Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP)


Abstract
:
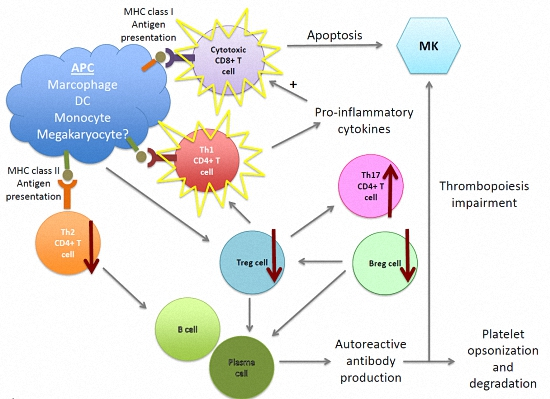{kind=link}
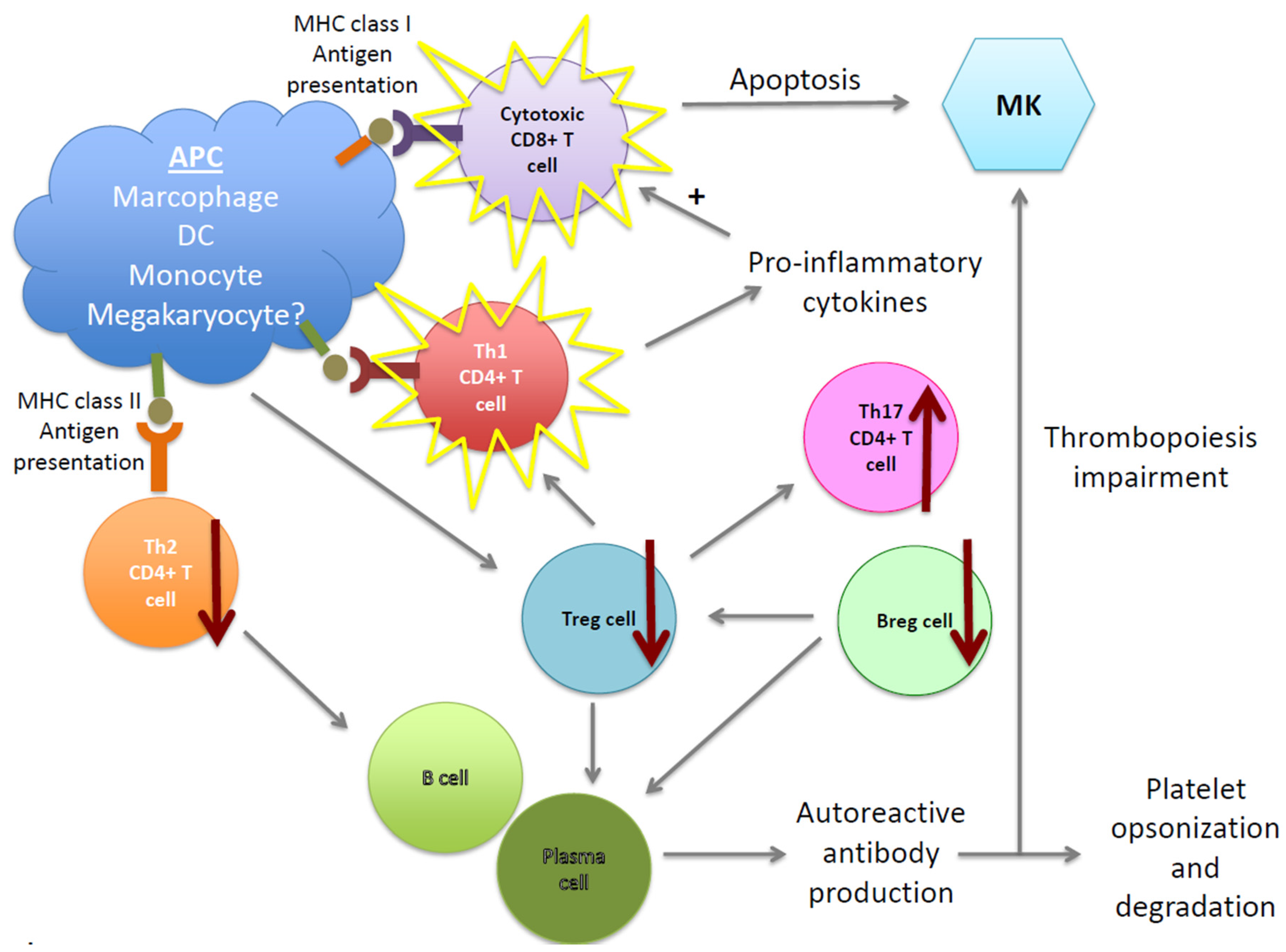{kind=link}
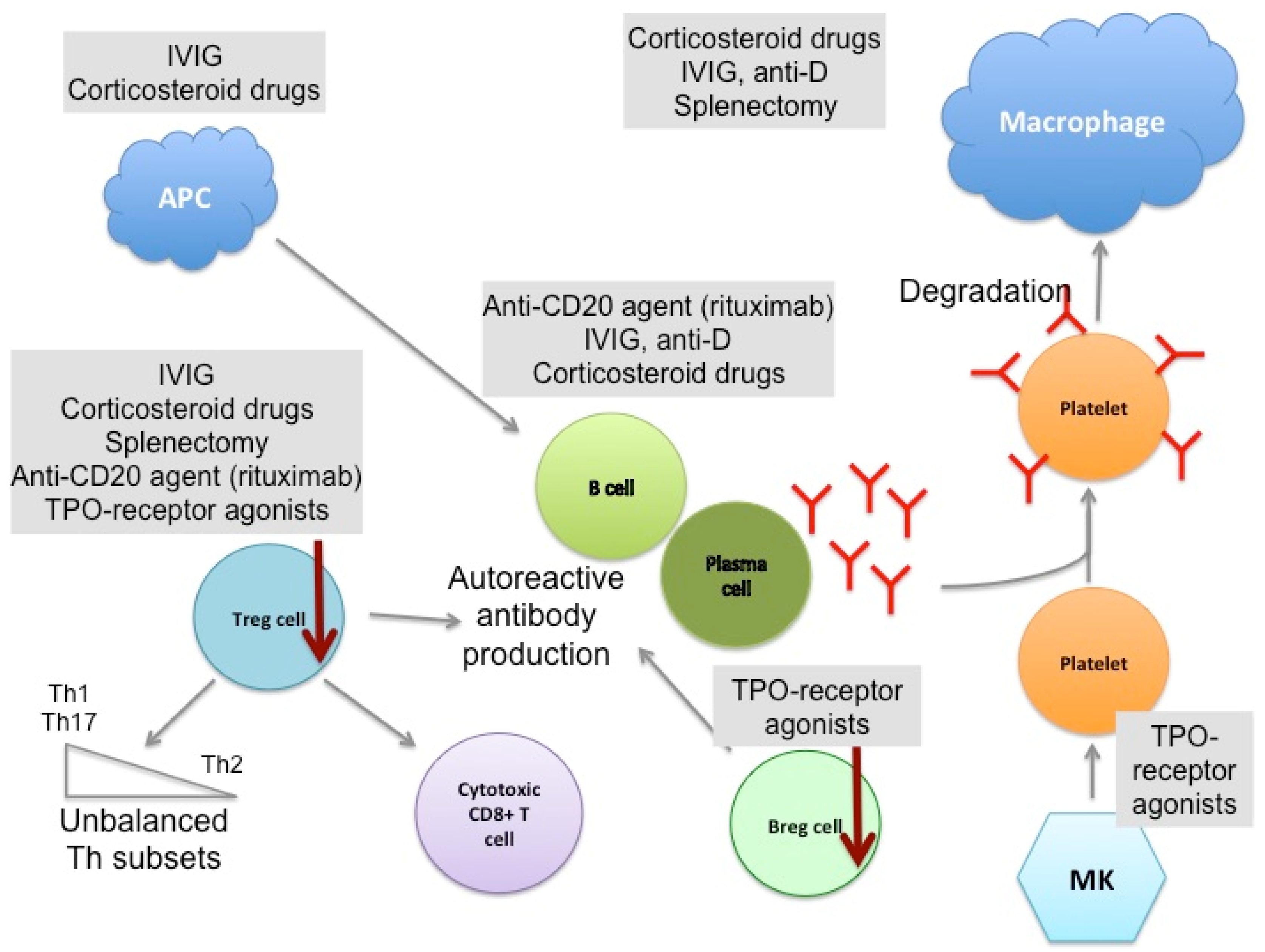{kind=link}
1. Introduction
2. Molecular and Cellular Mechanisms of the Pathogenesis of ITP
2.1. B Cells and Autoantibodies
2.2. T-Cell Imbalance in ITP
2.3. Dendritic Cells in ITP
2.4. Megakaryocytes in ITP
3. Therapies of ITP
3.1. First-Line Treatments
3.2. Second-Line Treatments
3.3. Third-Line Treatment
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rodeghiero, F.; Stasi, R.; Gernsheimer, T.; Michel, M.; Provan, D.; Arnold, D.M.; Bussel, J.B.; Cines, D.B.; Chong, B.H.; Cooper, N.; et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009, 113, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Harrington, W.J.; Minnich, V.; Hollingsworth, J.W.; Moore, C.V. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J. Lab. Clin. Med. 1951, 38, 1–10. [Google Scholar] [PubMed]
- Shulman, N.R.; Marder, V.J.; Weinrach, R.S. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann. N. Y. Acad. Sci. 1965, 124, 499–542. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Andersson, P.O.; Jernas, M.; Jacobsson, S.; Carlsson, B.; Carlsson, L.M.; Wadenvik, H. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat. Med. 2003, 9, 1123–1124. [Google Scholar] [CrossRef] [PubMed]
- Khodadi, E.; Asnafi, A.A.; Shahrabi, S.; Shahjahani, M.; Saki, N. Bone marrow niche in immune thrombocytopenia: A focus on megakaryopoiesis. Ann. Hematol. 2016, 95, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Dameshek, W.; Miller, E.B. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946, 1, 27–50. [Google Scholar] [PubMed]
- Pisciotta, A.V.; Stefanini, M.; Dameshek, W. Studies on platelets. X. Morphologic characteristics of megakaryocytes by phase contrast microscopy in normals and in patients with idiopathic thrombocytopenic purpura. Blood 1953, 8, 703–723. [Google Scholar] [PubMed]
- Neylon, A.J.; Saunders, P.W.; Howard, M.R.; Proctor, S.J.; Taylor, P.R.; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: A prospective study of a population-based cohort of 245 patients. Br. J. Haematol. 2003, 122, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.; Garrido, T. Advances in the pathophysiology of primary immune thrombocytopenia. Hematology 2016, 22, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Gaedicke, G. Immune thrombocytopenia in children and adults: What’s the same, what’s different? Haematologica 2011, 96, 1739–1741. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Newland, A.C. Current Management of Primary Immune Thrombocytopenia. Adv. Ther. 2015, 32, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Nardi, M.A.; Borkowsky, W.; Li, Z.; Karpatkin, S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009, 113, 4086–4093. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.F.; Blanchette, V.S.; Wang, H.; Arya, N.; Petric, M.; Semple, J.W.; Chia, W.K.; Freedman, J. Characterization of platelet-reactive antibodies in children with varicella-associated acute immune thrombocytopenic purpura (ITP). Br. J. Haematol. 1996, 95, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yujiri, T.; Shinohara, K.; Inoue, Y.; Sato, Y.; Fujii, Y.; Okubo, M.; Zaitsu, Y.; Ariyoshi, K.; Nakamura, Y.; et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br. J. Haematol. 2004, 124, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nardi, M.A.; Karpatkin, S. Role of molecular mimicry to HIV-1 peptides in HIV-1-related immunologic thrombocytopenia. Blood 2005, 106, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Bussel, J.B.; Liebman, H.A.; Luning Prak, E.T. The ITP syndrome: Pathogenic and clinical diversity. Blood 2009, 113, 6511–6521. [Google Scholar] [CrossRef] [PubMed]
- Terrell, D.R.; Beebe, L.A.; Vesely, S.K.; Neas, B.R.; Segal, J.B.; George, J.N. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am. J. Hematol. 2010, 85, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Schmidt, K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood 1999, 94, 909–913. [Google Scholar] [PubMed]
- Fogarty, P.F.; Segal, J.B. The epidemiology of immune thrombocytopenic purpura. Curr. Opin. Hematol. 2007, 14, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, A.H.; Semple, J.W.; Cines, D.B. Innate and adaptive immunity in immune thrombocytopenia. Semin. Hematol. 2013, 50, S68–S70. [Google Scholar] [CrossRef] [PubMed]
- Heitink-Polle, K.M.; Haverman, L.; Annink, K.V.; Schep, S.J.; de Haas, M.; Bruin, M.C. Health-related quality of life in children with newly diagnosed immune thrombocytopenia. Haematologica 2014, 99, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Mathias, S.D.; Go, R.S.; Guo, M.; Henry, D.H.; Lyons, R.; Redner, R.L.; Rice, L.; Schipperus, M.R. Improved quality of life for romiplostim-treated patients with chronic immune thrombocytopenic purpura: Results from two randomized, placebo-controlled trials. Br. J. Haematol. 2009, 144, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Efficace, F.; Mandelli, F.; Fazi, P.; Santoro, C.; Gaidano, G.; Cottone, F.; Borchiellini, A.; Carpenedo, M.; Simula, M.P.; Di Giacomo, V.; et al. Health-related quality of life and burden of fatigue in patients with primary immune thrombocytopenia by phase of disease. Am. J. Hematol. 2016, 91, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Newland, A.C. Fatigue in immune thrombocytopenia. Br. J. Haematol. 2015, 170, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.L.; Reese, J.A.; Watson, S.I.; Vesely, S.K.; Bolton-Maggs, P.H.; George, J.N.; Terrell, D.R. Fatigue in adult patients with primary immune thrombocytopenia. Eur. J. Haematol. 2011, 86, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Lim, W.; Crowther, M.; Cohen, A.; Solberg, L., Jr.; Crowther, M.A.; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011, 117, 4190–4207. [Google Scholar] [CrossRef] [PubMed]
- Karpatkin, S.; Siskind, G.W. In vitro detection of platelet antibody in patients with idiopathic thrombocytopenic purpura and systemic lupus erythematosus. Blood 1969, 33, 795–812. [Google Scholar] [PubMed]
- Karpatkin, S.; Strick, N.; Karpatkin, M.B.; Siskind, G.W. Cumulative experience in the detection of antiplatelet antibody in 234 patients with idiopathic thrombocytopenic purpura, systemic lupus erythematosus and other clinical disorders. Am. J. Med. 1972, 52, 776–785. [Google Scholar] [CrossRef]
- Lightsey, A.L., Jr.; McMillan, R.; Koenig, H.M.; Schanberger, J.E.; Lang, J.E. In vitro production of platelet-binding IgG in childhood idiopathic thrombocytopenic purpura. J. Pediatr. 1976, 88, 415–418. [Google Scholar] [CrossRef]
- He, R.; Reid, D.M.; Jones, C.E.; Shulman, N.R. Spectrum of Ig classes, specificities, and titers of serum antiglycoproteins in chronic idiopathic thrombocytopenic purpura. Blood 1994, 83, 1024–1032. [Google Scholar] [PubMed]
- Boylan, B.; Chen, H.; Rathore, V.; Paddock, C.; Salacz, M.; Friedman, K.D.; Curtis, B.R.; Stapleton, M.; Newman, D.K.; Kahn, M.L.; et al. Anti-GPVI-associated ITP: An acquired platelet disorder caused by autoantibody-mediated clearance of the GPVI/FcRgamma-chain complex from the human platelet surface. Blood 2004, 104, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Moore, D.L.; Lee, J.Y.; LoBuglio, A.F. Monocyte-platelet interaction in immune and nonimmune thrombocytopenia. Blood 1989, 74, 1328–1331. [Google Scholar] [PubMed]
- Chow, L.; Aslam, R.; Speck, E.R.; Kim, M.; Cridland, N.; Webster, M.L.; Chen, P.; Sahib, K.; Ni, H.; Lazarus, A.H.; et al. A murine model of severe immune thrombocytopenia is induced by antibody- and CD8+ T cell-mediated responses that are differentially sensitive to therapy. Blood 2010, 115, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Nakagawa, P.A.; Williams, S.A.; Schwartz, M.R.; Imfeld, K.L.; Buzby, J.S.; Nugent, D.J. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood 2003, 102, 887–895. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Wang, L.; Tomer, A.; Nichol, J.; Pistillo, J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004, 103, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Aledort, L.M.; Hayward, C.P.; Chen, M.G.; Nichol, J.L.; Bussel, J.; Group, I.T.P.S. Prospective screening of 205 patients with ITP, including diagnosis, serological markers, and the relationship between platelet counts, endogenous thrombopoietin, and circulating antithrombopoietin antibodies. Am. J. Hematol. 2004, 76, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, K. The molecular mechanisms that control thrombopoiesis. J. Clin. Investig. 2005, 115, 3339–3347. [Google Scholar] [CrossRef] [PubMed]
- Ballem, P.J.; Segal, G.M.; Stratton, J.R.; Gernsheimer, T.; Adamson, J.W.; Slichter, S.J. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J. Clin. Investig. 1987, 80, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Emmons, R.V.; Reid, D.M.; Cohen, R.L.; Meng, G.; Young, N.S.; Dunbar, C.E.; Shulman, N.R. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood 1996, 87, 4068–4071. [Google Scholar] [PubMed]
- Semple, J.W.; Freedman, J. Increased antiplatelet T helper lymphocyte reactivity in patients with autoimmune thrombocytopenia. Blood 1991, 78, 2619–2625. [Google Scholar] [PubMed]
- Stasi, R. Immune thrombocytopenia: Pathophysiologic and clinical update. Semin. Thromb. Hemost. 2012, 38, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Ridell, B.; Carlsson, L.; Jacobsson, S.; Wadenvik, H. Recruitment of T cells into bone marrow of ITP patients possibly due to elevated expression of VLA-4 and CX3CR1. Blood 2008, 112, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Houwerzijl, E.J.; Blom, N.R.; van der Want, J.J.; Esselink, M.T.; Koornstra, J.J.; Smit, J.W.; Louwes, H.; Vellenga, E.; de Wolf, J.T. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004, 103, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Houwerzijl, E.J.; Blom, N.R.; van der Want, J.J.; Vellenga, E.; de Wolf, J.T. Megakaryocytic dysfunction in myelodysplastic syndromes and idiopathic thrombocytopenic purpura is in part due to different forms of cell death. Leukemia 2006, 20, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Milev, Y.; Cosgrave, D.; Mody, M.; Hornstein, A.; Blanchette, V.; Freedman, J. Differences in serum cytokine levels in acute and chronic autoimmune thrombocytopenic purpura: Relationship to platelet phenotype and antiplatelet T-cell reactivity. Blood 1996, 87, 4245–4254. [Google Scholar] [PubMed]
- Feng, X.; Scheinberg, P.; Samsel, L.; Rios, O.; Chen, J.; McCoy, J.P., Jr.; Ghanima, W.; Bussel, J.B.; Young, N.S. Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. J. Thromb. Haemost. 2012, 10, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Talaat, R.M.; Elmaghraby, A.M.; Barakat, S.S.; El-Shahat, M. Alterations in immune cell subsets and their cytokine secretion profile in childhood idiopathic thrombocytopenic purpura (ITP). Clin. Exp. Immunol. 2014, 176, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Prak, E.T.; Cines, D.B. Can immune thrombocytopenia be cured with medical therapy? Semin. Thromb. Hemost. 2015, 41, 395–404. [Google Scholar] [PubMed]
- Nomura, S. Advances in Diagnosis and Treatments for Immune Thrombocytopenia. Clin. Med. Insights Blood Disord. 2016, 9, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Tuckuviene, R.; Nielsen, K.R.; Rosthoj, S. Flow cytometric measurement of platelet-associated immunoglobulin in children with newly diagnosed Immune Thrombocytopenia. Eur. J. Haematol. 2016, 96, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Arnason, J.E.; Campigotto, F.; Neuberg, D.; Bussel, J.B. Abnormalities in IgA and IgM are associated with treatment-resistant ITP. Blood 2012, 119, 5016–5020. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Cuker, A.; Semple, J.W. Pathogenesis of immune thrombocytopenia. Presse Med. 2014, 43, e49–e59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Hou, M.; Zhang, X.H.; Guan, X.H.; Sun, G.Z. The diagnostic value of platelet glycoprotein-specific autoantibody detection in idiopathic thrombocytopenic purpura. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2004, 12, 204–206. [Google Scholar] [PubMed]
- Fujisawa, K.; O’Toole, T.E.; Tani, P.; Loftus, J.C.; Plow, E.F.; Ginsberg, M.H.; McMillan, R. Autoantibodies to the presumptive cytoplasmic domain of platelet glycoprotein IIIa in patients with chronic immune thrombocytopenic purpura. Blood 1991, 77, 2207–2213. [Google Scholar] [PubMed]
- Kuwana, M.; Kaburaki, J.; Ikeda, Y. Autoreactive T cells to platelet GPIIb-IIIa in immune thrombocytopenic purpura. Role in production of anti-platelet autoantibody. J. Clin. Investig. 1998, 102, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.Q.; Dong, H.X.; Cheng, P.P.; Zhou, J.W.; Zheng, B.Y.; Liu, F. Antioxidant status and oxidative stress in patients with chronic ITP. Scand. J. Immunol. 2013, 77, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Bergmeier, W.; Rackebrandt, K.; Gessner, J.E.; Zirngibl, H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood 2000, 96, 2520–2527. [Google Scholar] [PubMed]
- Webster, M.L.; Sayeh, E.; Crow, M.; Chen, P.; Nieswandt, B.; Freedman, J.; Ni, H. Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbalpha antibodies. Blood 2006, 108, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Leytin, V.; Mykhaylov, S.; Starkey, A.F.; Allen, D.J.; Lau, H.; Ni, H.; Semple, J.W.; Lazarus, A.H.; Freedman, J. Intravenous immunoglobulin inhibits anti-glycoprotein IIb-induced platelet apoptosis in a murine model of immune thrombocytopenia. Br. J. Haematol. 2006, 133, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Yu, S.; Li, Q.; He, Y.; Liang, W.; Yu, L.; Xu, D. Investigation of platelet apoptosis in adult patients with chronic immune thrombocytopenia. Hematology 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Urbanus, R.T.; van der Wal, D.E.; Koekman, C.A.; Huisman, A.; van den Heuvel, D.J.; Gerritsen, H.C.; Deckmyn, H.; Akkerman, J.N.; Schutgens, R.E.G.; Gitz, E. Patient autoantibodies induce platelet destruction signals via raft-associated glycoprotein Ibalpha and Fc RIIa in immune thrombocytopenia. Haematologica 2013, 98, e70–e72. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Heitink-Polle, K.M.; Porcelijn, L.; Bentlage, A.E.; Bruin, M.C.; Visser, R.; Roos, D.; Schasfoort, R.B.; de Haas, M.; van der Schoot, C.E.; et al. C-reactive protein enhances IgG-mediated phagocyte responses and thrombocytopenia. Blood 2015, 125, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Rosove, M.H.; Lages, B.A.; Kaplan, K.L. Acquired storage pool deficiency with increased platelet-associated IgG. Report of five cases. Am. J. Med. 1980, 69, 711–717. [Google Scholar] [CrossRef]
- Sarpatwari, A.; Bennett, D.; Logie, J.W.; Shukla, A.; Beach, K.J.; Newland, A.C.; Sanderson, S.; Proven, D. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010, 95, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Severinsen, M.T.; Engebjerg, M.C.; Farkas, D.K.; Jensen, A.O.; Norgaard, M.; Zhao, S.; Sorensen, H.T. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: A Danish population-based cohort study. Br. J. Haematol. 2011, 152, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, M.; Severinsen, M.T.; Lund Maegbaek, M.; Jensen, A.O.; Cha, S.; Sorensen, H.T. Risk of arterial thrombosis in patients with primary chronic immune thrombocytopenia: A Danish population-based cohort study. Br. J. Haematol. 2012, 159, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Jy, W.; Horstman, L.L.; Arce, M.; Ahn, Y.S. Clinical significance of platelet microparticles in autoimmune thrombocytopenias. J. Lab. Clin. Med. 1992, 119, 334–345. [Google Scholar] [PubMed]
- Alvarez-Roman, M.T.; Fernandez-Bello, I.; Jimenez-Yuste, V.; Martin-Salces, M.; Arias-Salgado, E.G.; Rivas Pollmar, M.I.; Justo Sanz, R.; Butta, N.V. Procoagulant profile in patients with immune thrombocytopenia. Br. J. Haematol. 2016, 175, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Yanabu, M.; Nomura, S.; Fukuroi, T.; Suzuki, M.; Kawakatsu, T.; Kido, H.; Yamaguchi, K. Platelet activation induced by an antiplatelet autoantibody against CD9 antigen and its inhibition by another autoantibody in immune thrombocytopenic purpura. Br. J. Haematol. 1993, 84, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Yang, L.H.; Chang, L.X.; Feng, J.J.; Liu, J.Q. The clinical significance of circulating B cells secreting anti-glycoprotein IIb/IIIa antibody and platelet glycoprotein IIb/IIIa in patients with primary immune thrombocytopenia. Hematology 2012, 17, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, F.; Bal, G.; Barakat, A.; Milz, J.; Muhle, C.; Martinez-Gamboa, L.; Dorner, T.; Salama, A. High-level serum B-cell activating factor and promoter polymorphisms in patients with idiopathic thrombocytopenic purpura. Br. J. Haematol. 2007, 136, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Zhuang, L.; Li, X.Y.; Li, J.; Luo, S.K. The role of B cell-activating factor secreted by peripheral blood monocyte-derived dendritic cell in chronic idiopathic thrombocytopenic purpura. Zhonghua Xue Ye Xue Za Zhi 2010, 31, 599–602. [Google Scholar] [PubMed]
- Yang, Q.; Xu, S.; Li, X.; Wang, B.; Wang, X.; Ma, D.; Yang, L.; Peng, J.; Hou, M. Pathway of Toll-like receptor 7/B cell activating factor/B cell activating factor receptor plays a role in immune thrombocytopenia in vivo. PLoS ONE 2011, 6, e22708. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, Y.; Han, J.; Yang, Z.; Sheng, W.; Dai, H.; Wang, Y.; Xia, T.; Hou, M. TLR7 regulates dendritic cell-dependent B-cell responses through BlyS in immune thrombocytopenic purpura. Eur. J. Haematol. 2011, 86, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Ridell, B.; Jernas, M.; Wadenvik, H. Increased number of B-cells in the red pulp of the spleen in ITP. Ann. Hematol. 2012, 91, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Daridon, C.; Loddenkemper, C.; Spieckermann, S.; Kuhl, A.A.; Salama, A.; Burmester, G.R.; Lipsky, P.E.; Dörner, T. Splenic proliferative lymphoid nodules distinct from germinal centers are sites of autoantigen stimulation in immune thrombocytopenia. Blood 2012, 120, 5021–5031. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, H.; Bao, W.; Boulad, N.; Evangelista, J.; Haider, M.A.; Bussel, J.; Yazdanbakhsh, K. Defective regulatory B-cell compartment in patients with immune thrombocytopenia. Blood 2012, 120, 3318–3325. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W. Bregging rights in ITP. Blood 2012, 120, 3169. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T.; Kuwana, M. CD4+CD25+Foxp3+ regulatory T cells in the pathophysiology of immune thrombocytopenia. Semin. Hematol. 2013, 50, S43–S49. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Segel, G.B.; Burack, R.; Spence, S.A.; Speck, E.R.; Guo, L.; Semple, J.W. Splenic lymphocyte subtypes in immune thrombocytopenia: Increased presence of a subtype of B-regulatory cells. Br. J. Haematol. 2016, 173, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.H.; Renier, W.O.; de Vaan, G.; Reekers, P.; Vingerhoets, D.M.; Gabreels, F.J. Effect of thymectomy on myasthenia gravis and autoimmune thrombocytopenic purpura in a 13-year-old girl. Eur. J. Pediatr. 1987, 146, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.J.; Tomar, R.H.; Miller, M.L.; Davey, F.R. Chronic idiopathic thrombocytopenic purpura. A familial immunodeficiency syndrome? JAMA 1978, 239, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.H. Autoimmune thrombocytopenia: Clinical aspects. Semin. Hematol. 1992, 29, 18–25. [Google Scholar] [PubMed]
- Qiu, J.; Liu, X.; Li, X.; Zhang, X.; Han, P.; Zhou, H.; Shao, L.; Hou, Y.; Min, Y.; Kong, Z.; et al. CD8(+) T cells induce platelet clearance in the liver via platelet desialylation in immune thrombocytopenia. Sci. Rep. 2016, 6, 27445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chu, X.; Wang, L.; Zhu, Y.; Li, L.; Ma, D.; Peng, J.; Hou, M. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2006, 76, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, X.; Zhang, F.; Wang, L.; Peng, J.; Hou, M. Increased cytotoxic T-lymphocyte-mediated cytotoxicity predominant in patients with idiopathic thrombocytopenic purpura without platelet autoantibodies. Haematologica 2008, 93, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Cooper, N.; Del Poeta, G.; Stipa, E.; Laura Evangelista, M.; Abruzzese, E.; Amadori, S. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood 2008, 112, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Bussel, J.B.; Heck, S.; He, W.; Karpoff, M.; Boulad, N.; Yazdanbakhsh, K. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood 2010, 116, 4639–4645. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Samson, M.; Guy, J.; Janikashvili, N.; Fraszczak, J.; Trad, M.; Ciudad, M.; Leguy, V.; Berthier, S.; Petrella, T.; et al. Immunologic effects of rituximab on the human spleen in immune thrombocytopenia. Blood 2011, 118, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mou, W.; Lu, G.; Cao, J.; He, X.; Pan, X.; Xu, K. Low-dose rituximab combined with short-term glucocorticoids up-regulates Treg cell levels in patients with immune thrombocytopenia. Int. J. Hematol. 2011, 93, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ge, J.; Zhao, H.; Du, W.; Xu, J.; Sui, T.; Ma, L.; Zhou, Z.; Qi, A.; Yang, R. Association of cytotoxic T-lymphocyte antigen 4 gene polymorphisms with idiopathic thrombocytopenic purpura in a Chinese population. Platelets 2011, 22, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Fahim, N.M.; Monir, E. Functional role of CD4+CD25+ regulatory T cells and transforming growth factor-beta1 in childhood immune thrombocytopenic purpura. Egypt. J. Immunol. 2006, 13, 173–187. [Google Scholar] [PubMed]
- Ling, Y.; Cao, X.; Yu, Z.; Ruan, C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. Eur. J. Haematol. 2007, 79, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Cao, X.S.; Yu, Z.Q.; Luo, G.H.; Bai, X.; Su, J.; Dai, L.; Ruan, C.G. Alterations of CD4+ CD25+ regulatory T cells in patients with idiopathic thrombocytopenic purpura. Zhonghua Xue Ye Xue Za Zhi 2007, 28, 184–188. [Google Scholar] [PubMed]
- Sakakura, M.; Wada, H.; Tawara, I.; Nobori, T.; Sugiyama, T.; Sagawa, N.; Shiku, H. Reduced Cd4+Cd25+ T cells in patients with idiopathic thrombocytopenic purpura. Thromb. Res. 2007, 120, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, D.; Zhu, X.; Qu, X.; Ji, C.; Hou, M. Elevated profile of Th17, Th1 and Tc1 cells in patients with immune thrombocytopenic purpura. Haematologica 2009, 94, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Peng, J.; Sun, J.Z.; Liu, J.J.; Guo, C.S.; Wang, Z.G.; Yu, Y.; Shi, Y.; Qin, P.; Li, S.G.; et al. De novo induction of platelet-specific CD4(+)CD25(+) regulatory T cells from CD4(+)CD25(−) cells in patients with idiopathic thrombocytopenic purpura. Blood 2009, 113, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Abudureheman, A.; Yasen, H.; Zhao, F.; Zhang, X.; Ding, J.; Ma, X.; Guo, X. Expression of CD4+ CD25+ regulatory T cells and TGF-ss1 in patient with idiopathic thrombocytopenic purpura. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2010, 26, 895–897. [Google Scholar] [PubMed]
- Chang, D.Y.; Ouyang, J.; Zhou, R.F.; Xu, J.Y.; Chen, B.; Yang, Y.G.; Zhang, Q.G.; Shao, X.Y.; Guan, C.Y.; Xu, Y. Profiles of different subsets of CD(4)(+) T cells in chronic idiopathic thrombocytopenic purpura. Zhonghua Nei Ke Za Zhi 2010, 49, 213–216. [Google Scholar] [PubMed]
- Aboul-Fotoh Lel, M.; Abdel Raheem, M.M.; El-Deen, M.A.; Osman, A.M. Role of CD4+CD25+ T cells in children with idiopathic thrombocytopenic purpura. J. Pediatr. Hematol. Oncol. 2011, 33, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Qiu, Y.S.; Hao, G.P.; Zhu, L. Levels of regulatory T cells in peripheral blood of children with idiopathic thrombocytopenic purpura. Zhongguo Dang Dai Er Ke Za Zhi 2011, 13, 282–284. [Google Scholar] [PubMed]
- Park, S.H.; Kim, J.Y.; Kim, S.K.; Choe, J.Y.; Kim, S.G.; Ryoo, H.M. Regulatory T-cells in systemic lupus erythematosus-associated thrombocytopenia: A comparison with idiopathic thrombocytopenic purpura. Lupus 2010, 19, 888–889. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zhan, Y.; Hua, F.; Li, F.; Zou, S.; Wang, W.; Song, D. The ratio of Treg/Th17 cells correlates with the disease activity of primary immune thrombocytopenia. PLoS ONE 2012, 7, e50909. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Andersson, P.O.; Jacobsson, S.; Carlsson, L.; Wadenvik, H. Disturbed apoptosis of T-cells in patients with active idiopathic thrombocytopenic purpura. Thromb. Haemost. 2005, 93, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Gratz, I.K.; Rosenblum, M.D.; Abbas, A.K. The life of regulatory T cells. Ann. N. Y. Acad. Sci. 2013, 1283, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Provan, D.; Garvey, M.B.; Freedman, J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr. Opin. Hematol. 2010, 17, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Ikeda, Y. The role of autoreactive T-cells in the pathogenesis of idiopathic thrombocytopenic purpura. Int. J. Hematol. 2005, 81, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Andre, S.; Tough, D.F.; Lacroix-Desmazes, S.; Kaveri, S.V.; Bayry, J. Surveillance of antigen-presenting cells by CD4+ CD25+ regulatory T cells in autoimmunity: Immunopathogenesis and therapeutic implications. Am. J. Pathol. 2009, 174, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhao, H.; Poon, M.C.; Han, Z.; Gu, D.; Xu, M.; Jia, H.; Yang, R.; Han, Z.C. Abnormality of CD4(+)CD25(+) regulatory T cells in idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2007, 78, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Heck, S.; Patel, V.; Levan, J.; Yu, Y.; Bussel, J.B.; Yazdanbakhsh, K. Defective circulating CD25 regulatory T cells in patients with chronic immune thrombocytopenic purpura. Blood 2008, 112, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Hu, Y.; Gebremeskel, S.; Segel, G.B.; Speck, E.R.; Guo, L.; Kim, M. Thymic retention of CD4+CD25+FoxP3+ T regulatory cells is associated with their peripheral deficiency and thrombocytopenia in a murine model of immune thrombocytopenia. Blood 2012, 120, 2127–2132. [Google Scholar] [CrossRef] [PubMed]
- Catani, L.; Sollazzo, D.; Trabanelli, S.; Curti, A.; Evangelisti, C.; Polverelli, N.; Palandri, F. Decreased expression of indoleamine 2,3-dioxygenase 1 in dendritic cells contributes to impaired regulatory T cell development in immune thrombocytopenia. Ann. Hematol. 2013, 92, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, X.; Zhang, D.; Li, H.; Ma, L.; Xuan, M.; Wang, H.; Yang, R. Abnormal Distribution and Function of Monocyte Subsets in Patients With Primary Immune Thrombocytopenia. Clin. Appl. Thromb. Hemost. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Bao, W.; Li, X.; Miller, A.; Seery, C.; Haq, N.; Bussel, J.; Yazdanbakhsh, K. CD16+ monocytes control T-cell subset development in immune thrombocytopenia. Blood 2012, 120, 3326–3335. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; Howard, T.A. Phenotypic and clonal analysis of T lymphocytes in childhood immune thrombocytopenic purpura. Blood 1993, 82, 2137–2142. [Google Scholar] [PubMed]
- Filion, M.C.; Bradley, A.J.; Devine, D.V.; Decary, F.; Chartrand, P. Autoreactive T cells in healthy individuals show tolerance in vitro with characteristics similar to but distinct from clonal anergy. Eur. J. Immunol. 1995, 25, 3123–3127. [Google Scholar] [CrossRef] [PubMed]
- Coopamah, M.D.; Garvey, M.B.; Freedman, J.; Semple, J.W. Cellular immune mechanisms in autoimmune thrombocytopenic purpura: An update. Transfus. Med. Rev. 2003, 17, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, C.; Li, L.; Zeng, L.; Li, Z.; Yan, Z.; Chen, W.; Cheng, H.; Sang, W.; Xu, K. Effects of high-dose dexamethasone on regulating interleukin-22 production and correcting Th1 and Th22 polarization in immune thrombocytopenia. J. Clin. Immunol. 2012, 32, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, C.; Zeng, L.; Li, L.; Li, X.; Li, Z.; Xu, K. Elevated plasma IL-22 levels correlated with Th1 and Th22 cells in patients with immune thrombocytopenia. Clin. Immunol. 2011, 141, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, H.; Zhang, L.; Shan, B.; Xu, X.; Li, H.; Liu, X.; Xu, S.; Yu, S.; Ma, D.; et al. Elevated profiles of Th22 cells and correlations with Th17 cells in patients with immune thrombocytopenia. Hum. Immunol. 2012, 73, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.M.; Zhang, X.X.; Zhao, G.S.; Si, Y.J.; Lin, G.Q.; Zhang, Y.M.; He, G.S.; Wu, D.P. Change of Th22 cells in peripheral blood of patients with primary immune thrombocytopenia and clinical implication. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2012, 28, 1314–1316. [Google Scholar] [PubMed]
- Guo, N.H.; Shi, Q.Z.; Hua, J.Y.; Li, Z.J.; Li, J.; He, W.F.; Wu, Q. Expression of regulatory T cells and Th17 cells in idiopathic thrombocytopenic purpura and its significance. Zhonghua Xue Ye Xue Za Zhi 2010, 31, 610–612. [Google Scholar] [PubMed]
- Hu, Y.; Ma, D.X.; Shan, N.N.; Zhu, Y.Y.; Liu, X.G.; Zhang, L.; Yu, S. Increased number of Tc17 and correlation with Th17 cells in patients with immune thrombocytopenia. PLoS ONE 2011, 6, e26522. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.M.; Souza, C.; Rocha, G.A.; de Melo, F.F.; Clementino, N.C.; Marino, M.C.; Bozzi, A.; Silva, M.L.; Martins Filho, O.A.; Queiroz, D.M. The levels of IL-17A and of the cytokines involved in Th17 cell commitment are increased in patients with chronic immune thrombocytopenia. Haematologica 2011, 96, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.J.; Yang, L.H.; Zhang, L.; Ren, F.G.; Zhang, R.J.; Chen, J.F.; Qin, X.Y.; Liang, H.Z. Expressions of Th17 cells and interleukin 17 in patients with primary immune thrombocytopenia and their clinical significance. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2012, 20, 1154–1157. [Google Scholar] [PubMed]
- Yoh, K.; Morito, N.; Ojima, M.; Shibuya, K.; Yamashita, Y.; Morishima, Y.; Ishii, Y.; Kusakabe, M.; Nishikii, H.; Fujita, A.; et al. Overexpression of RORgammat under control of the CD2 promoter induces polyclonal plasmacytosis and autoantibody production in transgenic mice. Eur. J. Immunol. 2012, 42, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.L.; Kuchroo, V.K. How Cytokine networks fuel inflammation: Interleukin-17 and a tale of two autoimmune diseases. Nat. Med. 2013, 19, 824–825. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.X.; Chen, Z.P.; Zheng, C.L.; Jia, H.R.; Ge, J.; Gu, D.S.; Du, W.T.; Wang, X.Y.; Zhao, H.F.; Yang, R.C. The role of Th17 cells in adult patients with chronic idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2009, 82, 488–489. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhu, X.; Zhao, P.; Zhao, C.; Li, X.; Zhu, Y.; Li, L. Profile of Th17 cytokines (IL-17, TGF-beta, IL-6) and Th1 cytokine (IFN-gamma) in patients with immune thrombocytopenic purpura. Ann. Hematol. 2008, 87, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Sollazzo, D.; Trabanelli, S.; Curti, A.; Vianelli, N.; Lemoli, R.M.; Catani, L. Circulating CD4+CD161+CD196+ Th17 cells are not increased in immune thrombocytopenia. Haematologica 2011, 96, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Benacerraf, B.; Abbas, A.K. Antigen presentation by hapten-specific B lymphocytes. I. Role of surface immunoglobulin receptors. J. Exp. Med. 1984, 160, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Unanue, E.R. Antigen-presenting function of the macrophage. Annu. Rev. Immunol. 1984, 2, 395–428. [Google Scholar] [CrossRef] [PubMed]
- Amodio, G.; Gregori, S. Dendritic cells a double-edge sword in autoimmune responses. Front. Immunol. 2012, 3, 233. [Google Scholar] [CrossRef] [PubMed]
- Watts, C. Capture and processing of exogenous antigens for presentation on MHC molecules. Annu. Rev. Immunol. 1997, 15, 821–850. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–23. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Benson, R.A.; Bedaj, M.; Maffia, P. Antigen-Presenting Cells and Antigen Presentation in Tertiary Lymphoid Organs. Front. Immunol. 2016, 7, 481. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F.; Miossec, P. Altered dendritic cell functions in autoimmune diseases: Distinct and overlapping profiles. Nat. Rev. Rheumatol. 2016, 12, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Catani, L.; Fagioli, M.E.; Tazzari, P.L.; Ricci, F.; Curti, A.; Rovito, M.; Preda, P. Dendritic cells of immune thrombocytopenic purpura (ITP) show increased capacity to present apoptotic platelets to T lymphocytes. Exp. Hematol. 2006, 34, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yokohama, A.; Osaki, Y.; Ogawa, Y.; Nakahashi, H.; Toyama, K.; Mitsui, T.; Hashimoto, Y.; Koiso, H.; Uchiumi, H.; et al. Circulating plasmacytoid dendritic cells in patients with primary and Helicobacter pylori-associated immune thrombocytopenia. Eur. J. Haematol. 2012, 88, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Kolbeck, R.; Sanjuan, M.A. Plasmacytoid dendritic cells in autoimmunity. Curr. Opin. Immunol. 2016, 44, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Aslam, R.; Kim, M.; Speck, E.R.; Freedman, J. Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood 2007, 109, 4803–4805. [Google Scholar] [CrossRef] [PubMed]
- Machlus, K.R.; Thon, J.N.; Italiano, J.E., Jr. Interpreting the developmental dance of the megakaryocyte: A review of the cellular and molecular processes mediating platelet formation. Br. J. Haematol. 2014, 165, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Stahl, C.P.; Zucker-Franklin, D.; McDonald, T.P. Incomplete antigenic cross-reactivity between platelets and megakaryocytes: Relevance to ITP. Blood 1986, 67, 421–428. [Google Scholar] [PubMed]
- Nugent, D.; McMillan, R.; Nichol, J.L.; Slichter, S.J. Pathogenesis of chronic immune thrombocytopenia: Increased platelet destruction and/or decreased platelet production. Br. J. Haematol. 2009, 146, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Norol, F.; Vitrat, N.; Cramer, E.; Guichard, J.; Burstein, S.A.; Vainchenker, W.; Debili, N. Effects of cytokines on platelet production from blood and marrow CD34+ cells. Blood 1998, 91, 830–843. [Google Scholar] [PubMed]
- Hou, M.; Andersson, P.O.; Stockelberg, D.; Mellqvist, U.H.; Ridell, B.; Wadenvik, H. Plasma thrombopoietin levels in thrombocytopenic states: Implication for a regulatory role of bone marrow megakaryocytes. Br. J. Haematol. 1998, 101, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Iraqi, M.; Perdomo, J.; Yan, F.; Choi, P.Y.; Chong, B.H. Immune thrombocytopenia: Antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica 2015, 100, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, H.; Ma, L.; Zhang, X.; Xue, F.; Zhou, Z.; Chi, Y. The defective bone marrow-derived mesenchymal stem cells in patients with chronic immune thrombocytopenia. Autoimmunity 2014, 47, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takubo, K.; Kobayashi, H.; Suzuki-Inoue, K.; Suda, T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J. Exp. Med. 2015, 212, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Malara, A.; Abbonante, V.; Di Buduo, C.A.; Tozzi, L.; Currao, M.; Balduini, A. The secret life of a megakaryocyte: Emerging roles in bone marrow homeostasis control. Cell. Mol. Life Sci. 2015, 72, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Winter, O.; Moser, K.; Mohr, E.; Zotos, D.; Kaminski, H.; Szyska, M.; Roth, K.; Wong, D.M.; Dame, C.; Tarlinton, D.M.; et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood 2010, 116, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Immunoregulatory function of mesenchymal stem cells. Eur. J. Immunol. 2006, 36, 2566–2573. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Hu, Y.; Zhang, X.H.; Wang, Y.Z.; Mo, X.D.; Zhang, Y.Y.; Wang, Y.; Han, W.; Xu, L.P.; Chang, Y.J.; et al. Association between an impaired bone marrow vascular microenvironment and prolonged isolated thrombocytopenia after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2014, 20, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Kojouri, K.; Vesely, S.K.; Terrell, D.R.; George, J.N. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: A systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004, 104, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F.; Ruggeri, M. Short- and long-term risks of splenectomy for benign haematological disorders: Should we revisit the indications? Br. J. Haematol. 2012, 158, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, A.H. Monoclonal versus polyclonal anti-D in the treatment of ITP. Expert Opin. Biol. Ther. 2013, 13, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Crow, A.R.; Lazarus, A.H. Mechanistic properties of intravenous immunoglobulin in murine immune thrombocytopenia: Support for FcgammaRIIB falls by the wayside. Semin. Hematol. 2016, 53, S20–S22. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Stasi, R.; Newland, A.C.; Blanchette, V.S.; Bolton-Maggs, P.; Bussel, J.B.; Chong, B.H. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010, 115, 168–186. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Lee, C.S.; Seery, C.; Imahiyerobo, A.A.; Thompson, M.V.; Catellier, D.; Turenne, I.G. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica 2014, 99, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Zaja, F.; Baccarani, M.; Mazza, P.; Bocchia, M.; Gugliotta, L.; Zaccaria, A.; Vianelli, N.; Defina, M.; Tieghi, A.; Amadori, S.; et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010, 115, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005, 23, 487–513. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, D.; Canoui-Poitrine, F.; Croisille, L.; Lee, K.; Roudot-Thoraval, F.; Languille, L.; Khellaf, M. Antiplatelet antibodies detected by the MAIPA assay in newly diagnosed immune thrombocytopenia are associated with chronic outcome and higher risk of bleeding. Ann. Hematol. 2014, 93, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J. Glucocorticoid action and the development of selective glucocorticoid receptor ligands. Biotechnol. Annu. Rev. 2006, 12, 269–300. [Google Scholar] [PubMed]
- Li, J.; Wang, Z.; Dai, L.; Cao, L.; Su, J.; Zhu, M.; Yu, Z.; Bai, X.; Ruan, C. Effects of rapamycin combined with low dose prednisone in patients with chronic immune thrombocytopenia. Clin. Dev. Immunol. 2013, 2013, 548085. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Hu, S.; Zhao, X.; Cao, L. Correction of abnormal T cell subsets by high-dose dexamethasone in patients with chronic idiopathic thrombocytopenic purpura. Immunol. Lett. 2013, 154, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.H.; Zhao, F.; Shi, W.; Ma, X.M.; Xu, Q.; Patiguli, A.B.; Halida, Y.S. Detection and clinical significance of Th1/Th2 cytokines in patients with idiopathic thrombocytopenic purpura. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2012, 28, 1185–1187. [Google Scholar] [PubMed]
- Clynes, R. Immune complexes as therapy for autoimmunity. J. Clin. Investig. 2005, 115, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, S.Q.; Kuijpers, T.W. Immunomodulation by IVIg and the Role of Fc-Gamma Receptors: Classic Mechanisms of Action after all? Front. Immunol. 2014, 5, 674. [Google Scholar] [CrossRef] [PubMed]
- Gilardin, L.; Bayry, J.; Kaveri, S.V. Intravenous immunoglobulin as clinical immune-modulating therapy. CMAJ 2015, 187, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Park-Min, K.H.; Serbina, N.V.; Yang, W.; Ma, X.; Krystal, G.; Neel, B.G.; Nutt, S.L.; Hu, X.; Ivashkiv, L.B. FcgammaRIII-dependent inhibition of interferon-gamma responses mediates suppressive effects of intravenous immune globulin. Immunity 2007, 26, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.J.; Lazarus, A.H. Mechanisms of platelet recovery in ITP associated with therapy. Ann. Hematol. 2010, 89, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Crow, A.R.; Lazarus, A.H. The mechanisms of action of intravenous immunoglobulin and polyclonal anti-d immunoglobulin in the amelioration of immune thrombocytopenic purpura: What do we really know? Transfus. Med. Rev. 2008, 22, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Fehr, J.; Hofmann, V.; Kappeler, U. Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin. N. Engl. J. Med. 1982, 306, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, P.; Dale, G.L.; Tani, P.; McMillan, R. Inhibition of autoantibody binding to platelet glycoprotein IIb/IIIa by anti-idiotypic antibodies in intravenous gammaglobulin. Blood 1989, 74, 2414–2417. [Google Scholar] [PubMed]
- Hansen, R.J.; Balthasar, J.P. Intravenous immunoglobulin mediates an increase in anti-platelet antibody clearance via the FcRn receptor. Thromb. Haemost. 2002, 88, 898–899. [Google Scholar] [PubMed]
- Bayry, J.; Lacroix-Desmazes, S.; Carbonneil, C.; Misra, N.; Donkova, V.; Pashov, A.; Chevailler, A.; Mouthon, L.; Weill, B.; Bruneval, P.; et al. Inhibition of maturation and function of dendritic cells by intravenous immunoglobulin. Blood 2003, 101, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Siragam, V.; Crow, A.R.; Brinc, D.; Song, S.; Freedman, J.; Lazarus, A.H. Intravenous immunoglobulin ameliorates ITP via activating Fc gamma receptors on dendritic cells. Nat. Med. 2006, 12, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Wermeling, F.; Karlsson, M.C.; Ravetch, J.V. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc. Natl. Acad. Sci. USA 2008, 105, 19571–19578. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Kiefel, V.; Amberg, R.; Mueller-Eckhardt, C. Treatment of autoimmune thrombocytopenic purpura with rhesus antibodies (anti-Rho(D)). Blut 1984, 49, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Crow, A.R.; Siragam, V.; Freedman, J.; Lazarus, A.H. Monoclonal antibodies that mimic the action of anti-D in the amelioration of murine ITP act by a mechanism distinct from that of IVIg. Blood 2005, 105, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Ambriz-Fernandez, R.; Martinez-Murillo, C.; Quintana-Gonzalez, S.; Collazo-Jaloma, J.; Bautista-Juarez, J. Fc receptor blockade in patients with refractory chronic immune thrombocytopenic purpura with anti-D IgG. Arch. Med. Res. 2002, 33, 536–540. [Google Scholar] [CrossRef]
- Boughton, B.J.; Cooke, R.M.; Smith, N.A.; Simpson, A.W. Autoimmune thrombocytopenia: Anti-glycoprotein IIb/IIIa auto antibodies are reduced after human anti-D immunoglobulin treatment. Autoimmunity 1994, 18, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Okazaki, Y.; Kaburaki, J.; Kawakami, Y.; Ikeda, Y. Spleen is a primary site for activation of platelet-reactive T and B cells in patients with immune thrombocytopenic purpura. J. Immunol. 2002, 168, 3675–3682. [Google Scholar] [CrossRef] [PubMed]
- Knobl, P. Inherited and acquired thrombotic thrombocytopenic purpura (TTP) in adults. Semin. Thromb. Hemost. 2014, 40, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Wang, S.; Xue, F.; Liu, X.; Zhang, L.; Li, H.; Yang, R. Long-term results of splenectomy in adult chronic immune thrombocytopenia. Eur. J. Haematol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Manganini, M.; Facchinetti, V.; Gramigna, R.; Broady, R.; Borleri, G.; Rambaldi, A.; Introna, M. Rituximab-mediated antibody-dependent cellular cytotoxicity against neoplastic B cells is stimulated strongly by interleukin-2. Haematologica 2003, 88, 1002–1012. [Google Scholar] [PubMed]
- Guo, L.; Kapur, R.; Aslam, R.; Speck, E.R.; Zufferey, A.; Zhao, Y.; Kim, M.; Lazarus, A.H.; Ni, H.; Semple, J.W. CD20+ B-cell depletion therapy suppresses murine CD8+ T-cell-mediated immune thrombocytopenia. Blood 2016, 127, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, D.; Oszmiana, A.; Finch, D.K.; Strickland, I.; Schofield, D.J.; Lowe, D.C.; Sleeman, M.A. Rituximab causes a polarization of B cells that augments its therapeutic function in NK-cell-mediated antibody-dependent cellular cytotoxicity. Blood 2013, 121, 4694–4702. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Del Poeta, G.; Stipa, E.; Evangelista, M.L.; Trawinska, M.M.; Cooper, N.; Amadori, S. Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood 2007, 110, 2924–2930. [Google Scholar] [CrossRef] [PubMed]
- Eisenbeis, C.F.; Grainger, A.; Fischer, B.; Baiocchi, R.A.; Carrodeguas, L.; Roychowdhury, S.; Chen, L. Combination immunotherapy of B-cell non-Hodgkin’s lymphoma with rituximab and interleukin-2: A preclinical and phase I study. Clin. Cancer Res. 2004, 10, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Gluck, W.L.; Hurst, D.; Yuen, A.; Levine, A.M.; Dayton, M.A.; Gockerman, J.P.; Lucas, J. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin. Cancer Res. 2004, 10, 2253–2264. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Pagano, A.; Stipa, E.; Amadori, S. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood 2001, 98, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Chapin, J.; Lee, C.S.; Zhang, H.; Zehnder, J.L.; Bussel, J.B. Gender and duration of disease differentiate responses to rituximab-dexamethasone therapy in adults with immune thrombocytopenia. Am. J. Hematol. 2016, 91, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Marangon, M.; Vianelli, N.; Palandri, F.; Mazzucconi, M.G.; Santoro, C.; Barcellini, W.; Fattizzo, B.; Volpetti, S.; Lucchini, E.; Polverelli, N.; et al. Rituximab in immune thrombocytopenia: Gender, age and response as predictors of long-term response. Eur. J. Haematol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Reboursiere, E.; Fouques, H.; Maigne, G.; Johnson, H.; Chantepie, S.; Gac, A.C.; Reman, O.; Macro, M.; Benabed, K.; Troussard, X.; et al. Rituximab salvage therapy in adults with immune thrombocytopenia: Retrospective study on efficacy and safety profiles. Int. J. Hematol. 2016, 104, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.L.; Mahevas, M.; Lee, S.Y.; Stasi, R.; Cunningham-Rundles, S.; Godeau, B.; Kanter, J. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 2012, 119, 5989–5995. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W.; Khelif, A.; Waage, A.; Michel, M.; Tjonnfjord, G.E.; Romdhan, N.B.; Kahrs, J. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 1653–1661. [Google Scholar] [CrossRef]
- Erickson-Miller, C.L.; DeLorme, E.; Tian, S.S.; Hopson, C.B.; Stark, K.; Giampa, L.; Valoret, E.I.; Duffy, K.J.; Luengo, J.L.; Rosen, J.; et al. Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp. Hematol. 2005, 33, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cohn, C.S.; Bussel, J.B. Romiplostim: A second-generation thrombopoietin agonist. Drugs Today 2009, 45, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.; Godeau, B.; Priego, V.; Viallard, J.F.; Lopez Fernandez, M.F.; Orejudos, A.; Eisen, M. Remission and platelet responses with romiplostim in primary immune thrombocytopenia: Final results from a phase 2 study. Br. J. Haematol. 2016, 172, 262–273. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, C.G.; Guo, L.; Freedman, J.; Semple, J.W. Cellular immune dysfunction in immune thrombocytopenia (ITP). Br. J. Haematol. 2013, 163, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.H.; Jelkmann, W. Subcellular localization of thrombopoietin in human blood platelets and its release upon thrombin stimulation. Br. J. Haematol. 2001, 115, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Bosworth, J.; Rhodes, E.; Shannon, M.S.; Willis, F.; Gordon-Smith, E.C. Thrombopoietic agents. Blood Rev. 2010, 24, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Zufferey, A.; Boilard, E.; Semple, J.W. Nouvelle Cuisine: Platelets Served with Inflammation. J. Immunol. 2015, 194, 5579–5587. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Semple, J.W. The nonhemostatic immune functions of platelets. Semin. Hematol. 2016, 53, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Semple, J.W. Platelets as immune-sensing cells. Blood Adv. 2016, 1, 10–14. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zufferey, A.; Kapur, R.; Semple, J.W. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). J. Clin. Med. 2017, 6, 16. https://doi.org/10.3390/jcm6020016
Zufferey A, Kapur R, Semple JW. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). Journal of Clinical Medicine. 2017; 6(2):16. https://doi.org/10.3390/jcm6020016
Chicago/Turabian StyleZufferey, Anne, Rick Kapur, and John W. Semple. 2017. "Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP)" Journal of Clinical Medicine 6, no. 2: 16. https://doi.org/10.3390/jcm6020016