
Mammary Gland Involution Provides a Unique Model to Study the TGF-β Cancer Paradox

Abstract
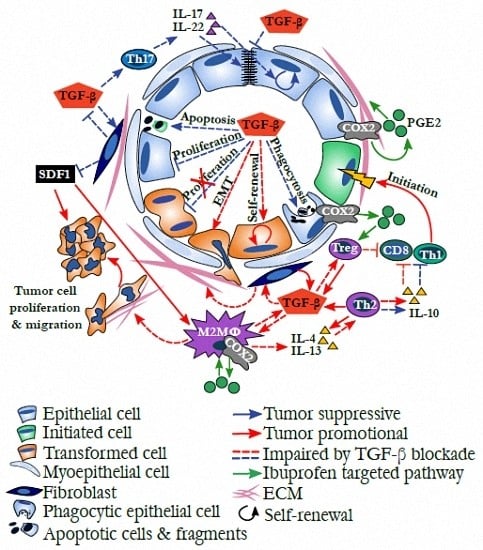
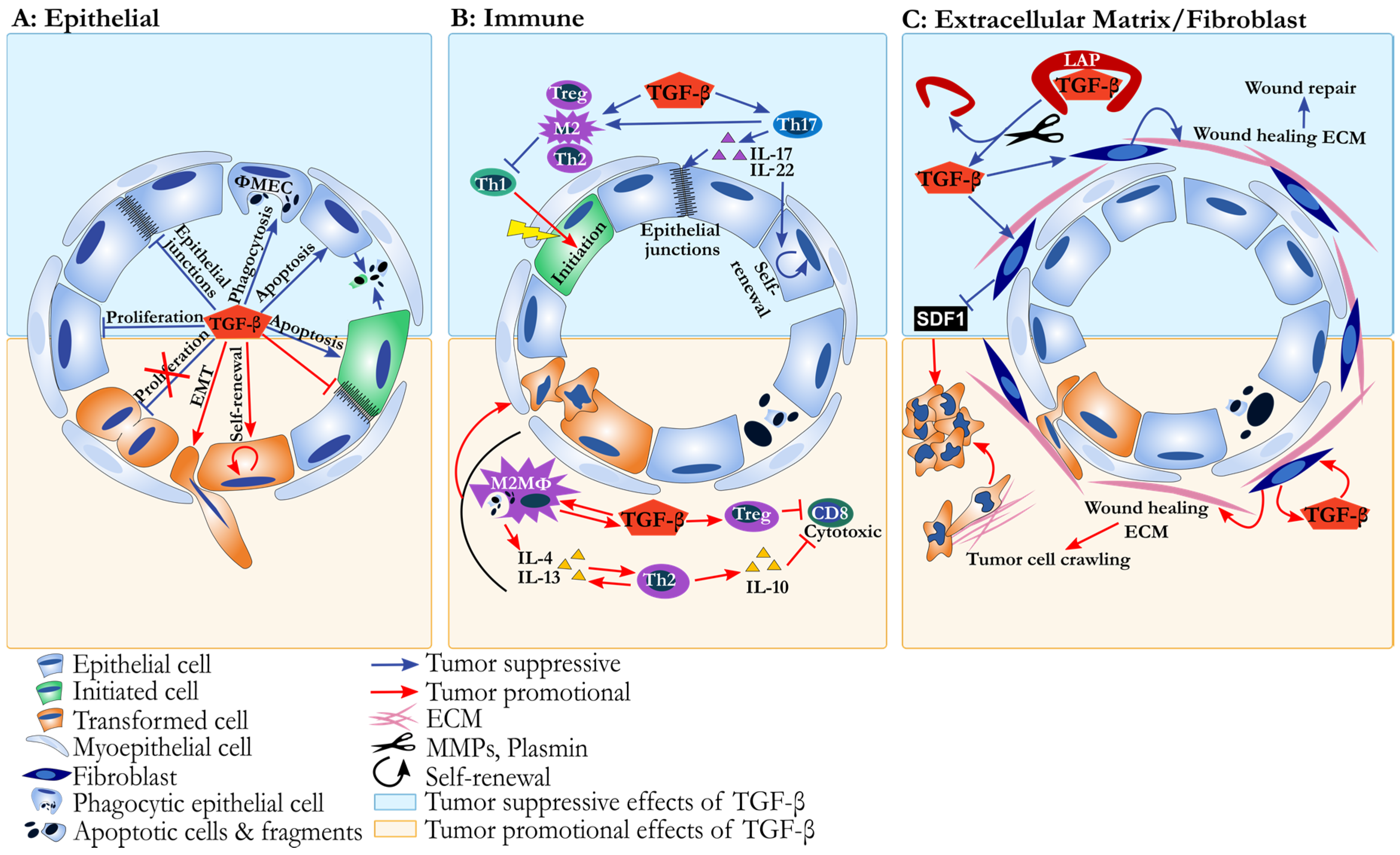
:
{kind=link}
{kind=link}
{kind=link}
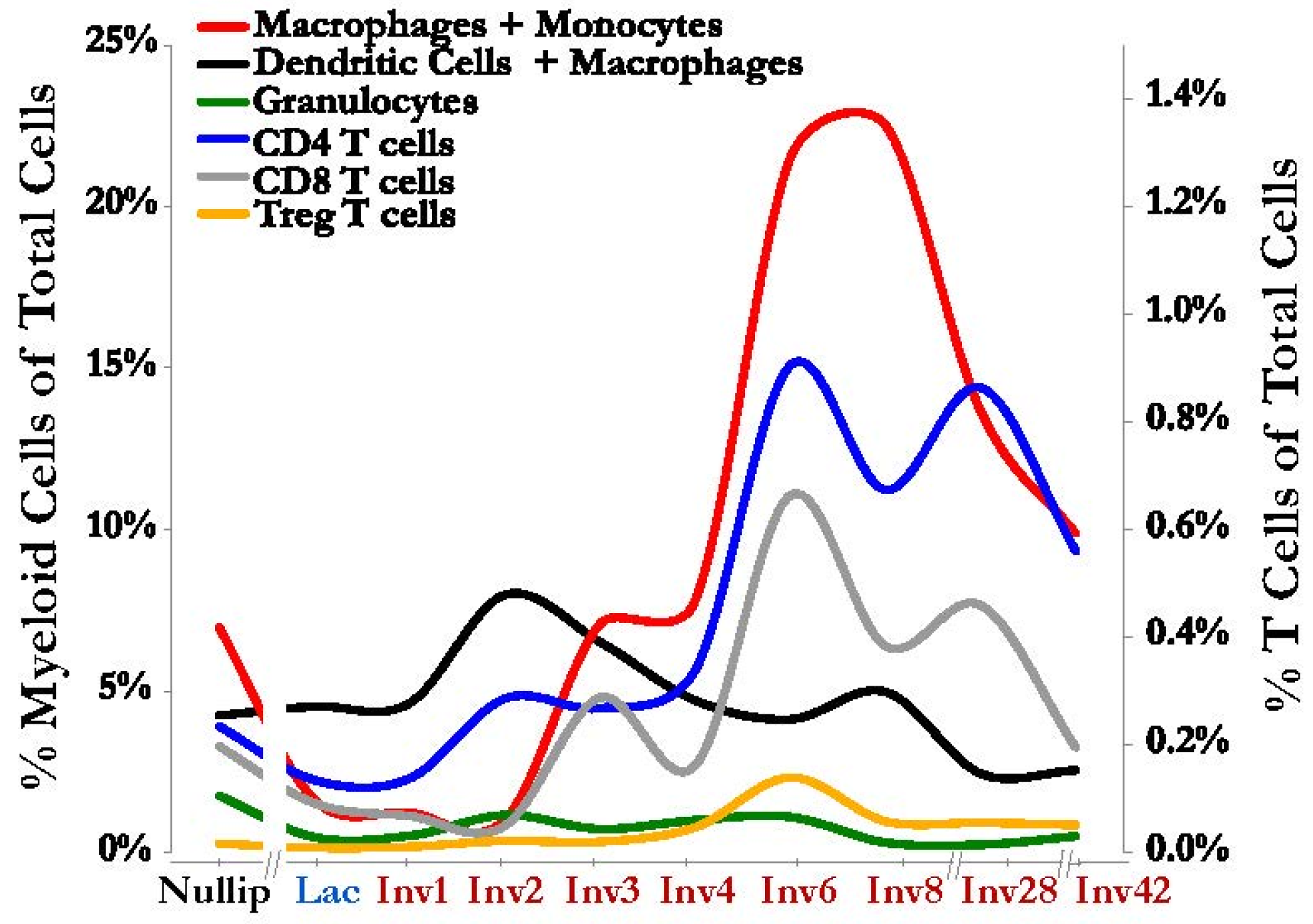
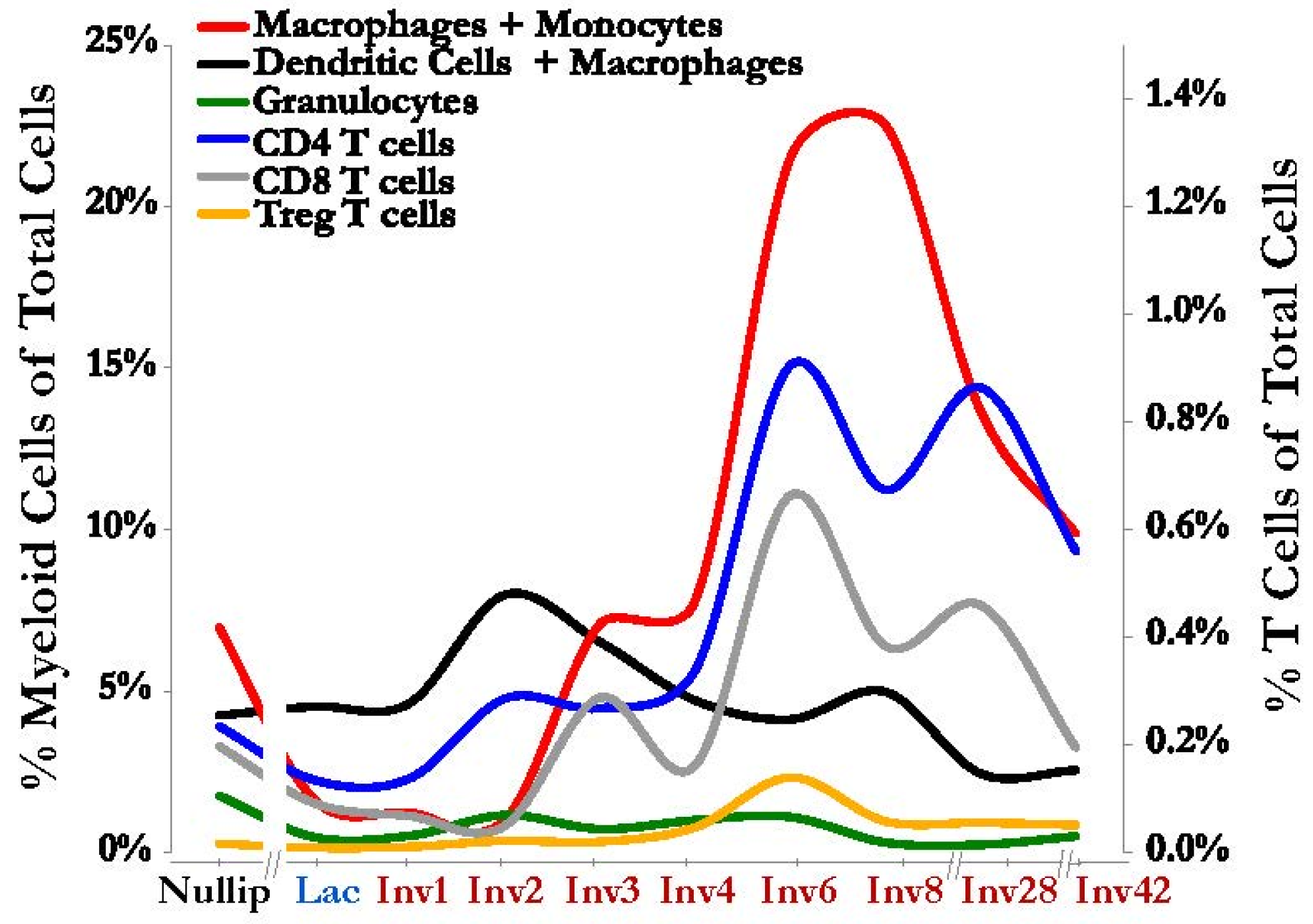{kind=link}
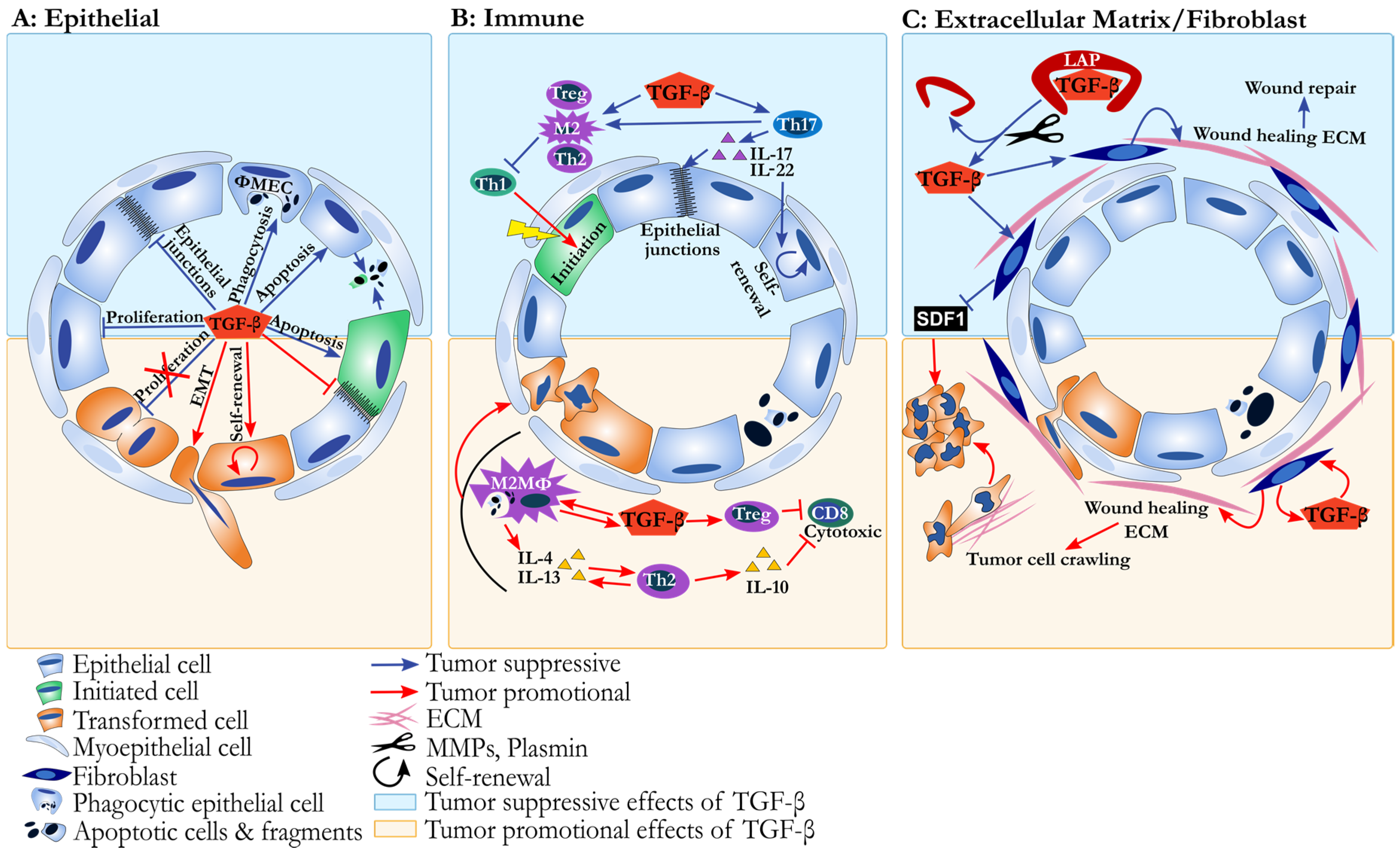
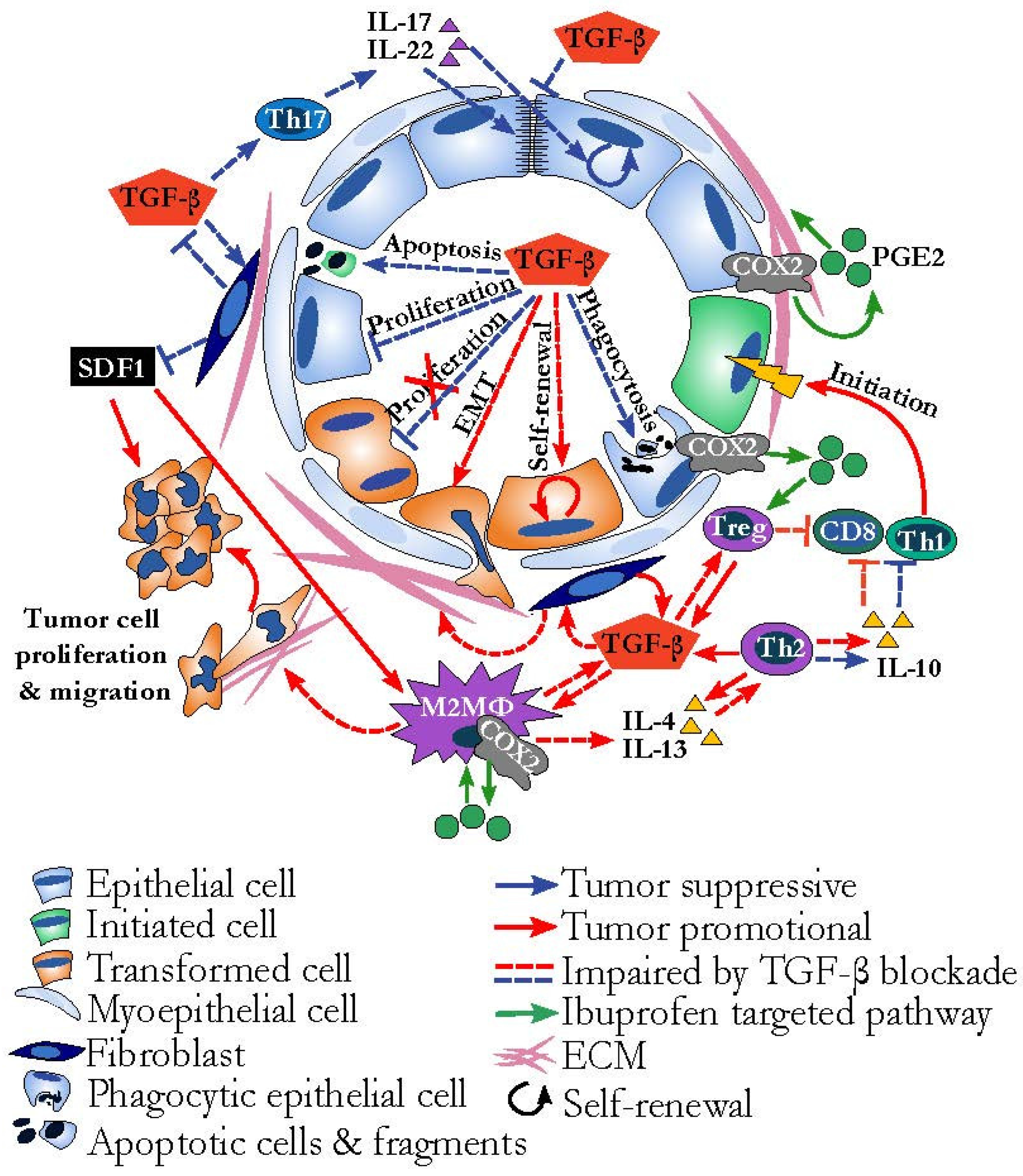{kind=link}
1. Introduction
2. The TGF-β Cancer Paradox
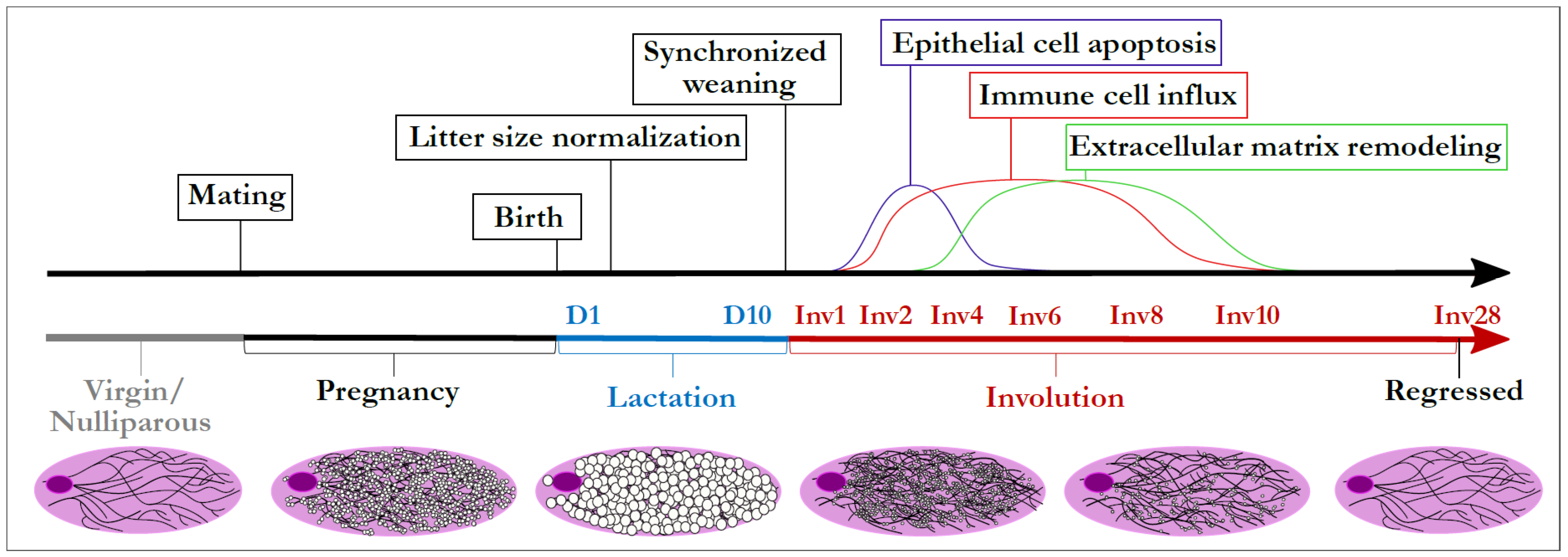
3. Mammary Gland Involution as a Model to Integrate Disparate TGF-β Biology
4. Evidence for TGF-β Orchestrating Weaning-Induced Mammary Gland Involution
5. TGF-β in Developmental Immune Tolerance and the Inhibition of Cytolytic Immunity
6. The Immune Environment of Involution Is Tumor Promotional and Targetable
7. TGF-β and Th17 Promote Barrier Function, Reducing Inflammation and Tumor Initiation
8. The Role of TGF-β in Tissue Repair and Remodeling
9. Dual Effects of ECM and Fibroblasts on Cancer Progression
10. Implications/Next Steps for Therapy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tian, M.; Schiemann, W.P. The tgf-beta paradox in human cancer: An update. Future Oncol. 2009, 5, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Jakowlew, S.B. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006, 25, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L. TGF-beta regulation of epithelial cell proliferation. Mol. Reprod. Dev. 1992, 32, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; McCoy, G.A.; Folkvord, J.M.; McPherson, J.M. TGF-beta 1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: A fibronectin matrix-dependent event. J. Cell. Physiol. 1997, 170, 69–80. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Pollard, J.W. Transforming growth factor beta3 induces cell death during the first stage of mammary gland involution. Development 2000, 127, 3107–3118. [Google Scholar] [PubMed]
- Fornetti, J.; Flanders, K.C.; Henson, P.M.; Tan, A.C.; Borges, V.F.; Schedin, P. Mammary epithelial cell phagocytosis downstream of TGF-beta3 is characterized by adherens junction reorganization. Cell Death Differ. 2016, 23, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta signaling through the smad pathway: Role in extracellular matrix gene expression and regulation. J. Invest. Dermatol. 2002, 118, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-smad pathways in tgf-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Chen, Y.G. TGF-beta signaling from receptors to smads. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Biswas, T.; Gu, X.; Yang, J.; Ellies, L.G.; Sun, L.Z. Attenuation of tgf-beta signaling supports tumor progression of a mesenchymal-like mammary tumor cell line in a syngeneic murine model. Cancer Lett. 2014, 346, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Rotello, R.J.; Lieberman, R.C.; Purchio, A.F.; Gerschenson, L.E. Coordinated regulation of apoptosis and cell proliferation by transforming growth factor beta 1 in cultured uterine epithelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 3412–3415. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Howe, P.H. Mechanisms of tgf-beta-induced cell cycle arrest. Miner. Electrolyte Metab. 1998, 24, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-beta induces apoptosis through smad-mediated expression of dap-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Inbal, B.; Kissil, J.L.; Raveh, T.; Berissi, H.; Spivak-Kroizaman, T.; Feinstein, E.; Kimchi, A. Dap-kinase participates in tnf-alpha- and fas-induced apoptosis and its function requires the death domain. J. Cell Biol. 1999, 146, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. Dap-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberhammer, F.A.; Pavelka, M.; Sharma, S.; Tiefenbacher, R.; Purchio, A.F.; Bursch, W.; Schulte-Hermann, R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc. Natl. Acad. Sci. USA 1992, 89, 5408–5412. [Google Scholar] [CrossRef] [PubMed]
- Mithani, S.K.; Balch, G.C.; Shiou, S.R.; Whitehead, R.H.; Datta, P.K.; Beauchamp, R.D. Smad3 has a critical role in TGF-beta-mediated growth inhibition and apoptosis in colonic epithelial cells. J. Surg. Res. 2004, 117, 296–305. [Google Scholar] [CrossRef]
- Kolek, O.; Gajkowska, B.; Godlewski, M.M.; Motyl, T. Molecular mechanism of tgf-beta1-induced apoptosis in hc11 mouse mammary epithelial cells (mec). Cell Mol. Biol. 2001, 47, OL197–208. [Google Scholar] [PubMed]
- Engle, S.J.; Hoying, J.B.; Boivin, G.P.; Ormsby, I.; Gartside, P.S.; Doetschman, T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999, 59, 3379–3386. [Google Scholar] [PubMed]
- Matsushita, M.; Matsuzaki, K.; Date, M.; Watanabe, T.; Shibano, K.; Nakagawa, T.; Yanagitani, S.; Amoh, Y.; Takemoto, H.; Ogata, N.; et al. Down-regulation of tgf-beta receptors in human colorectal cancer: Implications for cancer development. Br. J. Cancer 1999, 80, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. Smad2, smad3 and smad4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Musci, T.; Derynck, R. The tumor suppressor smad4/dpc 4 as a central mediator of smad function. Curr. Biol. 1997, 7, 270–276. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a rhoa-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, G.; Tirino, V.; Camerlingo, R.; Franco, R.; La Rocca, A.; Liguori, E.; Martucci, N.; Paino, F.; Normanno, N.; Rocco, G. Epithelial to mesenchymal transition by tgfbeta-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PloS ONE 2011, 6, e21548. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Fabra, A.; Nakajima, M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 7678–7682. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the tgfbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-beta targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhasz, E.; Ramlau, R.; van den Heuvel, M.M.; Lal, R.; Kloecker, G.H.; Eaton, K.D.; et al. A phase iii study of belagenpumatucel-l, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Cohn, A.; Lahn, M.M.; Williams, K.E.; Cleverly, A.L.; Pitou, C.; Kadam, S.K.; Farmen, M.W.; Desaiah, D.; Raju, R.; Conkling, P.; et al. A phase i dose-escalation study to a predefined dose of a transforming growth factor-beta1 monoclonal antibody (tbetam1) in patients with metastatic cancer. Int. J. Oncol. 2014, 45, 2221–2231. [Google Scholar]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef] [PubMed]
- Quarrie, L.H.; Addey, C.V.; Wilde, C.J. Programmed cell death during mammary tissue involution induced by weaning, litter removal, and milk stasis. J. Cell. Physiol. 1996, 168, 559–569. [Google Scholar] [CrossRef]
- Lund, L.R.; Romer, J.; Thomasset, N.; Solberg, H.; Pyke, C.; Bissell, M.J.; Dano, K.; Werb, Z. Two distinct phases of apoptosis in mammary gland involution: Proteinase-independent and -dependent pathways. Development 1996, 122, 181–193. [Google Scholar] [PubMed]
- Strange, R.; Li, F.; Saurer, S.; Burkhardt, A.; Friis, R.R. Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development 1992, 115, 49–58. [Google Scholar] [PubMed]
- Stein, T.; Morris, J.S.; Davies, C.R.; Weber-Hall, S.J.; Duffy, M.A.; Heath, V.J.; Bell, A.K.; Ferrier, R.K.; Sandilands, G.P.; Gusterson, B.A. Involution of the mouse mammary gland is associated with an immune cascade and an acute-phase response, involving lbp, cd14 and stat3. Breast Cancer Res. 2004, 6, R75–91. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, R.W.; Wayland, M.T.; Lee, J.; Freeman, T.; Watson, C.J. Gene expression profiling of mammary gland development reveals putative roles for death receptors and immune mediators in post-lactational regression. Breast Cancer Res. 2004, 6, R92–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, N.I.; Bennett, R.E.; Kerr, J.F. Cell death by apoptosis during involution of the lactating breast in mice and rats. Am. J. Anat. 1989, 185, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.M.; Selvarajan, S.; Mudgett, J.; Werb, Z. Stromelysin-1 regulates adipogenesis during mammary gland involution. J. Cell Biol. 2001, 152, 693–703. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Lyons, T.; Monks, J.; Lucia, M.S.; Wilson, R.S.; Hines, L.; Man, Y.G.; Borges, V.; Schedin, P. Alternatively activated macrophages and collagen remodeling characterize the postpartum involuting mammary gland across species. Am. J. Pathol. 2010, 176, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.H.; Vanderlinden, L.A.; Schedin, P.J.; Hansen, K.C. Rat mammary extracellular matrix composition and response to ibuprofen treatment during postpartum involution by differential gelc-ms/ms analysis. J. Proteome Res. 2012, 11, 4894–4905. [Google Scholar] [CrossRef] [PubMed]
- Martinson, H.A.; Jindal, S.; Durand-Rougely, C.; Borges, V.F.; Schedin, P. Wound healing-like immune program facilitates postpartum mammary gland involution and tumor progression. Int. J. Cancer 2015, 136, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Meier-Abt, F.; Brinkhaus, H.; Bentires-Alj, M. Early but not late pregnancy induces lifelong reductions in the proportion of mammary progesterone sensing cells and epithelial wnt signaling. Breast Cancer Res. 2014, 16, 402. [Google Scholar] [CrossRef] [PubMed]
- Fornetti, J.; Martinson, H.A.; Betts, C.B.; Lyons, T.R.; Jindal, S.; Guo, Q.; Coussens, L.M.; Borges, V.F.; Schedin, P. Mammary gland involution as an immunotherapeutic target for postpartum breast cancer. J. Mammary Gland Biol. Neoplasia 2014, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.R.; O’Brien, J.; Borges, V.F.; Conklin, M.W.; Keely, P.J.; Eliceiri, K.W.; Marusyk, A.; Tan, A.C.; Schedin, P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and cox-2. Nat. Med. 2011, 17, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Schedin, P. Pregnancy-associated breast cancer and metastasis. Nat. Rev. Cancer 2006, 6, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Sargeant, T.J.; Lloyd-Lewis, B.; Resemann, H.K.; Ramos-Montoya, A.; Skepper, J.; Watson, C.J. Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization. Nat. Cell Biol. 2014, 16, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Marti, A.; Ritter, P.M.; Jager, R.; Lazar, H.; Baltzer, A.; Schenkel, J.; Declercq, W.; Vandenabeele, P.; Jaggi, R. Mouse mammary gland involution is associated with cytochrome c release and caspase activation. Mech. Dev. 2001, 104, 89–98. [Google Scholar] [CrossRef]
- Sakamoto, K.; Wehde, B.L.; Yoo, K.H.; Kim, T.; Rajbhandari, N.; Shin, H.Y.; Triplett, A.A.; Radler, P.D.; Schuler, F.; Villunger, A.; et al. Janus kinase 1 is essential for inflammatory cytokine signaling and mammary gland remodeling. Mol. Cell. Biol. 2016, 36, 1673–1690. [Google Scholar] [CrossRef] [PubMed]
- Schere-Levy, C.; Buggiano, V.; Quaglino, A.; Gattelli, A.; Cirio, M.C.; Piazzon, I.; Vanzulli, S.; Kordon, E.C. Leukemia inhibitory factor induces apoptosis of the mammary epithelial cells and participates in mouse mammary gland involution. Exp. Cell Res. 2003, 282, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Bagci, H.; Laurin, M.; Huber, J.; Muller, W.J.; Cote, J.F. Impaired cell death and mammary gland involution in the absence of dock1 and rac1 signaling. Cell Death Dis. 2014, 5, e1375. [Google Scholar] [CrossRef] [PubMed]
- Baxter, F.O.; Came, P.J.; Abell, K.; Kedjouar, B.; Huth, M.; Rajewsky, K.; Pasparakis, M.; Watson, C.J. Ikkbeta/2 induces tweak and apoptosis in mammary epithelial cells. Development 2006, 133, 3485–3494. [Google Scholar] [CrossRef] [PubMed]
- Llobet-Navas, D.; Rodriguez-Barrueco, R.; Castro, V.; Ugalde, A.P.; Sumazin, P.; Jacob-Sendler, D.; Demircan, B.; Castillo-Martin, M.; Putcha, P.; Marshall, N.; et al. The mir-424(322)/503 cluster orchestrates remodeling of the epithelium in the involuting mammary gland. Genes Dev. 2014, 28, 765–782. [Google Scholar] [CrossRef] [PubMed]
- Llobet-Navas, D.; Rodriguez-Barrueco, R.; de la Iglesia-Vicente, J.; Olivan, M.; Castro, V.; Saucedo-Cuevas, L.; Marshall, N.; Putcha, P.; Castillo-Martin, M.; Bardot, E.; et al. The microrna 424/503 cluster reduces cdc25a expression during cell cycle arrest imposed by transforming growth factor beta in mammary epithelial cells. Mol. Cell. Biol. 2014, 34, 4216–4231. [Google Scholar] [CrossRef] [PubMed]
- Dunker, N.; Krieglstein, K. Targeted mutations of transforming growth factor-beta genes reveal important roles in mouse development and adult homeostasis. Eur. J. Biochem. 2000, 267, 6982–6988. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking tgf-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Gorska, A.E.; Stover, D.G.; Moses, H.L. TGF-beta promotes cell death and suppresses lactation during the second stage of mammary involution. J. Cell. Physiol. 2009, 219, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Monks, J.; Smith-Steinhart, C.; Kruk, E.R.; Fadok, V.A.; Henson, P.M. Epithelial cells remove apoptotic epithelial cells during post-lactation involution of the mouse mammary gland. Biol. Reprod. 2008, 78, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.J.; Sakamoto, T.; Osusky, R.; Wang, H.M.; Ogden, T.E.; Ryan, S.J.; Hinton, D.R.; Gopalakrishna, R. Transforming growth factor-beta regulates human retinal pigment epithelial cell phagocytosis by influencing a protein kinase c-dependent pathway. Graefe’s Arch. Clin. Exp. Ophthalmol. 1994, 232, 695–701. [Google Scholar] [CrossRef]
- Maderna, P.; Godson, C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim. Biophys. Acta 2003, 1639, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, pge2, and paf. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Frasch, S.C.; Thomas, S.M.; Bratton, D.L.; Henson, P.M. Induction of tgf-beta1 synthesis by macrophages in response to apoptotic cells requires activation of the scavenger receptor cd36. PloS ONE 2013, 8, e72772. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Martinson, H.; Durand-Rougely, C.; Schedin, P. Macrophages are crucial for epithelial cell death and adipocyte repopulation during mammary gland involution. Development 2012, 139, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Laouar, Y.; Li, M.O.; Green, E.A.; Flavell, R.A. TGF-beta regulates in vivo expansion of foxp3-expressing cd4+cd25+ regulatory T cells responsible for protection against diabetes. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4572–4577. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Abrogation of tgfbeta signaling in T cells leads to spontaneous t cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Wahl, S.M.; Hunt, D.A.; Wakefield, L.M.; McCartney-Francis, N.; Wahl, L.M.; Roberts, A.B.; Sporn, M.B. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 5788–5792. [Google Scholar] [CrossRef] [PubMed]
- Turley, J.M.; Falk, L.A.; Ruscetti, F.W.; Kasper, J.J.; Francomano, T.; Fu, T.; Bang, O.S.; Birchenall-Roberts, M.C. Transforming growth factor beta 1 functions in monocytic differentiation of hematopoietic cells through autocrine and paracrine mechanisms. Cell Growth Differ. 1996, 7, 1535–1544. [Google Scholar] [PubMed]
- Gong, D.; Shi, W.; Yi, S.J.; Chen, H.; Groffen, J.; Heisterkamp, N. Tgfbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Sozzani, S.; Stine, J.T.; Luini, W.; D’mico, G.; Allavena, P.; Chantry, D.; Mantovani, A. Divergent effects of interleukin-4 and interferon-gamma on macrophage-derived chemokine production: An amplification circuit of polarized T helper 2 responses. Blood 1998, 92, 2668–2671. [Google Scholar] [PubMed]
- Zheng, W.; Flavell, R.A. The transcription factor gata-3 is necessary and sufficient for th2 cytokine gene expression in cd4 T cells. Cell 1997, 89, 587–596. [Google Scholar] [CrossRef]
- Kanhere, A.; Hertweck, A.; Bhatia, U.; Gokmen, M.R.; Perucha, E.; Jackson, I.; Lord, G.M.; Jenner, R.G. T-bet and gata3 orchestrate th1 and th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat. Commun. 2012, 3, 1268. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.C.; Young, C.; Hicks, D.; Owens, P.; Williams, A.; Vaught, D.B.; Morrison, M.M.; Lim, J.; Williams, M.; Brantley-Sieders, D.M.; et al. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Investig. 2014, 124, 4737–4752. [Google Scholar] [CrossRef] [PubMed]
- Cools, N.; Van Tendeloo, V.F.; Smits, E.L.; Lenjou, M.; Nijs, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Ponsaerts, P. Immunosuppression induced by immature dendritic cells is mediated by TGF-beta/IL-10 double-positive cd4+ regulatory T cells. J. Cell. Mol. Med. 2008, 12, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into tgf-beta-secreting cells inducing cd4+cd25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.R.; Borges, V.F.; Betts, C.B.; Guo, Q.; Kapoor, P.; Martinson, H.A.; Jindal, S.; Schedin, P. Cyclooxygenase-2-dependent lymphangiogenesis promotes nodal metastasis of postpartum breast cancer. J. Clin. Investig. 2014, 124, 3901–3912. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hansen, K.; Barkan, D.; Green, J.; Schedin, P.; O’Brien, J.; Hansen, K.; Barkan, D.; Green, J.; Schedin, P. Non-steroidal anti-inflammatory drugs target the pro-tumorigenic extracellular matrix of the postpartum mammary gland. Int. J. Dev. Biol. 2011, 55, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Engle, S.J.; Ormsby, I.; Pawlowski, S.; Boivin, G.P.; Croft, J.; Balish, E.; Doetschman, T. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 2002, 62, 6362–6366. [Google Scholar] [PubMed]
- Liu, J.Z.; Pezeshki, M.; Raffatellu, M. Th17 cytokines and host-pathogen interactions at the mucosa: Dichotomies of help and harm. Cytokine 2009, 48, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Li, C.; Qiao, W.; Mamura, M.; Kasprzak, B.; Anver, M.; Wolfraim, L.; Hong, S.; Mushinski, E.; Potter, M.; et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006, 441, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Frutos Rde, L.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific microbiota direct the differentiation of IL-17-producing t-helper cells in the mucosa of the small intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor rorgammat directs the differentiation program of proinflammatory il-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the t(h)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Low, E.N.; Zagieboylo, L.; Martino, B.; Wilson, E. Iga asc accumulation to the lactating mammary gland is dependent on vcam-1 and alpha4 integrins. Mol. Immunol. 2010, 47, 1608–1612. [Google Scholar] [CrossRef] [PubMed]
- Morteau, O.; Gerard, C.; Lu, B.; Ghiran, S.; Rits, M.; Fujiwara, Y.; Law, Y.; Distelhorst, K.; Nielsen, E.M.; Hill, E.D.; et al. An indispensable role for the chemokine receptor ccr10 in iga antibody-secreting cell accumulation. J. Immunol. 2008, 181, 6309–6315. [Google Scholar] [CrossRef] [PubMed]
- Rainard, P.; Cunha, P.; Bougarn, S.; Fromageau, A.; Rossignol, C.; Gilbert, F.B.; Berthon, P. T helper 17-associated cytokines are produced during antigen-specific inflammation in the mammary gland. PLoS ONE 2013, 8, e63471. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, S.J.; Atsuta, R.; McIntyre, J.O.; Hackett, T.L.; Singhera, G.K.; Dorscheid, D.R. Il-13 and th2 cytokine exposure triggers matrix metalloproteinase 7-mediated fas ligand cleavage from bronchial epithelial cells. J. Allergy Clin. Immunol. 2010, 126, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, C.; Rezzonico, R.; De Luca, C.; Burger, D.; Dayer, J.M. Th2 cell membrane factors in association with il-4 enhance matrix metalloproteinase-1 (mmp-1) while decreasing mmp-9 production by granulocyte-macrophage colony-stimulating factor-differentiated human monocytes. J. Immunol. 2000, 164, 5952–5960. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.A.; Hewitson, T.D. Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytol. 2007, 257, 143–179. [Google Scholar] [PubMed]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-b1 induces a-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef]
- Evans, R.; Tian, Y.; Steadman, R.; Phillips, A. Tgf-b1-mediated fibroblast–myofibroblast terminal differentiation—The role of smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Annes, J.; Munger, J.; Rifkin, D. Making sense of latent TGF-β activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lyons, R.M.; Keski-Oja, J.; Moses, H.L. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J. Cell Biol. 1988, 106, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Van De Water, L.; Varney, S.; Tomasek, J.J. Mechanoregulation of the myofibroblast in wound contraction, scarring, and fibrosis: Opportunities for new therapeutic intervention. Adv. Wound Care 2013, 2, 122–141. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Muro, A.F.; Chauhan, A.K.; Gajovic, S.; Iaconcig, A.; Porro, F.; Stanta, G.; Baralle, F.E. Regulated splicing of the fibronectin eda exon is essential for proper skin wound healing and normal lifespan. J. Cell Biol. 2003, 162, 149–160. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, S.M.; Rumer, K.K.; Biroc, S.L.; Metz, R.P.; Singh, M.; Porter, W.; Schedin, P. Remodeling of the mammary microenvironment after lactation promotes breast tumor cell metastasis. Am. J. Pathol. 2006, 168, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Goddard, E.T.; Hill, R.C.; Barrett, A.; Betts, C.; Guo, Q.; Maller, O.; Borges, V.F.; Hansen, K.C.; Schedin, P. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. Int. J. Biochem. Cell Biol. 2016, 81, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Schedin, P.; Strange, R.; Mitrenga, T.; Wolfe, P.; Kaeck, M. Fibronectin fragments induce mmp activity in mouse mammary epithelial cells: Evidence for a role in mammary tissue remodeling. J. Cell Sci. 2000, 113, 795–806. [Google Scholar] [PubMed]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Keely, P.J. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 2011, 16, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Bhowmick, N.A.; Chytil, A.; Gorksa, A.E.; Brown, K.A.; Muraoka, R.; Arteaga, C.L.; Neilson, E.G.; Hayward, S.W.; Moses, H.L. Loss of TGF-beta type ii receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, msp- and hgf-mediated signaling networks. Oncogene 2005, 24, 5053–5068. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.B.; Jokar, I.; Chytil, A.; Moses, H.L.; Abel, T.; Cheng, N. Loss of one tgfbr2 allele in fibroblasts promotes metastasis in mmtv: Polyoma middle t transgenic and transplant mouse models of mammary tumor progression. Clin. Exp. Metastasis 2011, 28, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zheng, M.Q.; Lu, J.W.; Jiang, Q.; Wang, T.H.; Huang, X.E. Cxcl12-cxcr4 promotes proliferation and invasion of pancreatic cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 5403–5408. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Long, P.; Huang, Y.; Sun, F.; Wang, Z. Cxcl12/cxcr4 axis induces proliferation and invasion in human endometrial cancer. Am. J. Transl. Res. 2016, 8, 1719–1729. [Google Scholar] [PubMed]
- Bleul, C.C.; Fuhlbrigge, R.C.; Casasnovas, J.M.; Aiuti, A.; Springer, T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (sdf-1). J. Exp. Med. 1996, 184, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; von Ungern-Sternberg, S.N.; Seizer, P.; Schlegel, F.; Buttcher, M.; Sindhu, N.A.; Muller, S.; Mack, A.; Gawaz, M. Platelet-derived cxcl12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of cxcr4-cxcr7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Bader, D.A.; Robles, A.I.; Wangsa, D.; Harris, C.C.; Ried, T.; Yang, L. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genet. 2013, 9, e1003251. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S., 2nd; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Digiacomo, G.; Ziche, M.; Dello Sbarba, P.; Donnini, S.; Rovida, E. Prostaglandin e2 transactivates the colony-stimulating factor-1 receptor and synergizes with colony-stimulating factor-1 in the induction of macrophage migration via the mitogen-activated protein kinase erk1/2. FASEB J. 2015, 29, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The cox-2/pge2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Zaslona, Z.; Serezani, C.H.; Okunishi, K.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin e2 restrains macrophage maturation via e prostanoid receptor 2/protein kinase a signaling. Blood 2012, 119, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Montrose, D.C.; Zhou, X.K.; McNally, E.M.; Sue, E.; Yantiss, R.K.; Gross, S.S.; Leve, N.D.; Karoly, E.D.; Suen, C.S.; Ling, L.; et al. Celecoxib alters the intestinal microbiota and metabolome in association with reducing polyp burden. Cancer Prev. Res. 2016, 9, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Choi, H.K.; Lee, K.J.; Jung, J.Y.; Hur, G.Y.; Jung, K.H.; Kim, J.H.; Shin, C.; Shim, J.J.; In, K.H.; et al. The immune tolerance of cancer is mediated by ido that is inhibited by cox-2 inhibitors through regulatory T cells. J. Immunother. 2009, 32, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Yoon, Y.N.; Son, D.I.; Seok, S.H. Cyclooxygenase-2 inhibition blocks m2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS ONE 2013, 8, e63451. [Google Scholar] [CrossRef] [PubMed]
- Esbona, K.; Inman, D.; Saha, S.; Jeffery, J.; Schedin, P.; Wilke, L.; Keely, P. Cox-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 2016, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Callihan, E.B.; Gao, D.; Jindal, S.; Lyons, T.R.; Manthey, E.; Edgerton, S.; Urquhart, A.; Schedin, P.; Borges, V.F. Postpartum diagnosis demonstrates a high risk for metastasis and merits an expanded definition of pregnancy-associated breast cancer. Breast Cancer Res. Treat. 2013, 138, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Gelber, S.; Marotti, J.D.; White, S.; Ruddy, K.; Brachtel, E.F.; Schapira, L.; Come, S.E.; Borges, V.F.; Schedin, P.; et al. Molecular phenotype of breast cancer according to time since last pregnancy in a large cohort of young women. Oncologist 2015, 20, 713–718. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Q.; Betts, C.; Pennock, N.; Mitchell, E.; Schedin, P. Mammary Gland Involution Provides a Unique Model to Study the TGF-β Cancer Paradox. J. Clin. Med. 2017, 6, 10. https://doi.org/10.3390/jcm6010010
Guo Q, Betts C, Pennock N, Mitchell E, Schedin P. Mammary Gland Involution Provides a Unique Model to Study the TGF-β Cancer Paradox. Journal of Clinical Medicine. 2017; 6(1):10. https://doi.org/10.3390/jcm6010010
Chicago/Turabian StyleGuo, Qiuchen, Courtney Betts, Nathan Pennock, Elizabeth Mitchell, and Pepper Schedin. 2017. "Mammary Gland Involution Provides a Unique Model to Study the TGF-β Cancer Paradox" Journal of Clinical Medicine 6, no. 1: 10. https://doi.org/10.3390/jcm6010010