
Tapping Stem Cells to Target AMD: Challenges and Prospects

Abstract
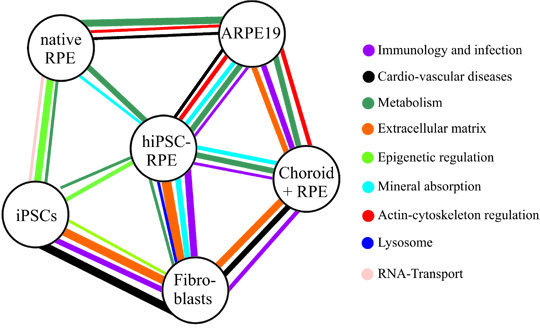
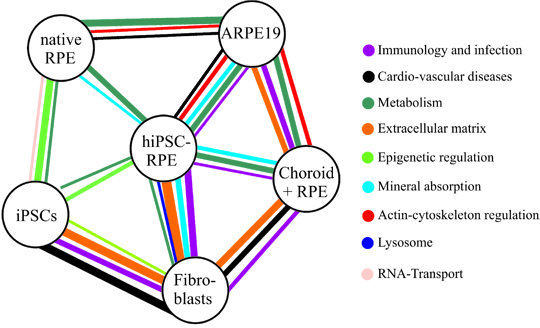
:
1. Introduction
2. Stem Cells: Numerous Types, Infinite Potential
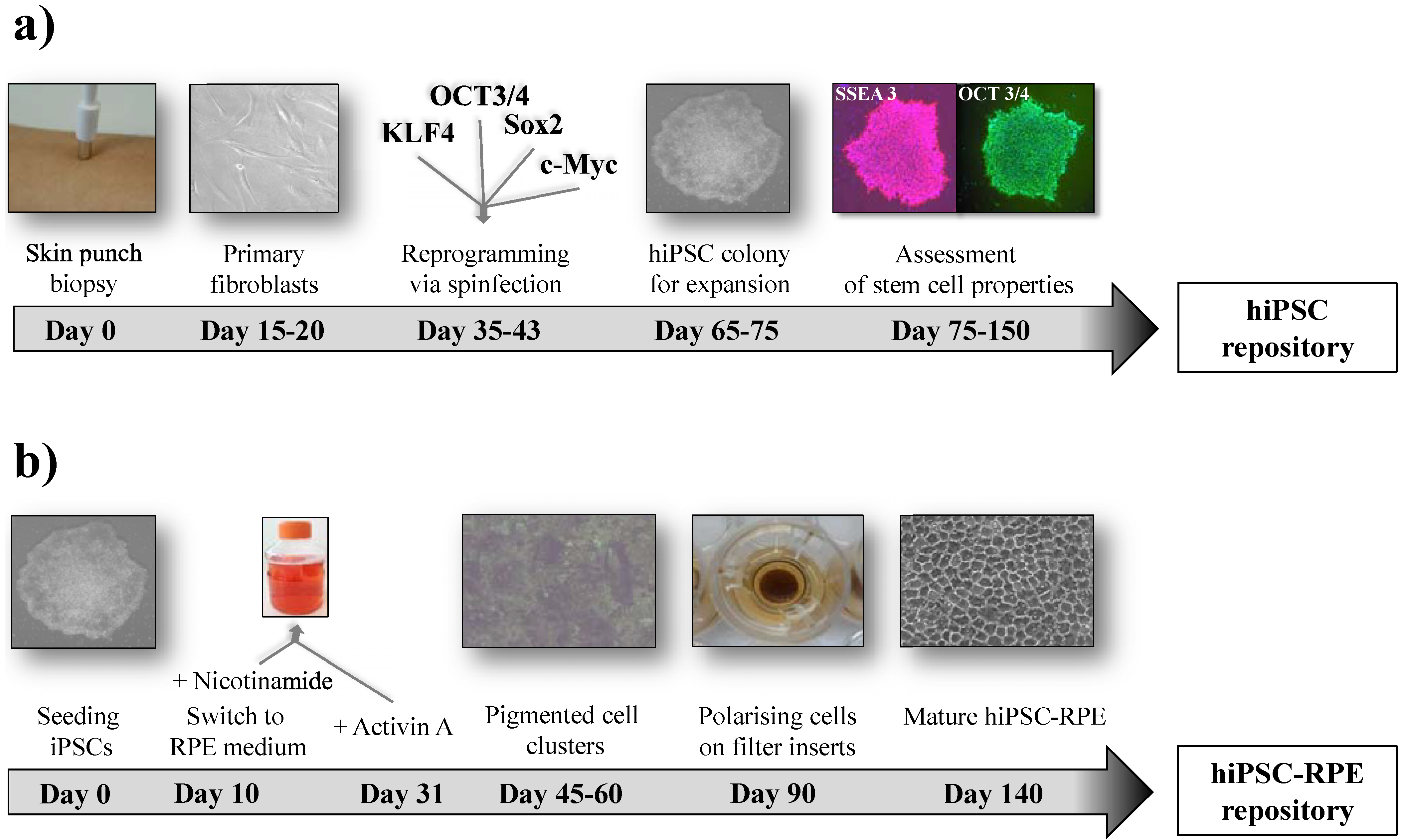
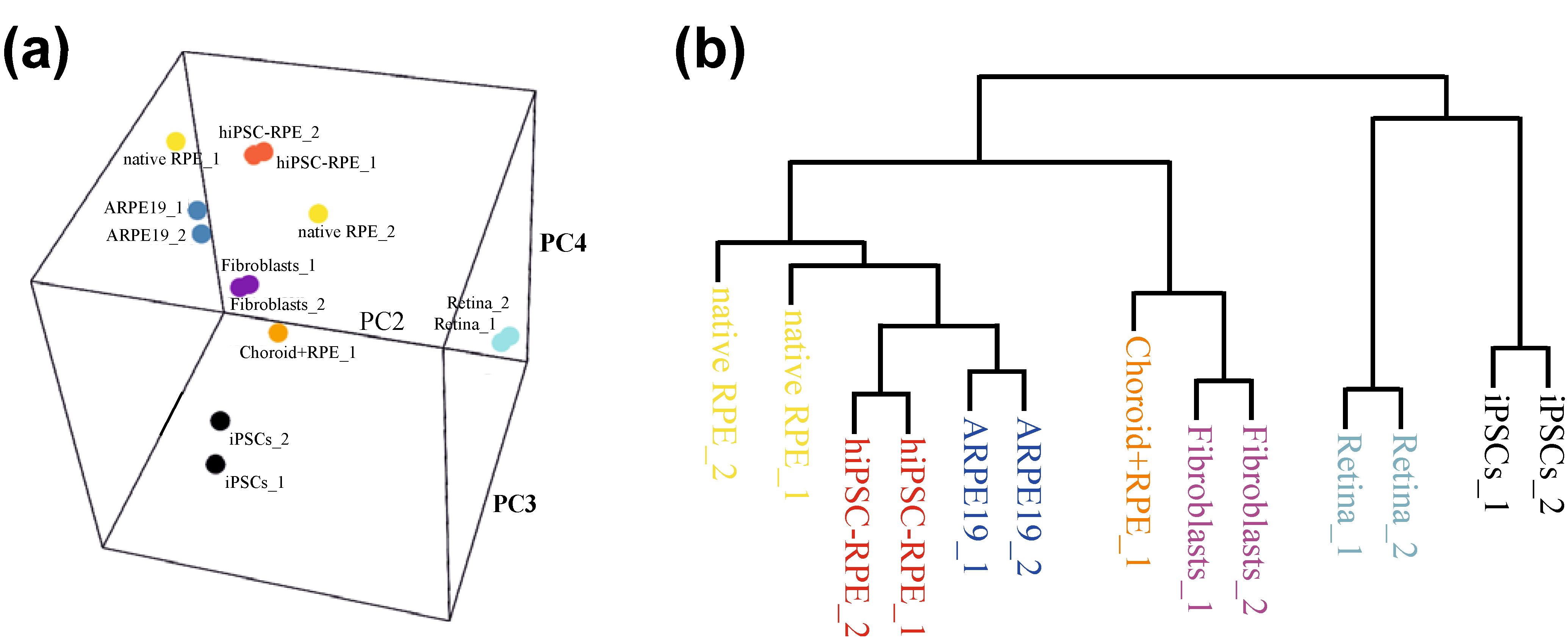
3. iPSC: The Stem Cell of the Future?
4. Disease Modelling of AMD: Current Status

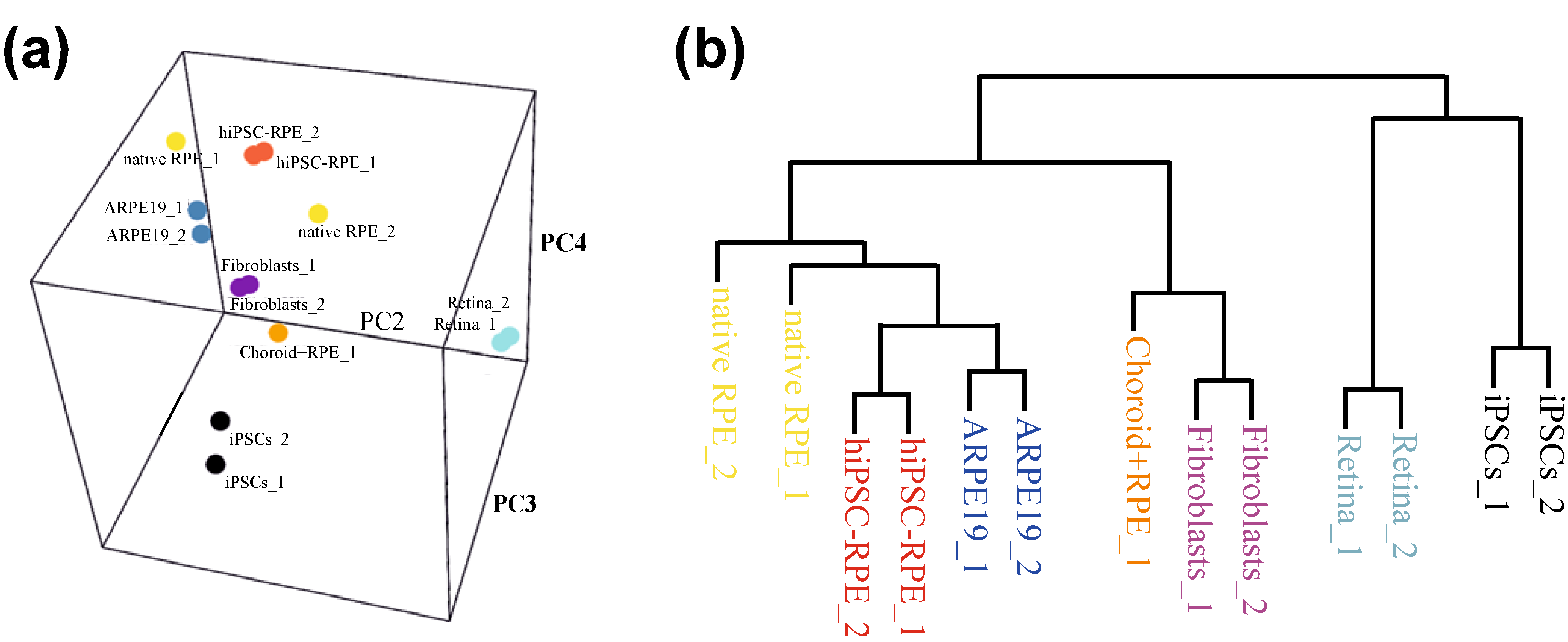

5. Disease Modelling of AMD: Future Possibilities
6. Cell-Based Therapy in AMD: Current and Projected Clinical Trials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Centre | Year of Launch/Status | (Stem) Cell Type Used | Main Facts | Publications/Sources (NCT = ClinicalTrials.gov Identifier) |
|---|---|---|---|---|
| A | ||||
| Jules Stein Eye Institute at University of California Los Angeles (UCLA), USA; Advanced Cell Technology, Inc., Marlborough, Massachusetts, MA, USA | 2011/preliminary report published in 2012 | hESC-derived RPE suspension |
| Schwartz et al., 2012 [106]; NCT01345006; NCT01344993 |
| Multi center USA (Jules Stein Eye Institute at UCLA, Los Angeles, LA, USA; Bascom Palmer Eye Institute, Miami, FL, USA; Wills Eye Institute-Mid Atlantic Retina, Philadelphia, PA, USA; Mass Eye and Ear, Boston, USA); Advanced Cell Technology, Inc., Marlborough, Massachusetts, MA, USA | 2011/report published in 2014 | hESC-derived RPE suspension |
| Schwartz et al., 2014 [32]; NCT01345006; NCT01344993 |
| University College London, Moorfields Eye Hospital, London, U.K.; Pfizer, Walton Oaks, U.K. | 2007/stem cell transplantation trial approved in 2013, ongoing | hESC-derived RPE sheets |
| Carr et al., 2013 [109]; NCT01691261 |
| Riken Institute, Kobe, Japan | 2013/ongoing | autologous hiPSC-derived RPE sheets |
| Kamao et al., 2014 [110] |
| B | ||||
| Hollywood Eye Institute, Cooper City, Florida, FL, USA; Bioheart, Inc., Sunrise, Florida, FL, USA | 2013/completion 2016 (estimated) | autologous adipose-derived stem cells (ASCs) |
| NCT02024269 |
| University of California; Davis Eye Center, Sacramento, California, CA, USA | 2012/completion 2014 (estimated) | autologous CD34+ bone marrow stem cells (BMSCs) |
| Park et al., 2012 [112]; NCT01736059 |
| Multi center USA; Stem Cells, Inc., Newark, California, CA, USA | 2012/completion 2015 (estimated) | human central nervous system stem cells (HuCNS-SC) |
| McGill et al., 2012 [113]; NCT01632527 |
| Rubens Siqueira Research Centre, São Paulo, Brazil; University of Sao Paulo, São Paulo, Brazil | 2011/completion January, 2014 (estimated) | autologous BMSC |
| Siqueira et al., 2011 [114]; NCT01518127 |
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2. [Google Scholar] [CrossRef]
- Brown, D.M.; Kaiser, P.K.; Michels, M.; Soubrane, G.; Heier, J.S.; Kim, R.Y.; Sy, J.P.; Schneider, S. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J. Bevacizumab versus ranibizumab for AMD. N. Engl. J. Med. 2011, 364, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Augood, C.A.; Vingerling, J.R.; de Jong, P.T.; Chakravarthy, U.; Seland, J.; Soubrane, G.; Tomazzoli, L.; Topouzis, F.; Bentham, G.; Rahu, M.; et al. Prevalence of age-related maculopathy in older Europeans: The European Eye Study (EUREYE). Arch. Ophthalmol. 2006, 124, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Klein, B.E.; Knudtson, M.D.; Meuer, S.M.; Swift, M.; Gangnon, R.E. Fifteen-year cumulative incidence of age-related macular degeneration: The Beaver Dam Eye Study. Ophthalmology 2007, 114, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-Related Macular Degeneration: Genetics and Biology Coming Together. Annu. Rev. Genomics Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Buitendijk, G.H.; Rochtchina, E.; Myers, C.; van Duijn, C.M.; Lee, K.E.; Klein, B.E.; Meuer, S.M.; de Jong, P.T.; Holliday, E.G.; Tan, A.G.; et al. Prediction of age-related macular degeneration in the general population: The Three Continent AMD Consortium. Ophthalmology 2013, 120, 2644–2655. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Wong, T.Y.; Fletcher, A.; Piault, E.; Evans, C.; Zlateva, G.; Buggage, R.; Pleil, A.; Mitchell, P. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 2010, 10. [Google Scholar] [CrossRef]
- Tomany, S.C.; Wang, J.J.; van, L.R.; Klein, R.; Mitchell, P.; Vingerling, J.R.; Klein, B.E.; Smith, W.; de Jong, P.T. Risk factors for incident age-related macular degeneration: Pooled findings from 3 continents. Ophthalmology 2004, 111, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Sperduto, R.D.; Sangiovanni, J.P.; Kurinij, N.; Davis, M.D. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report No. 35. Ophthalmology 2013, 120, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Clemons, T.E.; Sangiovanni, J.P.; Danis, R.P.; Ferris, F.L., III; Elman, M.J.; Antoszyk, A.N.; Ruby, A.J.; Orth, D.; Bressler, S.B.; et al. Secondary Analyses of the Effects of Lutein/Zeaxanthin on Age-Related Macular Degeneration Progression: AREDS2 Report No. 3. JAMA Ophthalmol. 2014, 132, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Buitendijk, G.H.; Rochtchina, E.; Lee, K.E.; Klein, B.E.; van Duijn, C.M.; Flood, V.M.; Meuer, S.M.; Attia, J.; Myers, C.; et al. Genetic Susceptibility, Dietary Antioxidants, and Long-term Incidence of Age-related Macular Degeneration in Two Populations. Ophthalmology 2014, 121, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmann, F.; Fritsche, L.G.; Keilhauer, C.N.; Heid, I.M.; Weber, B.H. Modelling the genetic risk in age-related macular degeneration. PLoS One 2012, 7, e37979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Cote, J.; Page, W.F.; Aggen, S.H.; Neale, M.C. The US twin study of age-related macular degeneration: Relative roles of genetic and environmental influences. Arch. Ophthalmol. 2005, 123, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Lauer, N.; Skerka, C. The role of complement in AMD. Adv. Exp. Med. Biol. 2010, 703, 9–24. [Google Scholar] [PubMed]
- De Jong, P.T. Age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, M.A. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004, 122, 598–614. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509. [Google Scholar] [CrossRef]
- Adhi, M.; Duker, J.S. Optical coherence tomography—Current and future applications. Curr. Opin. Ophthalmol. 2013, 24, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Kay, D.B.; Scoles, D.; Dubra, A.; Lombardo, M. Adaptive optics retinal imaging—Clinical opportunities and challenges. Curr. Eye Res. 2013, 38, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Eter, N. Molecular imaging in the eye. Br. J. Ophthalmol. 2010, 94, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Stein-Streilein, J. Mechanisms of immune privilege in the posterior eye. Int. Rev. Immunol. 2013, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Caspi, R.R. Ocular immune privilege. F1000 Biol. Rep. 2010, 2. [Google Scholar] [CrossRef]
- Ramsden, C.M.; Powner, M.B.; Carr, A.J.; Smart, M.J.; Da, C.L.; Coffey, P.J. Stem cells in retinal regeneration: Past, present and future. Development 2013, 140, 2576–2585. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Mullins, R.F.; Stone, E.M. Stem cells for investigation and treatment of inherited retinal disease. Hum. Mol. Genet. 2014, 23. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Induced pluripotent stem cells: Past, present, and future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Kim, C. Disease modeling and cell based therapy with iPSC: Future therapeutic option with fast and safe application. Blood Res. 2014, 49, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Vazin, T.; Freed, W.J. Human embryonic stem cells: Derivation, culture, and differentiation: A review. Restor. Neurol. Neurosci. 2010, 28, 589–603. [Google Scholar] [PubMed]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2014. [Google Scholar] [CrossRef]
- Chung, Y.; Klimanskaya, I.; Becker, S.; Li, T.; Maserati, M.; Lu, S.J.; Zdravkovic, T.; Ilic, D.; Genbacev, O.; Fisher, S.; et al. Human embryonic stem cell lines generated without embryo destruction. Cell Stem Cell 2008, 2, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Korbling, M.; Estrov, Z. Adult stem cells for tissue repair—A new therapeutic concept? N. Engl. J. Med. 2003, 349, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Frenette, P.S.; Pinho, S.; Lucas, D.; Scheiermann, C. Mesenchymal stem cell: Keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu. Rev. Immunol. 2013, 31, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Gimble, J.M.; Katz, A.J.; Bunnell, B.A. Adipose-derived stem cells for regenerative medicine. Circ. Res. 2007, 100, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Dhamodaran, K.; Subramani, M.; Ponnalagu, M.; Shetty, R.; Das, D. Ocular stem cells: A status update! Stem Cell Res. Ther. 2014, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Bhatia, B.; Jayaram, H.; Becker, S.; Jones, M.F.; Cottrill, P.B.; Khaw, P.T.; Salt, T.E.; Limb, G.A. Human Muller glia with stem cell characteristics differentiate into retinal ganglion cell (RGC) precursors in vitro and partially restore RGC function in vivo following transplantation. Stem Cells Transl. Med. 2012, 1, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Health Quality Ontario. Limbal stem cell transplantation: An evidence-based analysis. Ont. Health Technol. Assess. Ser. 2008, 8, 1–58. [Google Scholar]
- Li, D.Q.; Wang, Z.; Yoon, K.C.; Bian, F. Characterization, isolation, expansion and clinical therapy of human corneal epithelial stem/progenitor cells. J. Stem Cells 2014, 9, 79–91. [Google Scholar] [PubMed]
- Reh, T.A.; Fischer, A.J. Retinal stem cells. Methods Enzymol. 2006, 419, 52–73. [Google Scholar] [PubMed]
- Mayer, E.J.; Carter, D.A.; Ren, Y.; Hughes, E.H.; Rice, C.M.; Halfpenny, C.A.; Scolding, N.J.; Dick, A.D. Neural progenitor cells from postmortem adult human retina. Br. J. Ophthalmol. 2005, 89, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Zhao, X.; Ahmad, I. Stem cell therapy for retinal degeneration: Retinal neurons from heterologous sources. Semin. Ophthalmol. 2005, 20, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; ntosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Nagata, N.; Kurokawa, H.; Yamanaka, S. iPS cells: A game changer for future medicine. EMBO J. 2014, 33, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Zeng, F. Integration-free methods for generating induced pluripotent stem cells. Genomics Proteomics Bioinform. 2013, 11, 284–287. [Google Scholar] [CrossRef]
- Heng, B.C.; Fussenegger, M. Integration-free reprogramming of human somatic cells to induced pluripotent stem cells (iPSCs) without viral vectors, recombinant DNA, and genetic modification. Methods Mol. Biol. 2014, 1151, 75–94. [Google Scholar] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Slukvin, I. Generation of transgene-free iPSC lines from human normal and neoplastic blood cells using episomal vectors. Methods Mol. Biol. 2013, 997, 163–176. [Google Scholar] [PubMed]
- Narsinh, K.H.; Jia, F.; Robbins, R.C.; Kay, M.A.; Longaker, M.T.; Wu, J.C. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 2011, 6, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Christoforou, N.; Fattahi, A.; Herzog, R.W.; Leong, K.W. A robust strategy for negative selection of Cre-loxP recombination-based excision of transgenes in induced pluripotent stem cells. PLoS One 2013, 8, e64342. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.W.; Kim, W.H.; Williams, D.R. Reprogram or reboot: Small molecule approaches for the production of induced pluripotent stem cells and direct cell reprogramming. ACS Chem. Biol. 2014, 9, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Heng, B.C.; Richards, M. Induced Pluripotent Stem Cells (iPSC)—Can direct delivery of transcription factors into the cytosol overcome the perils of permanent genetic modification? Minim. Invasive. Ther. Allied Technol. 2008, 17, 326–327. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Ohnishi, H.; Oda, Y.; Tadokoro, M.; Sasao, M.; Kato, H.; Hattori, K.; Ohgushi, H. Generation of induced pluripotent stem cells from human adipose-derived stem cells without c-MYC. Tissue Eng. Part A 2010, 16, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Shen, Y.; Xue, Z.; Bibikova, M.; April, C.; Liu, Z.; Cheng, L.; Nagy, A.; Pellegrini, M.; Fan, J.B.; et al. A panel of CpG methylation sites distinguishes human embryonic stem cells and induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 36–43. [Google Scholar] [CrossRef]
- Ruiz, S.; Diep, D.; Gore, A.; Panopoulos, A.D.; Montserrat, N.; Plongthongkum, N.; Kumar, S.; Fung, H.; Giorgetti, A.; Bilic, J.; et al. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2012, 109, 16196–16201. [Google Scholar] [PubMed]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Ng, S.H.; Sharma, V.; Neculai, D.; Hussein, S.; Sam, M.; Trinh, Q.; Church, G.M.; McPherson, J.D.; Nagy, A.; et al. Elevated coding mutation rate during the reprogramming of human somatic cells into induced pluripotent stem cells. Stem Cells 2012, 30, 435–40. [Google Scholar] [CrossRef]
- Young, M.A.; Larson, D.E.; Sun, C.W.; George, D.R.; Ding, L.; Miller, C.A.; Lin, L.; Pawlik, K.M.; Chen, K.; Fan, X.; et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell 2012, 10, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Yeo, G.W.; Kainohana, O.; Marsala, M.; Gage, F.H.; Muotri, A.R. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PLoS One 2009, 4, e7076. [Google Scholar] [CrossRef] [PubMed]
- Vitaloni, M.; Pulecio, J.; Bilic, J.; Kuebler, B.; Laricchia-Robbio, L.; Izpisua Belmonte, J.C. MicroRNAs contribute to induced pluripotent stem cell somatic donor memory. J. Biol. Chem. 2014, 289, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Anguera, M.C.; Sadreyev, R.; Zhang, Z.; Szanto, A.; Payer, B.; Sheridan, S.D.; Kwok, S.; Haggarty, S.J.; Sur, M.; Alvarez, J.; et al. Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell 2012, 11, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.L.; Byron, M.; Butler, J.; Becker, K.A.; Nelson, A.; Amit, M.; Itskovitz-Eldor, J.; Stein, J.; Stein, G.; Ware, C.; et al. X-inactivation reveals epigenetic anomalies in most hESC but identifies sublines that initiate as expected. J. Cell. Physiol. 2008, 216, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Jeon, Y.; Anguera, M.C.; Lee, J.T. X-chromosome epigenetic reprogramming in pluripotent stem cells via noncoding genes. Semin. Cell Dev. Biol. 2011, 22, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Nazor, K.L.; Altun, G.; Lynch, C.; Tran, H.; Harness, J.V.; Slavin, I.; Garitaonandia, I.; Muller, F.J.; Wang, Y.C.; Boscolo, F.S.; et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 2012, 10, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Matsuno, Y.; Fouse, S.D.; Rao, N.; Root, S.; Xu, R.; Pellegrini, M.; Riggs, A.D.; Fan, G. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc. Natl. Acad. Sci. USA 2008, 105, 4709–4714. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.S.; Rowntree, R.K.; Mekhoubad, S.; Lee, J.T. X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4820–4825. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Kokaia, Z.; Lindvall, O.; Brustle, O. Emerging concepts in neural stem cell research: Autologous repair and cell-based disease modelling. Lancet Neurol. 2009, 8, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Silva, C.L.; Cabral, J.M. Genomic instability in human stem cells: Current status and future challenges. Stem Cells 2014, 32, 2824–2832. [Google Scholar] [CrossRef]
- Adams, W.J.; Zhang, Y.; Cloutier, J.; Kuchimanchi, P.; Newton, G.; Sehrawat, S.; Aird, W.C.; Mayadas, T.N.; Luscinskas, F.W.; Garcia-Cardena, G.; et al. Functional vascular endothelium derived from human induced pluripotent stem cells. Stem Cell Rep. 2013, 1, 105–113. [Google Scholar] [CrossRef]
- Tucker, B.A.; Mullins, R.F.; Streb, L.M.; Anfinson, K.; Eyestone, M.E.; Kaalberg, E.; Riker, M.J.; Drack, A.V.; Braun, T.A.; Stone, E.M.; et al. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. Elife 2013, 2, e00824. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.B.; Okamoto, S.; Osakada, F.; Homma, K.; Assawachananont, J.; Hirami, Y.; Iwata, T.; Takahashi, M. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS One 2011, 6, e17084. [Google Scholar] [CrossRef] [PubMed]
- Cramer, A.O.; MacLaren, R.E. Translating induced pluripotent stem cells from bench to bedside: Application to retinal diseases. Curr. Gene Ther. 2013, 13, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.S.; Phillips, M.J.; Pinilla, I.; Hei, D.; Gamm, D.M. Induced pluripotent stem cells as custom therapeutics for retinal repair: Progress and rationale. Exp. Eye Res. 2014, 123, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, S.; Balasubramanian, S.; Babai, N.; Qiu, F.; Eudy, J.D.; Thoreson, W.B.; Ahmad, I. Induced pluripotent stem cells generate both retinal ganglion cells and photoreceptors: Therapeutic implications in degenerative changes in glaucoma and age-related macular degeneration. Stem Cells 2010, 28, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Klimanskaya, I.; Hipp, J.; Rezai, K.A.; West, M.; Atala, A.; Lanza, R. Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using transcriptomics. Cloning Stem Cells 2004, 6, 217–245. [Google Scholar] [CrossRef] [PubMed]
- Vugler, A.; Carr, A.J.; Lawrence, J.; Chen, L.L.; Burrell, K.; Wright, A.; Lundh, P.; Semo, M.; Ahmado, A.; Gias, C.; et al. Elucidating the phenomenon of HESC-derived RPE: Anatomy of cell genesis, expansion and retinal transplantation. Exp. Neurol. 2008, 214, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Lim, S.L.; Grob, S.; Zhang, K. Induced pluripotent stem cell therapies for geographic atrophy of age-related macular degeneration. Semin. Ophthalmol. 2011, 26, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Shen, W.; Kuai, D.; Martin, J.M.; Guo, X.; Smith, M.A.; Perez, E.T.; Phillips, M.J.; Simonett, J.M.; Wallace, K.A.; et al. iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum. Mol. Genet. 2013, 22, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, D.E.; Pennington, B.O.; Croze, R.H.; Hinman, C.R.; Coffey, P.J.; Clegg, D.O. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl. Med. 2013, 2, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Krohne, T.U.; Westenskow, P.D.; Kurihara, T.; Friedlander, D.F.; Lehmann, M.; Dorsey, A.L.; Li, W.; Zhu, S.; Schultz, A.; Wang, J.; et al. Generation of retinal pigment epithelial cells from small molecules and OCT4 reprogrammed human induced pluripotent stem cells. Stem Cells Transl. Med. 2012, 1, 96–109. [Google Scholar] [CrossRef]
- Rowland, T.J.; Blaschke, A.J.; Buchholz, D.E.; Hikita, S.T.; Johnson, L.V.; Clegg, D.O. Differentiation of human pluripotent stem cells to retinal pigmented epithelium in defined conditions using purified extracellular matrix proteins. J. Tissue Eng. Regen. Med. 2013, 7, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Phillips, M.J.; Kuai, D.; Meyer, J.; Martin, J.M.; Smith, M.A.; Perez, E.T.; Shen, W.; Wallace, K.A.; Capowski, E.E.; et al. Functional analysis of serially expanded human iPS cell-derived RPE cultures. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6767–6778. [Google Scholar] [CrossRef]
- Zhu, Y.; Carido, M.; Meinhardt, A.; Kurth, T.; Karl, M.O.; Ader, M.; Tanaka, E.M. Three-dimensional neuroepithelial culture from human embryonic stem cells and its use for quantitative conversion to retinal pigment epithelium. PLoS One 2013, 8, e54552. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Zimmermann, S.J.; Milenkovic, V.M.; Rosendahl, S.M.; Grassmann, F.; Milenkovic, A.; Hehr, U.; Federlin, M.; Wetzel, C.H.; Helbig, H.; et al. In-Depth Characterisation of Retinal Pigment Epithelium (RPE) Cells Derived from Human Induced Pluripotent Stem Cells (hiPSC). Neuromol. Med. 2014, 16, 551–564. [Google Scholar] [CrossRef]
- Bharti, K.; Miller, S.S.; Arnheiter, H. The new paradigm: Retinal pigment epithelium cells generated from embryonic or induced pluripotent stem cells. Pigment Cell Melanoma Res. 2011, 24, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Croze, R.H.; Buchholz, D.E.; Radeke, M.J.; Thi, W.J.; Hu, Q.; Coffey, P.J.; Clegg, D.O. ROCK Inhibition Extends Passage of Pluripotent Stem Cell-Derived Retinal Pigmented Epithelium. Stem Cells Transl. Med. 2014, 3, 1066–1078. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.C.; Otaki-Keen, A.E.; Putkey, F.R.; Hjelmeland, L.M. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 1996, 62, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chang, W.C.; Hung, K.H.; Yang, D.M.; Cheng, Y.H.; Liao, Y.W.; Woung, L.C.; Tsai, C.Y.; Hsu, C.C.; Lin, T.C.; et al. The generation of induced pluripotent stem cells for macular degeneration as a drug screening platform: Identification of curcumin as a protective agent for retinal pigment epithelial cells against oxidative stress. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23. [Google Scholar] [CrossRef] [PubMed]
- Vogel, G. Stem cells. Diseases in a dish take off. Science 2010, 330, 1172–1173. [Google Scholar] [CrossRef] [PubMed]
- Birch, D.G.; Liang, F.Q. Age-related macular degeneration: A target for nanotechnology derived medicines. Int. J. Nanomed. 2007, 2, 65–77. [Google Scholar] [CrossRef]
- Yu, A.L.; Birke, K.; Burger, J.; Welge-Lussen, U. Biological effects of cigarette smoke in cultured human retinal pigment epithelial cells. PLoS One 2012, 7, e48501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, M.; Marin-Castano, M.E. Nicotine increases the VEGF/PEDF ratio in retinal pigment epithelium: A possible mechanism for CNV in passive smokers with AMD. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3842–3853. [Google Scholar] [CrossRef]
- Cano, M.; Thimmalappula, R.; Fujihara, M.; Nagai, N.; Sporn, M.; Wang, A.L.; Neufeld, A.H.; Biswal, S.; Handa, J.T. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vis. Res. 2010, 50, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Dardik, R.; Livnat, T.; Nisgav, Y.; Weinberger, D. Enhancement of angiogenic potential of endothelial cells by contact with retinal pigment epithelial cells in a model simulating pathological conditions. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6188–6195. [Google Scholar] [CrossRef]
- Baglio, S.R.; Pegtel, D.M.; Baldini, N. Mesenchymal stem cell secreted vesicles provide novel opportunities in (stem) cell-free therapy. Front. Physiol 2012, 3, 359. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.L.; Nichols, J.; Evans, E.P.; Smith, A.G. Changing potency by spontaneous fusion. Nature 2002, 416, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.D.; Hubschman, J.P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R.; et al. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, V.K.; Sugino, I.K.; Van, P.Y.; Shah, S.; Zarbin, M.A. Impaired RPE survival on aged submacular human Bruch’s membrane. Exp. Eye Res. 2005, 80, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Sugino, I.K.; Rapista, A.; Sun, Q.; Wang, J.; Nunes, C.F.; Cheewatrakoolpong, N.; Zarbin, M.A. A method to enhance cell survival on Bruch’s membrane in eyes affected by age and age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9598–9609. [Google Scholar] [CrossRef]
- Carr, A.J.; Smart, M.J.; Ramsden, C.M.; Powner, M.B.; Da, C.L.; Coffey, P.J. Development of human embryonic stem cell therapies for age-related macular degeneration. Trends Neurosci. 2013, 36, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef]
- Nakano-Okuno, M.; Borah, B.R.; Nakano, I. Ethics of iPSC-Based Clinical Research for Age-Related Macular Degeneration: Patient-Centered Risk-Benefit Analysis. Stem Cell Rev. 2014, 10, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Caballero, S.; Bauer, G.; Shibata, B.; Roth, A.; Fitzgerald, P.G.; Forward, K.I.; Zhou, P.; McGee, J.; Telander, D.G.; et al. Long-term effects of intravitreal injection of GMP-grade bone-marrow-derived CD34+ cells in NOD-SCID mice with acute ischemia-reperfusion injury. Investig. Ophthalmol. Vis. Sci. 2012, 53, 986–994. [Google Scholar] [CrossRef]
- McGill, T.J.; Cottam, B.; Lu, B.; Wang, S.; Girman, S.; Tian, C.; Huhn, S.L.; Lund, R.D.; Capela, A. Transplantation of human central nervous system stem cells—Neuroprotection in retinal degeneration. Eur. J. Neurosci. 2012, 35, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, R.C. Stem cell therapy for retinal diseases: Update. Stem Cell Res.Ther. 2011, 2. [Google Scholar] [CrossRef]
- Sterneckert, J.L.; Reinhardt, P.; Scholer, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandl, C.; Grassmann, F.; Riolfi, J.; Weber, B.H.F. Tapping Stem Cells to Target AMD: Challenges and Prospects. J. Clin. Med. 2015, 4, 282-303. https://doi.org/10.3390/jcm4020282
Brandl C, Grassmann F, Riolfi J, Weber BHF. Tapping Stem Cells to Target AMD: Challenges and Prospects. Journal of Clinical Medicine. 2015; 4(2):282-303. https://doi.org/10.3390/jcm4020282
Chicago/Turabian StyleBrandl, Caroline, Felix Grassmann, Julia Riolfi, and Bernhard H. F. Weber. 2015. "Tapping Stem Cells to Target AMD: Challenges and Prospects" Journal of Clinical Medicine 4, no. 2: 282-303. https://doi.org/10.3390/jcm4020282