; La Cono, V.; Yakimov, M.M. Comparative analysis of deep-sea bacterioplankton OMICS revealed the occurrence of habitat-specific genomic attributes. Mar. Genom. 2014, 17, 1–8. [Google Scholar] [CrossRef]
- Martín-Cuadrado, A.B.; López-García, P.; Alba, J.C.; Moreira, D.; Monticelli, L.; Strittmatter, A.; Gottschalk, G.; Rodríguez-Valera, F. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS ONE 2007, 2, e914. [Google Scholar] [CrossRef] [PubMed]
- Smedile, F.; Messina, E.; La Cono, V.; Tsoy, O.; Monticelli, L.S.; Borghini, M.; Giuliano, L.; Golyshin, P.N.; Mushegian, A.; Yakimov, M.M. Metagenomic analysis of hadopelagic microbial assemblages thriving at the deepest part of Mediterranean Sea, Matapan-Vavilov Deep. Environ. Microbiol. 2013, 15, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, T.; De Corte, D.; Sintes, E.; Herndl, G.J. Spatial patterns of bacterial abundance, activity and community composition in relation to water masses in the eastern Mediterranean Sea. Aquat Microb. Ecol. 2010, 59, 185–195. [Google Scholar] [CrossRef]
- Techtmann, S.M.; Fortney, J.L.; Ayers, K.A.; Joyner, D.C.; Linley, T.D.; Pfiffner, S.M.; Hazen, T.C. The Unique Chemistry of Eastern Mediterranean Water Masses Selects for Distinct Microbial Communities by Depth. PLoS ONE 2015, 10, e0120605. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, D.; van Sebille, E.; Rintoul, S.R.; Lauro, F.M.; Cavicchioli, R. Advection shapes Southern Ocean microbial assemblages independent of distance and environment effects. Nat. Commun. 2013, 4, 2457. [Google Scholar] [CrossRef] [PubMed]
- Alves Junior, N.; Meirelles, P.; de Oliveira Santos, E.; Dutilh, B.; Silva, G.Z.; Paranhos, R.; Cabral, A.S.; Rezende, C.; Iida, T.; de Moura, R.L.; et al. Microbial community diversity and physical–chemical features of the Southwestern Atlantic Ocean. Arch. Microbiol. 2014, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.M.; van Aken, H.M.; Sintes, E.; Herndl, G.J. Latitudinal trends of Crenarchaeota and Bacteria in the meso- and bathypelagic water masses of the Eastern North Atlantic. Environ. Microbiol. 2008, 10, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Galand, P.E.; Lovejoy, C.; Hamilton, A.K.; Ingram, R.G.; Pedneault, E.; Carmack, E.C. Archaeal diversity and a gene for ammonia oxidation are coupled to oceanic circulation. Environ. Microbiol. 2009, 11, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Galand, P.E.; Casamayor, E.O.; Kirchman, D.L.; Potvin, M.; Lovejoy, C. Unique archaeal assemblages in the Arctic Ocean unveiled by massively parallel tag sequencing. ISME J. 2009, 3, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krom, M.D.; Emeis, K.C.; Van Cappellen, P. Why is the Eastern Mediterranean phosphorus limited? Prog. Oceanogr. 2010, 85, 236–244. [Google Scholar] [CrossRef]
- La Cono, V.; Tamburini, C.; Genovese, L.; La Spada, G.; Denaro, R.; Yakimov, M.M. Cultivation-independent assessment of the bathypelagic archaeal diversity of Tyrrhenian Sea: Comparative study of rDNA and rRNA-derived libraries and influence of sample decompression. Deep Sea Res. II Top. Studies Oceanogr. 2009, 56, 768–773. [Google Scholar] [CrossRef]
- Ribera d’Alcalà, M.; Brunet, C.; Conversano, F.; Corato, F.; Lavezza, R. Nutrient and pigment distributions in the southern Tyrrhenian Sea during mid-summer and late fall 2005. Deep Sea Res. II Top. Studies Oceanogr. 2009, 56, 676–686. [Google Scholar] [CrossRef]
- Yakimov, M.M.; La Cono, V.; Smedile, F.; Ferrer, M.; Golyshin, P.N.; Giuliano, L. Contribution of crenarchaeal autotrophic ammonia oxidizers to the dark primary production in Tyrrhenian Deep Waters (Central Mediterranean Sea). ISME J. 2011, 5, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Millot, C.; Taupier-Lepage, I. Circulation in the Mediterranean Sea. In The Handbook of Environmental Chemistry; Saliot, A., Ed.; Springer: Ouverney, Germany, 2005; pp. 29–66. [Google Scholar]
- Carpenter, J.H. The accuracy of the Winkler method for dissolved oxygen analysis. Limnol. Oceanogr. 1965, 10, 135–140. [Google Scholar] [CrossRef]
- Yakimov, M.M.; La Cono, V.; Smedile, F.; Crisafi, F.; Arcadi, E.; Leonardi, M.; Decembrini, F.; Catalfamo, M.; Bargiela, R.; Ferrer, M.; et al. Heterotrophic bicarbonate assimilation is the main process of de novo organic carbon synthesis in hadal zone of the Hellenic Trench, the deepest part of Mediterranean Sea. Environ. Microbiol. Rep. 2014, 6, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Cauwet, G.; Minster, J.F. Dissolved organic carbon in the 825 equatorial Atlantic Ocean. Mar. Chem. 1995, 49, 155–169. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackerbrandt, E., Goodfellow, M., Eds.; Wiley: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Hallam, S.J.; Girguis, P.R.; Preston, C.M.; Richardson, P.M.; De Long, E.F. Identification of methyl coenzyme MreductaseA (mcrA) genes associated with methane-oxidizingarchaea. Appl. Environ. Microbiol. 2003, 69, 5483–5491. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.; Ludwig, W.; Schleifer, K. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [PubMed]
- Tamburini, C. Life under Pressure: Deep-Sea Microbial Ecology. In Life as we know it. Series: Cellular Origin and Life in Extreme Habitats and Astrobiology; Springer: Dordrecht, The Netherlands, 2006; pp. 125–143. [Google Scholar]
- Teira, E.; Reinthaler, T.; Pernthaler, A.; Pernthaler, J.; Herndl, G.J. Combining catalyzed reporter deposition-fluorescence in situ hybridization and microautoradiography to detect substrate utilization by bacteria and Archaea in the deep ocean. Appl. Environ. Microbiol. 2004, 70, 4411–4414. [Google Scholar] [CrossRef] [PubMed]
- Pernthaler, A.; Pernthaler, J.; Amann, R. Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl. Environ. Microbiol. 2002, 68, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Wallner, G.; Amann, R.; Beisker, W. Optimizing fluorescent in situ hybridization of suspended cells with rRNA-targeted oligonucleotide probes for the flow cytometric identification of microorganisms. Cytometry 1993, 14, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.; Faulkner, G.; Hugenholtz, P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 2004, 20, 2317–2319. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar; Buchner, A.; Lai, T.; Steppi, S.; Jobb, G.; et al. ARB: A software environment for sequence data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.R.; Furlong, M.A.; Rathbun, S.L.; Whitman, W.B. Quantitative comparisons of 16S rDNA sequence libraries from environmental samples. Appl. Environ. Microbiol. 2001, 67, 4373–4376. [Google Scholar] [CrossRef]
- Gentile, G.; Giuliano, L.; D’Auria, G.; Smedile, F.; Azzaro, M.; De Domenico, M.; Yakimov, M.M. Study of bacterial communities in Antarctic coastal waters by a combination of 16S rRNA and 16S rDNA sequencing. Environ. Microbiol. 2006, 8, 2150–2161. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S. Evolutionary biology: Oceans of bacteria. Nature 2004, 430, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.F.; Aller, J.Y. Bacterial diversity in aquatic and other environments: What 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 2004, 47, 161–177. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Good, I.J. The population frequencies of species and the estimation of the population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.N. PRIMER v6: User Manual/Tutorial; PRIMER-E Ltd.: Plymouth, UK, 2006. [Google Scholar]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Budillon, G.; Gasparini, G.P.; Schröder, K. Persistence of an eddy signature in the central Tyrrhenian basin. Deep Sea Res. II Top. Studies Oceanogr. 2009, 56, 713–724. [Google Scholar] [CrossRef]
- Romanenko, L.A.; Tanaka, N.; Frolova, G.M. Umboniibacter marinipuniceus gen. nov., sp. nov., a marine gammaproteobacterium isolated from the mollusc Umbonium costatum from the Sea of Japan. Int. J. Syst. Evol. Microbiol. 2010, 60, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Garczarek, L.; Dufresne, A.; Rousvoal, S.; West, N.J.; Mazard, S.; Marie, D.; Claustre, H.; Raimbault, P.; Post, A.F.; Scanlan, D.J.; et al. High vertical and low horizontal diversity of Prochlorococcus ecotypes in the Mediterranean Sea in summer, FEMS Microbiol. Ecol. 2007, 60, 189–206. [Google Scholar] [CrossRef]
- Margheri, M.C.; Piccard, R.; Ventura, S.; Viti, C.; Giovannetti, L. Genotypic diversity of Oscillatoriacean strains belonging to the genera Geitlerinema and Spirulina determined by 16S rDNA restriction analysis. Curr. Microbiol. 2003, 46, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Zaikova, E.; Howes, C.G.; Song, Y.C.; Wright, J.J.; Tringe, S.G.; Tortell, P.D.; Hallam, S.J. Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science 2009, 23, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, C.; Garel, M.; Al Ali, B.; Mérigot, B.; Kriwy, P.; Charrière, B.; Budillon, G. Distribution and activity of Bacteria and Archaea in the different water masses of the Tyrrhenian Sea. Deep Sea Res. II Top. Studies Oceanogr. 2009, 56, 700–712. [Google Scholar] [CrossRef]
- Zaballos, M.; Lopez-Lopez, A.; Ovreas, L.; Bartual, S.G.; D’Auria, G.; Alba, J.C.; Legault, B.; Pushker, R.; Daae, F.L.; Rodríguez-Valera, F. Comparison of prokaryotic diversity at offshore oceanic locations reveals a different microbiota in the Mediterranean Sea. FEMS. Microbiol. Ecol. 2006, 56, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A.; Davis, A.A. Widespread Archaea and novel Bacteria from the deep sea as shown by 16S rRNA gene sequences. Mar. Ecol. Prog. Ser. 1997, 150, 275–285. [Google Scholar] [CrossRef]
- Massana, R.; De Long, E.F.; Pedrós-Alió, C. A few cosmopolitan phylotypes dominate planktonic archaeal assemblages in widely different oceanic provinces. Appl. Environ. Microbiol. 2000, 66, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Statist. 1984, 11, 265–270. [Google Scholar]
- Fuller, N.J.; Marie, D.; Partensky, F.; Vaulot, D.; Post, A.F.; Scalan, D.J. Clade-specific 16S rDNA oligonucleotides reveal the predominance of a single marine Synechococcus clade throughout a stratified water column in the Red Sea. Appl. Environ. Microbiol. 2003, 69, 2430–2443. [Google Scholar] [CrossRef] [PubMed]
- Tittel, J.; Bissinger, V.; Zippel, B.; Gaedke, U.; Bell, E.; Lorke, A.; Kamjunke, N. Mixotrophs combine resource use to outcompete specialists: Implications for aquatic food webs. Proc. Natl. Acad. Sci. 2003, 100, 12776–12781. [Google Scholar] [CrossRef] [PubMed]
- Allers, E.; Wright, J.J.; Konwar, K.M.; Howes, C.G.; Beneze, E.; Hallam, S.J.; Sullivan, M.B. Diversity and population structure of Marine Group A bacteria in the Northeast subarctic Pacific Ocean. ISME J. 2013, 7, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Borin, S.; Brusetti, L.; Mapelli, F.; D’Auria, G.; Brusa, T.; Marzorati, M.; Rizzi, A.; Yakimov, M.; Martyd, D.; De Lange, G.J.; et al. Sulfur cycling and methanogenesis primarily drive microbial colonization of the highly sulfidic Urania deep hypersaline basin. Proc. Natl. Acad. Sci. 2009, 106, 9151–9156. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Taylor, L.T.; Marsh, T.L.; Preston, C.M. Visualization and enumeration of marine planktonic archaea and bacteria by using polynucleotide probes and fluorescence in situ hybridization. Appl. Environ. Microbiol. 1999, 65, 5554–5563. [Google Scholar] [PubMed]
- Konstantinidis, K.T.; Braff, J.; Karl, D.M.; DeLong, E.F. Comparative metagenomic analysis of a microbial community residing at a depth of 4000 meters at station ALOHA in the North Pacific subtropical gyre. Appl. Environ. Microbiol. 2009, 75, 5345–5355. [Google Scholar] [CrossRef] [PubMed]
- Feller, I.C.; Whigham, D.F.; Mckee, K.L.; Lovelock, C.E. Nitrogen limitation of growth and nutrient dynamics in adisturbed man grove forest, Indian River Lagoon, Florida. Oecologia 2003, 134, 405–414. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, S.; Marx, J.C.; Gerday, C.; Feller, G. Activity-stability relationships in extremophilic enzymes. J. Biol. Chem. 2003, 278, 7891–7896. [Google Scholar] [CrossRef] [PubMed]
- Greaves, R.B.; Warwicker, J. Mechanisms for stabilisation and the maintenance of solubility in proteins from thermophiles. BMC. Struct. Biol. 2007, 7, 18. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smedile, F.; Scarfi, S.; De Domenico, E.; Garel, M.; Glanville, H.; Gentile, G.; Cono, V.L.; Tamburini, C.; Giuliano, L.; Yakimov, M. Variations in Microbial Community Structure through the Stratified Water Column in the Tyrrhenian Sea (Central Mediterranean). J. Mar. Sci. Eng. 2015, 3, 845-865. https://doi.org/10.3390/jmse3030845
Smedile F, Scarfi S, De Domenico E, Garel M, Glanville H, Gentile G, Cono VL, Tamburini C, Giuliano L, Yakimov M. Variations in Microbial Community Structure through the Stratified Water Column in the Tyrrhenian Sea (Central Mediterranean). Journal of Marine Science and Engineering. 2015; 3(3):845-865. https://doi.org/10.3390/jmse3030845
Chicago/Turabian StyleSmedile, Francesco, Simona Scarfi, Emilio De Domenico, Marc Garel, Helen Glanville, Gabriella Gentile, Violetta La Cono, Cristian Tamburini, Laura Giuliano, and Michail Yakimov. 2015. "Variations in Microbial Community Structure through the Stratified Water Column in the Tyrrhenian Sea (Central Mediterranean)" Journal of Marine Science and Engineering 3, no. 3: 845-865. https://doi.org/10.3390/jmse3030845