
Update of Endocrine Dysfunction following Pediatric Traumatic Brain Injury

Abstract
:1. Introduction
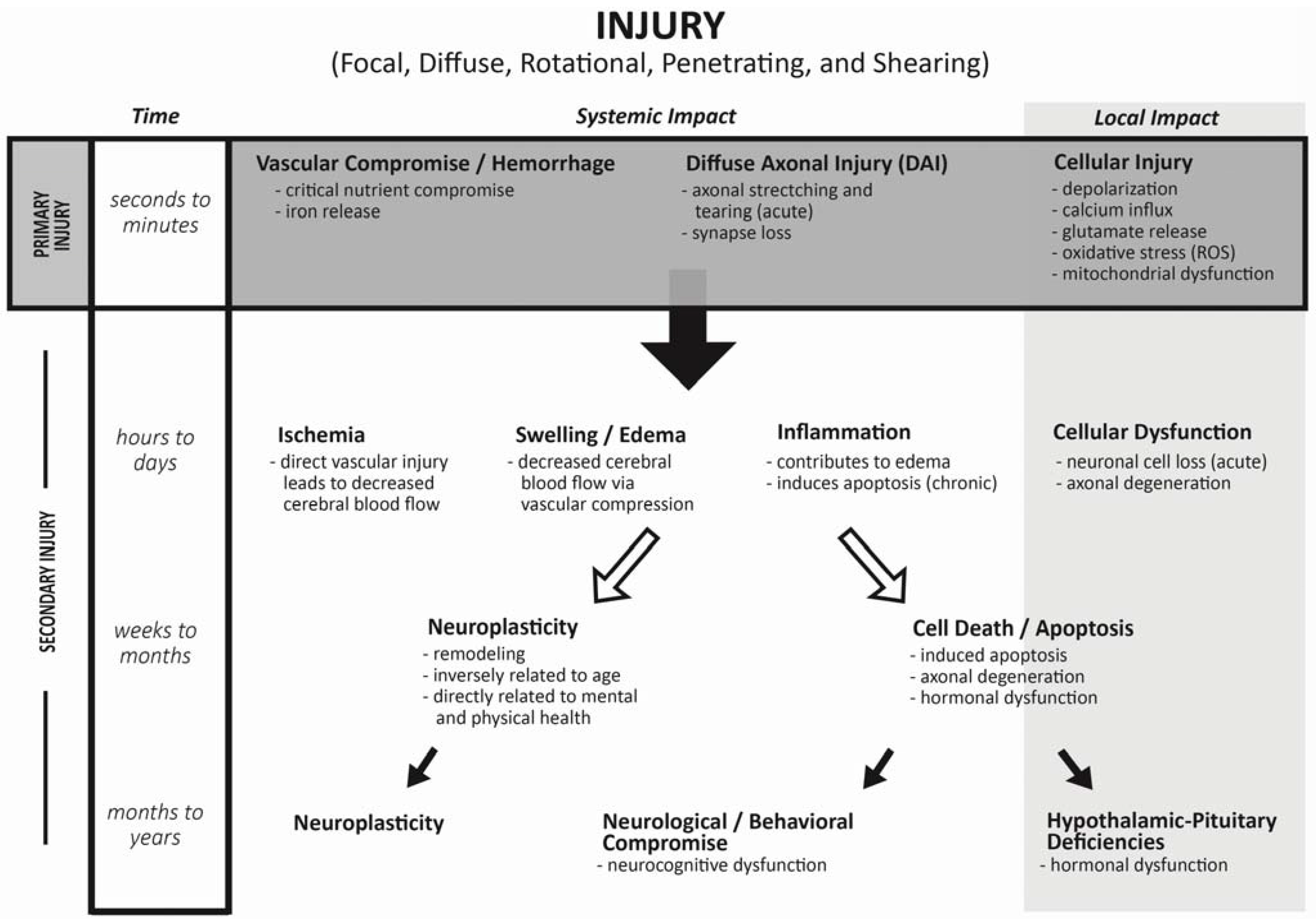
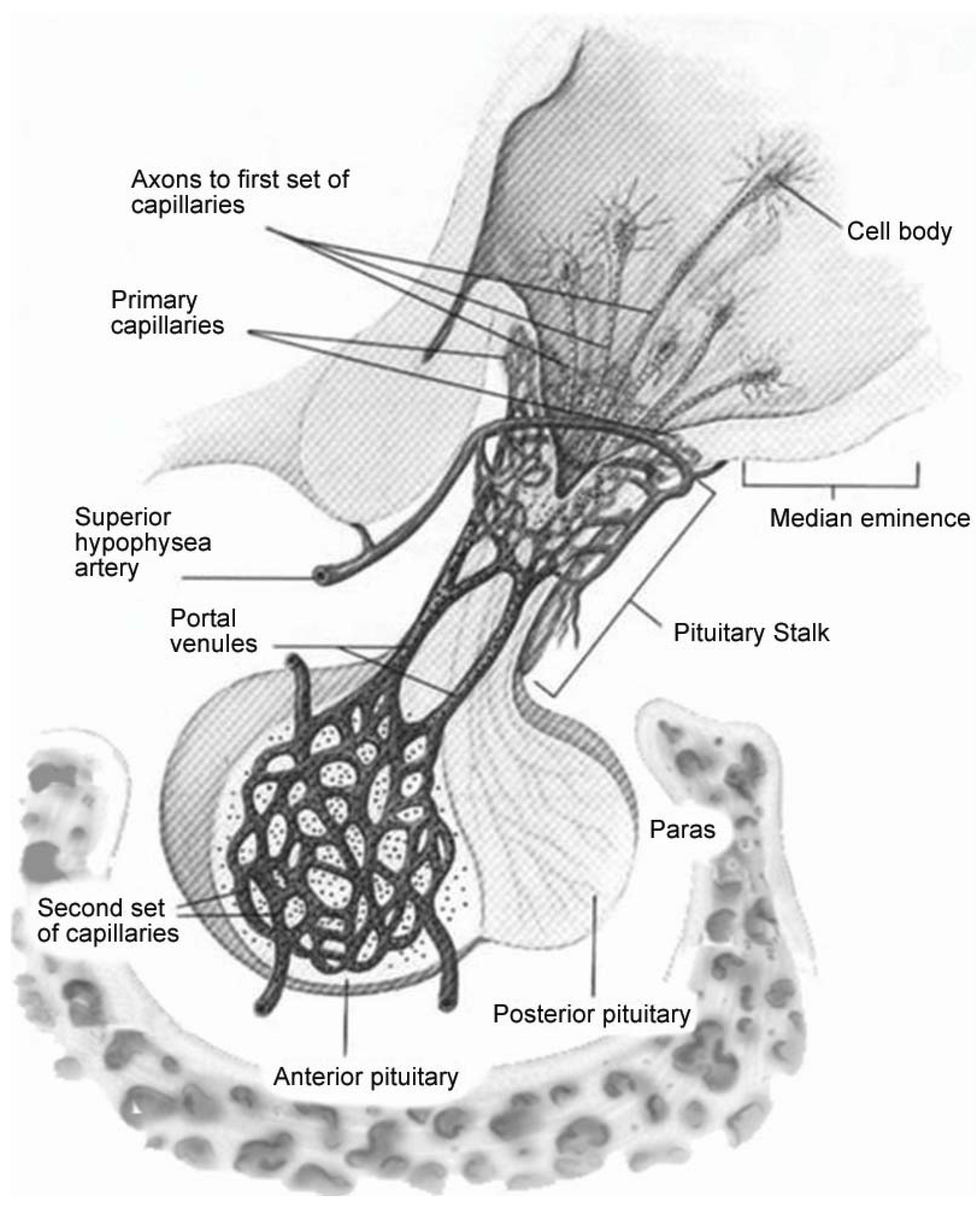
2. Mechanism of Injury during Traumatic Brain Injuries
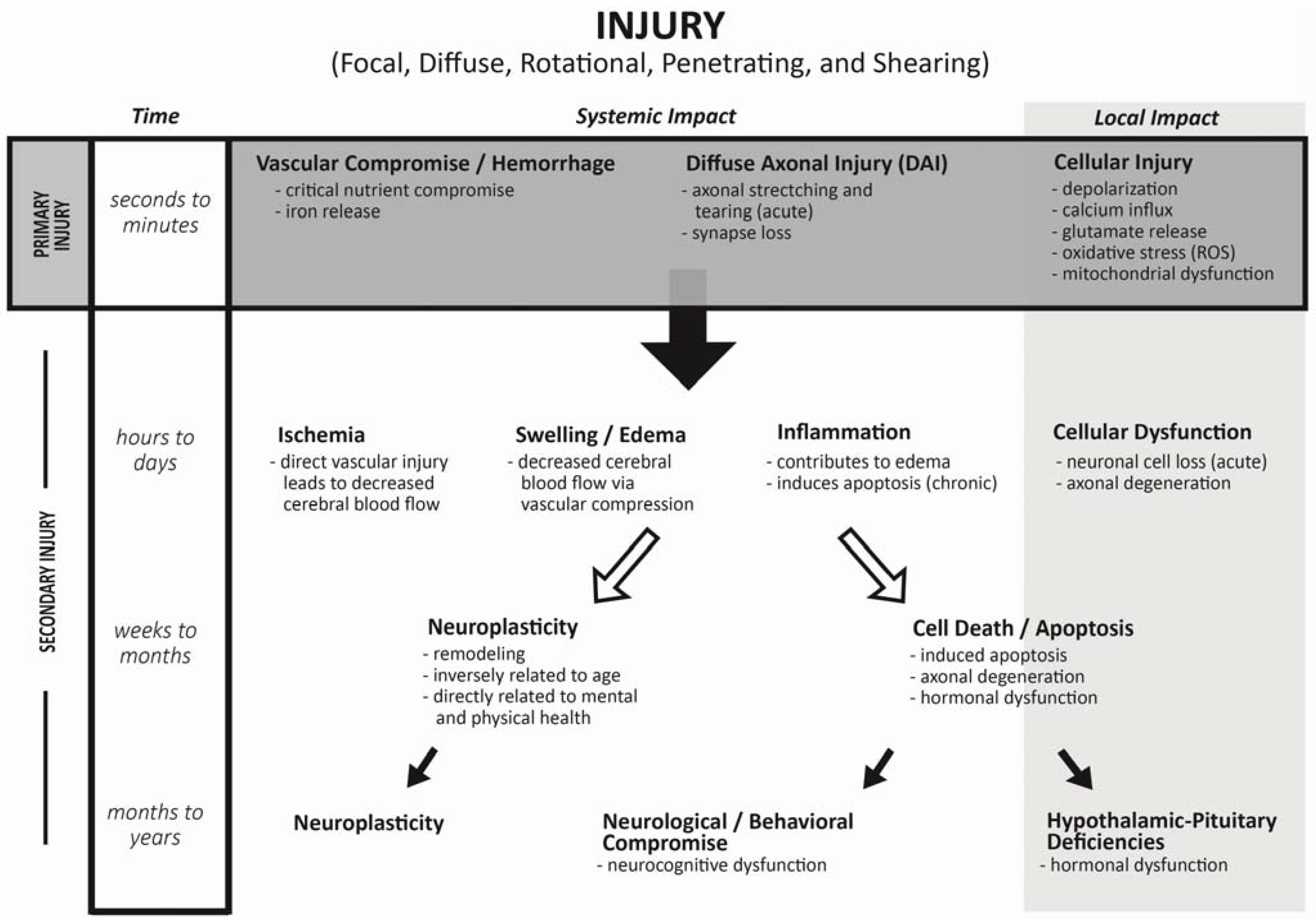

3. Incidence of Endocrine Changes Following TBI: Adult Literature
4. Acute Life Threatening Effects on Endocrine System in Children
5. Late Effects of TBI on Endocrine System in Children

{kind=link}
{kind=link}
| Author; year [reference] | Study Method # of patients | Age at injury (yo), Time after TBI until time of study (m or y) | TBI Severity (GSC) | Testing methods | Overall Prevalence of dysfunction | Pituitary Dysfunction by hormone |
|---|---|---|---|---|---|---|
| Einaudi 2006 [34] | Prospective 30 | Injury: 9.1 years old (0.25–15.5 yo) | 6 severe 9 moderate 15 mild | Baseline: T0, T6 & T12 GHRH + Arginine Glucagon | T0: 7 of 30 T6: 2 of 26 T12: 2 of 20 | T0: abnormal TFTs T6: low cortisol (2) T12: GHI (1), GHD (1) |
| Niederland 2007 [37] | Cross-sectional 26 | Injury: 8.9 yo Time to study: 30.6 ± 8.3m | Mixed | Screening TBI
vs. controls 1st GHST: L-DOPA 2nd GHST: ITT | 60% dysfunction 42% diminished GH | l-DOPA:GH 16.8 vs. 32 (p = 0.003) ITT:GH 20.5 vs. 27 (p = 0.06) Cortisol: 19 vs. 26 (p = 0.002) |
| Poomthavorn 2008 [38] | Cross-sectional (Questionaire) 54 | Injury: 9.7 yo (0.3–16.8) Time to study: 4.5 y (0.9–8.5) | All severe | Baseline: 29 of 54 Glucagon Stim if poor GV & low IGF (8 of 29) | 16.6% | 1 female precocious puberty 1 TSHD 2 gonadotropin def 3 partial ACTH def 2 GHD |
| Norwood 2010 [39] | Cross-sectional 32 pts | Injury: 12.7 yo Age at study (15.7 yo) | Mean: 5 Range: 3–15 | Overnight GH (<5 ng/mL) AND Arginine/glucagon (<7 ng/mL) | 34% failed either testing modality | 5 of 32 failed both 6 of 32 failed overnight 10 of 32 failed GHST |
| Kaulfers 2010 [47] | Prospective 31 | Injury: 11.6 yo | 24 severe | Screening: baseline, 3, 6 & 12 m 6m: overnight GH & TSH, ACTH stim 12m: GHST (Arg + Clon or GHRH) | Baseline: 2 of 3 DI resolved 21% abn TFTs 3m: all DI resolved 24% abn TFTs 5 of 9 oligomenorrhea | 6m: 13% low GH surge 46% low TSH surge 1 poor ACTH 12 m: 2 TSHD 1 GHD 3 males PP or rapid |
| Heather 2012 [40] | Cross-sectional 198 | Injury: 1.7 ±1.5 yo Time to Study: 6.5 ± 3.2 y | 27% severe 18% moderate 55% mild | Screening fasting Clonidine & Arginine (<5 mcg/L) | 33% GH peak < 10 mcg/L 8% GH peak < 5mcg/L 9% poor ACTH response 1% PP | No treatment initiated. All demonstrated normal growth 5 of 18 repeat GHST- only 1 failed 13 of 17 with AI passed retesting |
| Auble 2013 [41] | Cross-sectional 14 | Injury 0.5 yo (1–1.1) Time to Study: 2.5 y (2–9 y) | All severe: 11 required intubation 11 with seiz | Overnight TSH, GH sampling Fasting baseline and low-dose ACTH | 86% abnormal labs or height <10%ile | Most common: elev. Prolactin Blunted TSH surge (<50% rise) 2 with poor GH surge |
| Bellone 2013 [42] | Cross-sectional 70 | Injury: 8.1 ± 4.2 yo Time to Study: 1–9.1 y | 19 severe 11 moderate 40 mild | Baseline & 12m if poor GV GHST (GHRH + Arg) at 12m | Screening: 4 cases 6m: 20 of 70 poor GV 12m: 13 of 20 poor GV | Baseline: TSHD & ACTH def (1) FSH/LH def (1) ACTH def (1) PP (1) 12m: 4 of 13 GHD Total: 10% |
| Casano-Saucho 2013 [48] | Prospective 37 pts | 14 pts: age 0.2–2.3 yo 23 pts: age 7–19.9 yo | 22 severe 7 moderate 8 mild | <6 yo: baseline at 12 m >6 yo: baseline & 2 dynamic tests 3m & 12 m (glucagon/clonidine <10 ng/mL) | 3m: 11 of 23 GHD 10 of 23 ACTH 12m: 8 of 23 GHD 3 of 23 ACTH | <6 years old- no baseline or clinical abnormalities No sustained pubertal abnormality Transient thyroid 3 of 23 |
| Salomon-Estebanez 2014 [43] | Cross-sectional 36 | Injury: 3.3 yo Time to Study: 7.2 y | 36.6% severe & moderate 63.4% mild | Screening; provocative testing if abnormal | 4 low IGF markers 2 low cortisol | No dysfunction observed after clinical follow-up No provocative testing |
| Personnier 2014 [49] | Prospective 87 | Injury: 6.7 yo (0.8–15.2) | All severe | Baseline + 1st GHST (betaxolol, glucagon or glucagon only) 2nd GHST at 9 m after TBI if 1st <7 ng/mL (arginine, insulin) | 17% severe GHD 6 pts transient TFTs 1 with AI | 1st GHST: 35 of 87 failed 2nd GHST: 27 of 33 failed (22 with normal IGF values) Only 6 demonstrated poor growth |
6. Changes in Endocrine Function According to Specific Deficiency
6.1. GH Deficiency
6.2. Gonadotropin Deficiency
6.3. Precocious Puberty
6.4. ACTH Deficiency
6.5. Central Hypothyroidism
6.6. Hyperprolactinemia
6.7. Diabetes Insipidus (DI)
6.8. Predictors of Endocrinopathies
7. Time Course of Changes in Endocrine Function over Time after TBI
8. Conclusions/Recommendations
| Hormone test | Time of draw |
|---|---|
| Serum cortisol | 800 h |
| Free thyroxine (FT4) | 800 h |
| Thyrotropin (TSH) | 800 h and 1600 h |
| Insulin-like growth factor (IGF-I) | 800 h |
| Prolactin | 800 h |
| Persons in puberty or of pubertal age: Follicle-stimulating hormone (FSH) | 800 h |
| luteinizing hormone (LH), testosterone or estradiol | |
| Persons with polyuria: urine specific gravity, Na and plasma osmolality | After 12 h fasting |
| Clinical Assessment | |
| Height measurement and growth velocity (yearly) | |
| Pubertal Staging (yearly) | |
| Weight (yearly) | |
| Review of Systems (yearly): delayed puberty, lack of energy/stamina, reduced muscle mass, decreased bone density, changes in mood or scholastic decline | |
Acknowledgments
Author Contributions
Conflict of Interest
Abbreviations
| ACTH | adrenocorticotropic hormone |
| BMI | body mass index |
| CNS | central nervous system |
| DAI | diffuse axonal injury |
| DI | diabetes insipidus |
| FSH | follicle-stimulating hormone |
| FT4 | free thyroxine |
| GCS | Glasgow Coma Scale |
| GH | growth hormone |
| GHD | GH deficiency |
| GHRH | GH-releasing hormone |
| GHST | GH stimulation test |
| GnRH | gonadotropin-releasing hormone |
| GV | growth velocity |
| IGF-I | insulin-like growth factor-I |
| IGFBP3 | IGF-binding protein 3 |
| ITT | insulin tolerance test |
| LH | luteinizing hormone |
| PP | precocious puberty |
| PRL | prolactin |
| T3 | triiodothyronine |
| T4 | thyroxine |
| TBI | traumatic brain injury |
| TSH | thyroid stimulating hormone |
| TSHD | TSH deficiency or central hypothyroidism |
References
- Faul, M.; Xu, L.; Wald, M.M.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006; Centers for Disease Control and Prevention, National Center for Injury Prevention and Control: Atlanta, GA, USA, 2010.
- Olver, J.H.; Ponsford, J.L.; Curran, C.A. Outcome following TBI: A comparison between 2 and 5 years after injury. Brain Inj. 1996, 10, 841–848. [Google Scholar] [CrossRef] [PubMed]
- CDC. Available online: http://gov/traumaticbraininjury/statistics.html (accessed on 21 November 2014.
- McCarthy, M.L.; MacKenzie, E.J.; Durbin, D.R.; Aitken, M.E.; Jaffe, K.M.; Paidas, C.N.; Slomine, B.S.; Dorsch, A.M.; Christensen, J.R.; Ding, R.; et al. Health-related quality of life during the first year after traumatic brain injury. Arch. Pediatr. Adolesc. Med. 2006, 160, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Kreitschmann-Andermahr, I.; Ghigo, E.; Stalla, G.K.; Agha, A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. JAMA 2007, 298, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Phillips, J.; Thompson, C.J. Hypopituitarism following traumatic brain injury (TBI). Br. J. Neurosurg. 2007, 21, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Bushnik, T.; Englander, J.; Katznelson, L. Fatigue after TBI: Association with neuroendocrine abnormalities. Brain Inj. 2007, 21, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Tanrverdi, F.; De Bellis, A.; Bizzarro, A.; Sinisi, A.A.; Bellastella, G.; Pane, E.; Bellastella, A.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; et al. Antipituitary antibodies after traumatic brain injury: Is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur. J. Endocrinol. 2008, 159, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, D.; Pekic, S.; Stojanovic, M.; Zivkovic, V.; Djurovic, B.; Jovanovic, V.; Miljic, N.; Medic-Stojanoska, M.; Doknic, M.; Miljic, D.; et al. Chronic cognitive sequelae after traumatic brain injury are not related to growth hormone deficiency in adults. Eur. J. Neurol. 2010, 17, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.; Oeffner, A.; Schumm-Draeger, P.M.; Badorrek, F.; Brabant, G.; Gerbert, B.; Bornstein, S.; Zimmermann, A.; Weber, M.; Broecker-Preuss, M.; et al. Prevalence of anterior pituitary dysfunction in patients following traumatic brain injury in a German multi-centre screening program. Exp. Clin. Endocrinol. Diabetes 2010, 118, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, E.; Masel, B.; Aimaretti, G.; Leon-Carrion, J.; Casanueva, F.F.; Dominguez-Morales, M.R.; Elovic, E.; Perrone, K.; Stalla, G.; Thompson, C.; et al. Consensus guidelines on screening for hypopituitarism following traumatic brain injury. Brain Inj. 2005, 19, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- IUPUI. Available online: http://www.iupui.edu/ (accessed 23 July 2015).
- Hammoud, D.A.; Wasserman, B.A. Diffuse axonal injuries: Pathophysiology and imaging. Neuroimag. Clin. N. Am. 2002, 12, 205–216. [Google Scholar] [CrossRef]
- Munoz, A.; Urban, R. Neuroendocrine consequences of traumatic brain injury. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Marion, D.W.; Carlier, P.M. Problems with initial Glasgow Coma Scale assessment caused by prehospital treatment of patients with head injuries: Results of a national survey. J. Trauma 1994, 36, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.R.; Reiley, D.G.; Green, S.M. Interrater reliability of Glascow Coma Scale scores in the emergency department. Ann. Emerg. Med. 2004, 43, 215–223. [Google Scholar] [CrossRef]
- Jenny, C.; Hymel, K.P.; Ritzen, A.; Reinert, S.E.; Hay, T.C. Analysis of missed cases of abusive head trauma. JAMA 1999, 281, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.A.; Cockington, R.A.; Hanieh, A.; Raftos, J.; Reilly, P.L. Head injuries in infants and young children: The value of the Pediatric Coma Scale. Childs Nerv. Syst. 1991, 7, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Benvenga, S.; Campenni, A.; Ruggeri, R.M.; Trimarchi, F. Clinical Review 113: Hypopituitarism secondary to head trauma. J. Clin. Endocrinol. Metab. 2000, 85, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Senyurek, H.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. High risk of hypopituitarism after traumatic brain injury: A prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J. Clin. Endocrinol. Metab. 2009, 91, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Schneider, H.J.; Aimaretti, G.; Masel, B.E.; Casanueva, F.F.; Kelestimur, F. Pituitary dysfunction after traumatic brain injury: A clinical and pathophysiological approach. Endocr. Rev. 2015, 36, 305–342. [Google Scholar] [CrossRef] [PubMed]
- Aimaretti, G.; Ambrosio, M.R.; Di Somma, C.; Gasperi, M.; Cannavo, S.; Scaroni, C.; De Marinis, L.; Baldelli, R.; Bona, G.; Giordano, G.; et al. Hypopituitarism induced by traumatic brain injury in the transition phase. J. Endocrinol. Investig. 2005, 28, 984–989. [Google Scholar] [CrossRef]
- Nemes, O.; Kovacs, N.; Czeiter, E.; Kenyeres, P.; Tarjanyi, Z.; Bajnok, L.; Buki, A.; Doczi, T.; Mezosi, E. Predictors of post-traumatic pituitary failure during long-term follow-up. Hormones (Athens) 2014. [Google Scholar] [CrossRef] [PubMed]
- Bruce, D.A.; Schut, L.; Bruno, L.A.; Wood, J.H.; Sutton, L.N. Outcome following severe head injuries in children. J. Neurosurg. 1978, 48, 679–688. [Google Scholar] [CrossRef] [PubMed]
- High, W.M., Jr.; Briones-Galang, M.; Clark, J.A.; Gilkison, C.; Mossberg, K.A.; Zgaljardic, D.J.; Masel, B.E.; Urban, R.J. Effects of growth hormone replacement therapy on cognition after traumatic brain injury. J. Neurotrauma 2010, 27, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Reimunde, P.; Quintana, A.; Castañón, B.; Casteleiro, N.; Vilarnovo, Z.; Otero, A.; Devesa, A.; Otero-Cepeda, X.L.; Devesa, J. Effects of growth hormone (GH) replacement and cognitive rehabilitation after traumatic brain injury. Brain Inj. 2011, 25, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Morares, R.B.; Czepielewski, M.A.; Friedman, G.; de Borba, E.L. Diagnosis of adrenal failure in critically ill patients. Arq. Bras. Endocrinol. Metab. 2011, 55, 295–302. [Google Scholar] [CrossRef]
- Bolado, G.G.; Estebanez, M.; Arizkeuren, E.M.; Calcena, A.A.; Esteves, A.R.; Vela, A.; Rica, I. Assessment of Pituitary Function after Traumatic Brain Injury in Childhood; European Society for Pediatric Endocrinology (ESPE): Glasgow, UK, 2011. [Google Scholar]
- Barton, R.N.; Stoner, H.B.; Watson, S.M. Relationships among plasma cortisol, adrenocorticotrophin, and severity of injury in recently injured patients. J. Trauma 1987, 27, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Woolf, P.D.; Cox, C.; Kelly, M.; Nichols, D.; McDonald, J.V.; Hamill, R.W. The adrenocortical response to brain injury: Correlation with the severity of neurologic dysfunction, effects of intoxication, and patient outcome. Alcohol. Clin. Exp. Res. 1990, 14, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Cohan, P.; Wang, C.; McArthur, D.L.; Cook, S.W.; Dusick, J.R.; Armin, B.; Swerdloff, R.; Vespa, P.; Muizelaar, J.P.; Cryer, H.G.; et al. Acute secondary adrenal insufficiency after traumatic brain injury: A prospective study. Crit. Care Med. 2005, 33, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, R.; Brown, S.D.; Chang, Y.F.; Garcia-Fillion, P.; Adelson, P.D. Endocrine function in children acutely following severe traumatic brain injury. Childs Nerv. Syst. 2010, 26, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Einaudi, S.; Matarazzo, P.; Peretta, P.; Grossetti, R.; Giordano, F.; Altare, F.; Bondone, C.; Andreo, M.; Ivani, G.; Genitori, L.; et al. Hypothalamo-hypophysial dysfunction after traumatic brain injury in children and adolescents: A preliminary retrospective and prospective study. J. Pediatr. Endocrinol. Metab. 2006, 19, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Chiolero, R.L.; Lemarchand-Beraud, T.; Schutz, Y.; de Tribolet, N.; Bayer-Berger, M.; Freeman, J. Thyroid function in severely traumatized patients with or without head injury. Acta Endocrinol. (Copenh) 1988, 117, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.W.; Towbin, R.B.; Chernausek, S.D. Precocious puberty following severe head trauma. Am. J. Dis. Child. 1985, 139, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Niederland, T.; Makovi, H.; Gal, V.; Andreka, B.; Abraham, C.S.; Kovacs, J. Abnormalities of pituitary function after traumatic brain injury in children. J. Neurotrauma 2007, 24, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Poomthavorn, P.; Maixner, W.; Zacharin, M. Pituitary function in paediatric survivors of severe traumatic brain injury. Arch. Dis. Child. 2008, 93, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Norwood, K.W.; DeBoer, M.D.; Gurka, M.J.; Kuperminc, M.N.; Rogol, A.D.; Blackman, J.A.; Wamstad, J.B.; Buck, M.L.; Patrick, P.D. Traumatic brain injury in children and adolescents: Surveillance for pituitary dysfunction. Clin. Pediatr. 2010, 49, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Heather, N.L.; Jefferies, C.; Hofman, P.L.; Derraik, J.G.; Brennan, C.; Kelly, P.; Hamill, J.K.; Jones, R.G.; Rowe, D.L.; Cutfield, W.S. Permanent hypopituitarism is rare after structural brain injury in early childhood. J. Clin. Endocrinol. Metab. 2012, 97, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Auble, A.; Bollepalli, S.; Makoroff, K.; Weis, T.; Khoury, J.; Colliers, T.; Rose, S.R. Hypopituitarism in pediatric survivors of inflicted traumatic brain injury. J. Neurotrauma 2014, 31, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Bellone, S.; Einaudi, S.; Caputo, M.; Prodam, F.; Busti, A.; Belcastro, S.; Parlamento, S.; Zavattaro, M.; Verna, F.; Bondone, C.; et al. Measurement of height velocity is an useful marker for monitoring pituitary function in patients who had traumatic brain injury. Pituitary 2013, 16, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Estebanez, M.A.; Grau, G.; Vela, A.; Rodriguez, A.; Morterual, E.; Castano, L.; Rica, I. Is routine endocrine evaluation necessary after paediatric traumatic brain injury? J. Endocrinol. Investig. 2014, 37, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Medic-Stojanoska, M.; Pekic, S.; Curic, N.; Djilas-Ivanovic, D.; Popovic, V. Evolving hypopituitarism as a consequence of traumatic brain injury (TBI) in childhood—Call for attention. Endocrine 2007, 31, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Acerini, C.L.; Tasker, R.C. Endocrine sequelae of traumatic brain injury in childhood. Horm. Res. 2007, 68 (Suppl. S5), 14–17. [Google Scholar] [CrossRef] [PubMed]
- Kokshoorn, N.E.; Wassenaar, M.J.E.; Biermasz, N.R.; Roelfsema, F.; Smit, J.W.A.; Romijn, J.A.; Pereira, A.M. Hypopituitarism following traumatic brain injury: Prevalence is affected by the use of different dynamic tests and different normal values. Eur. J. Endocrinol. 2010, 162, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kaulfers, A.M.; Backeljauw, P.F.; Reifschneider, K.; Blum, S.; Michaud, L.; Weiss, M.; Rose, S.R. Endocrine dysfunction following traumatic brain injury in children. J. Pediatr. 2010, 157, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Casano-Sancho, P.; Suarez, L.; Ibanez, L.; Garcia-Fructuoso, G.; Medina, J.; Febrer, A. Pituitary dysfunction after traumatic brain injury in children: Is there a need for ongoing endocrine assessment? Clin. Endocrinol. 2013, 79, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Personnier, C.; Crosnier, H.; Meyer, P.; Chevignard, M.; Flechtner, I.; Boddaert, N.; Breton, S.; Mignot, C.; Dassa, Y.; Souberbielle, J.C.; et al. Prevalence of pituitary dysfunction after severe traumatic brain injury in children and adolescents: A large prospective study. J. Clin. Endocrinol. Metab. 2014, 99, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Thompson, C.J. High risk of hypogonadism after traumatic brain injury: Clinical implications. Pituitary 2005, 8, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Popii, V.; Baumann, G. Laboratory measurement of growth hormone. Clin. Chim. Acta 2004, 350, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nunez, S.B.; Municchi, G.; Barnes, K.M.; Rose, S.R. Insulin-like growth factor-1 (IGF-I) and IGF-binding protein-3 concentrations compared to stimulated and night growth hormone in the evaluation of short children: A clinical research center study. J. Clin. Endocrinol. Metab. 1996, 81, 1927–1932. [Google Scholar] [PubMed]
- Romshe, C.A.; Zipf, W.B.; Miser, A.; Miser, J.; Sotos, J.F.; Newton, W.A. Evaluation of growth hormone release and human growth hormone treatment in children with cranial irradiation-associated short stature. J. Pediatr. 1984, 104, 177–181. [Google Scholar] [CrossRef]
- Blatt, J.; Bercu, B.B.; Gillin, J.C.; Mendelson, W.B.; Poplack, D.G. Reduced pulsatile growth hormone secretion in children after therapy for acute lymphoblastic leukemia. J. Pediatr. 1984, 104, 182–186. [Google Scholar] [CrossRef]
- Rose, S.R.; Municchi, G. Six-hour and four-hour nocturnal sampling for growth hormone. J. Pediatr. Endocrinol. Metab. 1999, 12, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Bondanelli, M.; Ambrosio, M.R.; Cavazzini, L.; Bertocchi, A.; Zatelli, M.C.; Carli, A.; Valle, D.; Basaglia, N.; Uberti, E.C. Anterior pituitary function may predict functional and cognitive outcome in patients with traumatic brain injury undergoing rehabilitation. J. Neurotrauma 2007, 24, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Juul, A.; Struck, J.; Morgenthaler, N.G.; Kosteljanetz, M.; Feldt-Rasmussen, U. Acute and long-term pituitary insufficiency in traumatic brain injury: A prospective single-centre study. Clin. Endocrinol. (Oxf) 2007, 67, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Schneider, M.; Saller, B.; Petersenn, S.; Uhr, M.; Husemann, B.; von Rosen, F.; Stalla, G.K. Prevalence of anterior pituitary insufficiency 3 and 12 months after traumatic brain injury. Eur. J. Endocrinol. 2006, 154, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Bavisetty, S.; McArthur, D.L.; Dusick, J.R.; Wang, C.; Cohan, P.; Boscardin, W.J.; Swerdloff, R.; Levin, H.; Chang, D.J.; et al. Chronic hypopituitarism after traumatic brain injury: Risk assessment and relationship to outcome. Neurosurgery 2008, 62, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Stochholm, K.; Janukonyte, J.; Lehman Christensen, L.; Frystyk, J.; Anderson, M.; Laurberg, P.; Christiansen, J.S.; Feldt-Rasmussen, U. Prevalence of posttraumatic growth hormone deficiency is highly dependent on the diagnostic set-up: Results from Danish National Study on Posttraumatic Hypopituitarism. J. Clin. Endocrinol. Metab. 2014, 99, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Behan, L.A.; Agha, A. Endocrine consequences of adult traumatic brain injury. Horm. Res. 2007, 68 (Suppl. S5), 18–21. [Google Scholar] [CrossRef] [PubMed]
- Kokshoorn, N.E.; Smit, J.W.; Nieuwlaat, W.A.; Tiemensma, J.; Bisschop, P.H.; Groote Veldman, R.; Roelfsema, F.; Franken, A.A.; Wassenaar, M.J.; Biermasz, N.R.; et al. Low prevalence of hypopituitarism after traumatic brain injury: A multicenter study. Eur. J. Endocrinol. 2011, 165, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Grumbach, M.M. The neuroendocrinology of human puberty revisited. Horm. Res. 2002, 57 (Suppl. S2), 2–14. [Google Scholar] [CrossRef] [PubMed]
- Bourguinon, J.P.; Gerard, A.; Purnelle, G.; Czajkowski, V.; Yamanka, C.; Lemaitre, M.; Rigo, J.M.; Moonen, G.; Frainchimont, P. Duality of glutamatergic and GABAergic control of pulsatile GnRH secretion by rat hypothalamus explants: II. Reduced NR2C- and GABAA-receptor-mediated inhibition at the initiation of sexual maturation. J. Neuroendocrinol. 1997, 9, 193–199. [Google Scholar] [CrossRef]
- Kleindienst, A.; Brabant, G.; Bock, C.; Maser-Gluth, C.; Buchfelder, M. Neuroendocrine Function following Traumatic Brain Injury and Subsequent Intensive Care Treatment: A Prospective Longitudinal Evaluation. J. Neurotrauma 2009, 26, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, R.; Evans, A.T.; Villabona, C.V.; Abdu, T.A.; Ambrosi, B.; Atkinson, A.B.; Choi, C.H.; Clayton, R.N.; Courtney, C.H.; Gonc, E.N.; et al. Corticotropin tests for hypothalamic-pituitary-adrenal insufficiency: A metaanalysis. J. Clin. Endocrinol. Metab. 2008, 93, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Faglia, G.; Bitensky, L.; Pinchera, A.; Ferrari, C.; Paracchi, A.; Beck-Peccoz, P.; Ambrosi, B.; Spada, A. Thyrotropin secretion in patients with central hypothyroidism: Evidence for reduced biological activity of immunoreactive thyrotropin. J. Clin. Endocrinol. Metab. 1979, 48, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.R. Improved diagnosis of mild hypothyroidism using time-of-day normal ranges for thyrotropin. J. Pediatr. 2010, 157, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.R.; Nisula, B.C. Circadian variation of thyrotropin in childhood. J. Clin. Endocrinol. Metab. 1989, 68, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.R. Isolated central hypothyroidism in short stature. Pediatr. Res. 1995, 38, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.R. Cranial irradiation and central hypothyroidism. Trends Endocrinol. Metab. 2001, 12, 97–104. [Google Scholar] [CrossRef]
- Casanueva, F.F.; Leal, A.; Koltowska-Haggstrom, M.; Jonsson, P.; Goth, M.I. Traumatic brain injury as a relevant cause of growth hormone deficiency in adults: A KIMS-based study. Arch. Phys. Med. Rehabil. 2005, 86, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, D.B.; Held, K.; Wagner, P. Cognitive impairment following traumatic brain injury. Curr. Treat. Options Neurol. 2002, 4, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Acerini, C.L.; Tasker, R.C.; Bellone, S.; Bona, G.; Thompson, C.J.; Savage, M.O. Hypopituitarism in childhood and adolescence following traumatic brain injury: The case for prospective endocrine investigation. Eur. J. Endocrinol. 2006, 155, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.R.; Auble, B.A. Endocrine changes after pediatric traumatic brain injury. Pituitary 2012, 15, 267–275. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reifschneider, K.; Auble, B.A.; Rose, S.R. Update of Endocrine Dysfunction following Pediatric Traumatic Brain Injury. J. Clin. Med. 2015, 4, 1536-1560. https://doi.org/10.3390/jcm4081536
Reifschneider K, Auble BA, Rose SR. Update of Endocrine Dysfunction following Pediatric Traumatic Brain Injury. Journal of Clinical Medicine. 2015; 4(8):1536-1560. https://doi.org/10.3390/jcm4081536
Chicago/Turabian StyleReifschneider, Kent, Bethany A. Auble, and Susan R. Rose. 2015. "Update of Endocrine Dysfunction following Pediatric Traumatic Brain Injury" Journal of Clinical Medicine 4, no. 8: 1536-1560. https://doi.org/10.3390/jcm4081536