Importance of Beta Cell Function for the Treatment of Type 2 Diabetes


Department of Internal Medicine, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan
J. Clin. Med. 2014, 3(3), 923-943; https://doi.org/10.3390/jcm3030923
Submission received: 30 April 2014
/
Revised: 2 June 2014
/
Accepted: 24 June 2014
/
Published: 14 August 2014
(This article belongs to the Special Issue Obesity, Diabetes and Metabolic Syndrome)
Abstract
:Type 2 diabetes (T2DM) is characterized by insulin resistance and beta cell dysfunction. Recent evidence has emerged that beta cell dysfunction is a common pathogenetic feature of both type 1 and type 2 diabetes, and T2DM never develops without beta cell dysfunction. Therefore, treatment of T2DM should aim to restore beta cell function. Although the treatment of T2DM has greatly improved over the past few decades, remaining issues in the current treatment of T2DM include (1) hypoglycemia; (2) body weight gain; (3) peripheral hyperinsulinemia and (4) postprandial hyperglycemia, which are all associated with inappropriate insulin supplementation, again underpinning the important role of endogenous and physiological insulin secretion in the management of T2DM. This review summarizes the current knowledge on beta cell function in T2DM and discusses the treatment strategy for T2DM in relation to beta cell dysfunction.
1. Introduction
The incidence of diabetes is increasing worldwide. According to the International Diabetes Federation (IDF), there were estimated to be 382 million patients with diabetes in 2013, which is expected to increase to 592 million by 2035 [1]. Diabetes caused 5.1 million deaths, and health spending on diabetes accounted for 10.8% of total health expenditure worldwide in 2013 [1]. In Japan, diabetes causes ~4000 cases of blindness, and ~17,000 patients with diabetes commence hemodialysis annually. Of patients with diabetes, type 2 diabetes (T2DM) comprises 90%–95%; however, currently no effective strategy to prevent or cure T2DM is available.
T2DM is characterized by insulin resistance and beta cell dysfunction [2]. However, recent evidence has emerged that beta cell dysfunction is a core pathogenetic mechanism of diabetes and T2DM develops only when beta cell function is impaired [3]. This review summarizes the current knowledge on beta cell dysfunction in T2DM and discusses the treatment strategy for T2DM in relation to beta cell function.
2. Deficit of Beta Cell Function and Mass in T2DM
T2DM is characterized by insulin resistance and beta cell dysfunction [2]. Since plasma insulin level is often higher in patients with T2DM compared with non-diabetic individuals, T2DM is characterized by obesity, hyperinsulinemia, and insulin resistance; however, the significance of beta cell dysfunction in patients with T2DM has been often ignored.
However, recent studies have consistently shown the presence of beta cell dysfunction in patients with T2DM [4]. Since insulin secretion shows a compensatory increase in the presence of insulin resistance, true beta cell function should be evaluated based on insulin secretion adjusted by insulin sensitivity, the so-called disposition index [5]. Using the disposition index, DeFronzo et al. have reported that beta cell function is already decreased by approximately 80% in patients with impaired glucose tolerance (IGT) compared with non-diabetic subjects [4].
Moreover, recent studies have shown that not only beta cell function, but also beta cell mass is decreased in patients with T2DM. Butler et al. conducted histological analysis of beta cell mass in the pancreas obtained from autopsy subjects with or without T2DM [6]. As a result, they found that beta cell mass was decreased by approximately 40% and 65% in lean and obese individuals with T2DM, respectively, compared to age- and BMI-matched non-diabetic individuals. Other histological studies have also confirmed reduced beta cell mass in patients with T2DM [7,8,9].
Taken together, the current evidence consistently shows that there is reduced functional mass of beta cells in patients with T2DM. Since type 1 diabetes (T1DM) is characterized by beta cell loss due to autoimmune attack [10], the current evidence indicates that reduced beta cell mass is a common pathophysiological feature of both type 1 and type 2 diabetes.
3. Beta Cell Function and Glycemic Control
If reduced functional beta cell mass is a common pathophysiological feature of both type 1 and type 2 diabetes, what is the clinical relevance of beta cell dysfunction in T2DM? Since beta cell function is already reduced in patients with IGT, preceding the onset of T2DM, beta cell dysfunction has an important role in the deterioration of glucose tolerance [4]. Thus, preservation or recovery of beta cell function could be an effective therapeutic strategy to prevent T2DM [11].
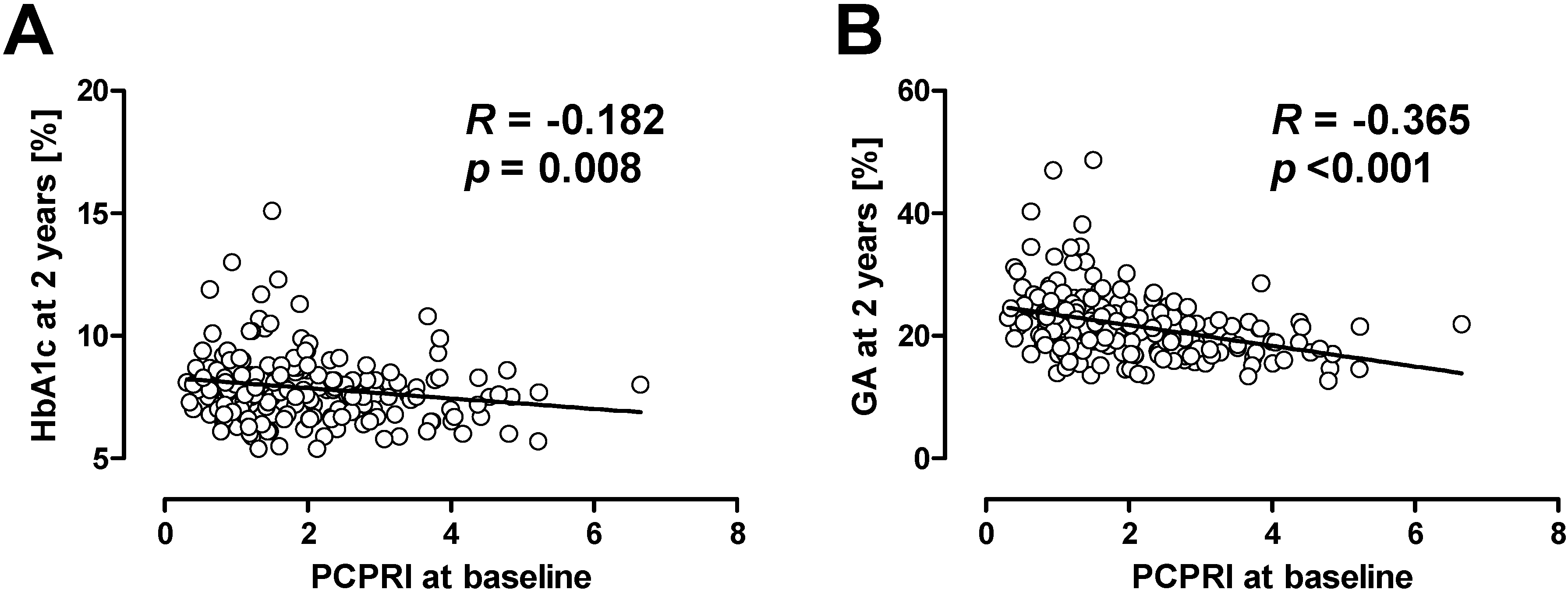
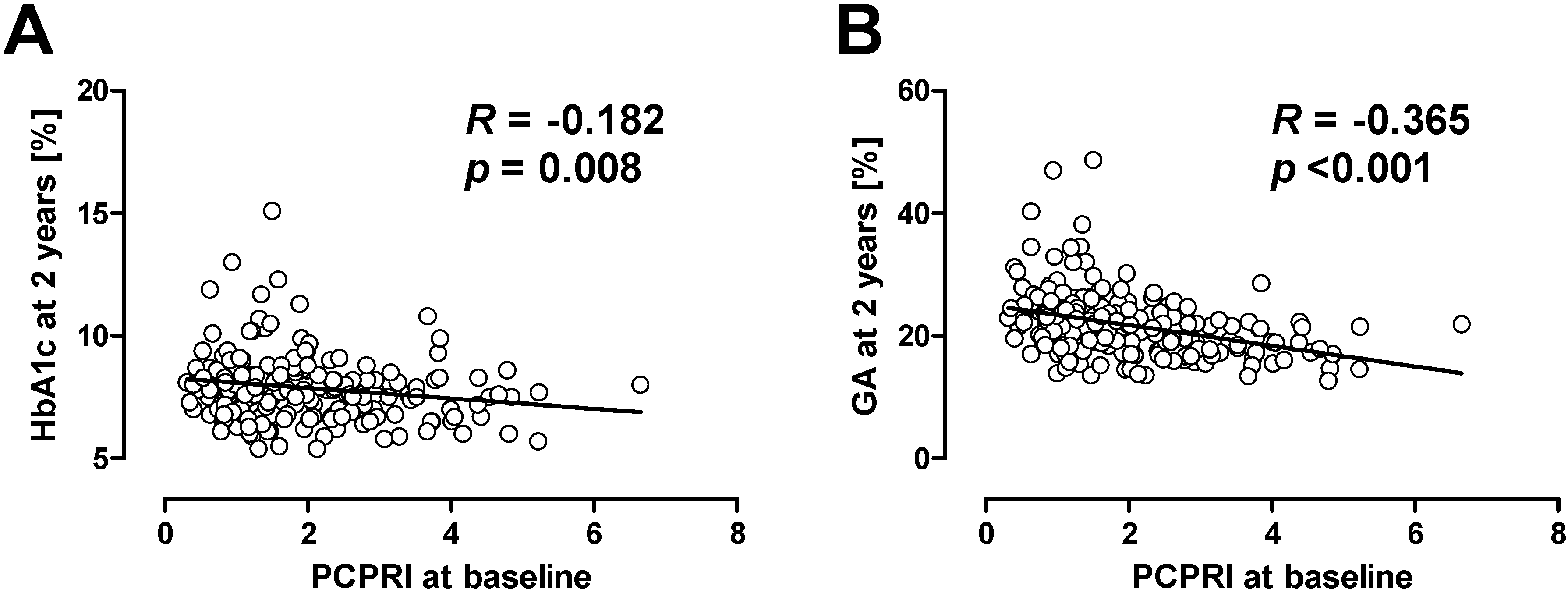
Recent studies also suggest the importance of beta cell function in the management of hyperglycemia in patients with T2DM. In the UK Prospective Diabetes Study (UKPDS) [12] and a Diabetes Outcome Progression Trial (ADOPT) [13], treatment failure was associated with a progressive decline of beta cell function [14,15]. The association between beta cell dysfunction and treatment failure is not only observed in adult patients, but was also shown in adolescent patients with T2DM. The Treatment Options for type 2 Diabetes in Adolescents and Youth (TODAY) trial showed that lower beta cell function at baseline was associated with poorer glycemic control after four years in adolescent patients with T2DM who were treated with metformin or metformin plus rosiglitazone [16]. In our retrospective cohort study, we also evaluated the association between beta cell function and clinical outcome in Japanese patients with T2DM [17,18]. Beta cell function was evaluated by serum or urinary C-peptide level. As a result, we found that patients with lower beta cell function at baseline were more likely to subsequently need insulin therapy compared with those with higher beta cell function. It is of note that among C-peptide indices, postprandial C-peptide index (i.e., 2 h postprandial serum C-peptide (ng/mL)/plasma glucose (mg/dL) × 100) was the best predictor of subsequent need for insulin therapy compared with fasting C-peptide index and urinary C-peptide [17,19]. More surprisingly, even with more frequent insulin therapy, patients with lower beta cell function at baseline showed higher glycated hemoglobin (HbA1c) and glycated albumin (GA) levels after 2 years (Figure 1) [18,20]. These results indicate that beta cell function is significantly associated with clinical outcome, i.e., glycemic control, in patients with T2DM.
Figure 1.
Correlation between baseline postprandial C-peptide index (PCPRI) and HbA1c (A) and glycated albumin (GA) (B) after two years. Reproduced with permission from [20].
Figure 1.
Correlation between baseline postprandial C-peptide index (PCPRI) and HbA1c (A) and glycated albumin (GA) (B) after two years. Reproduced with permission from [20].
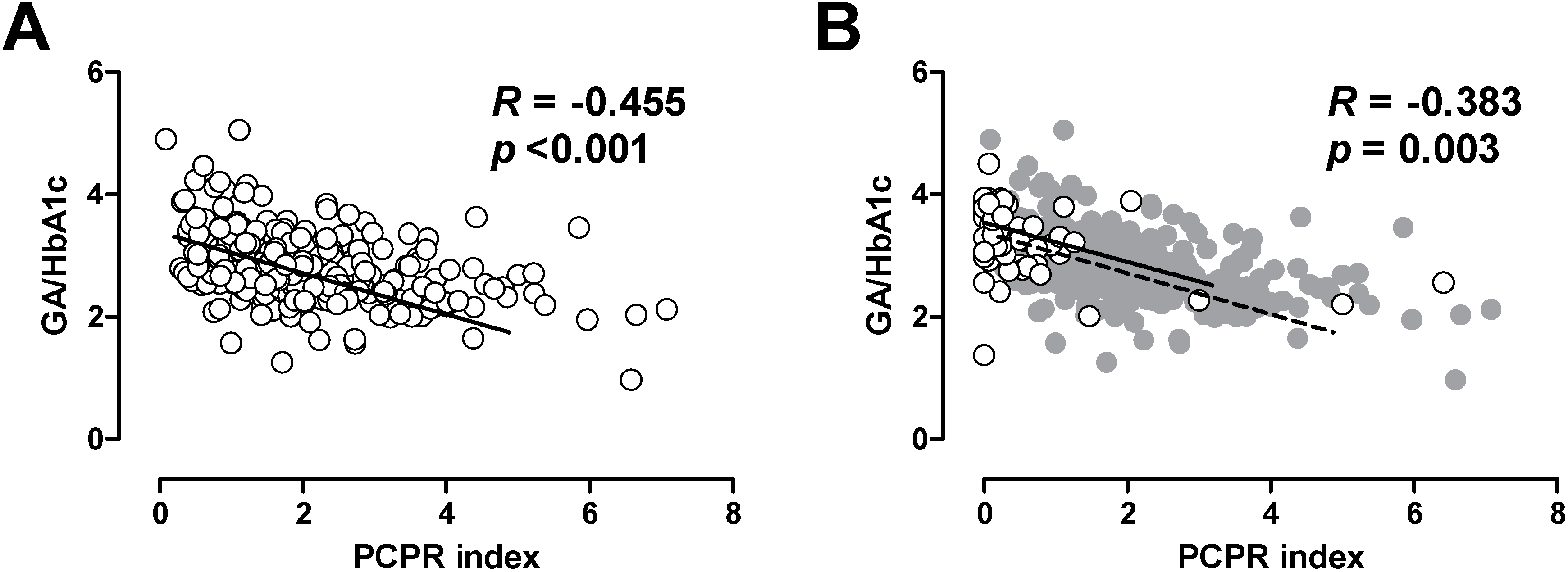
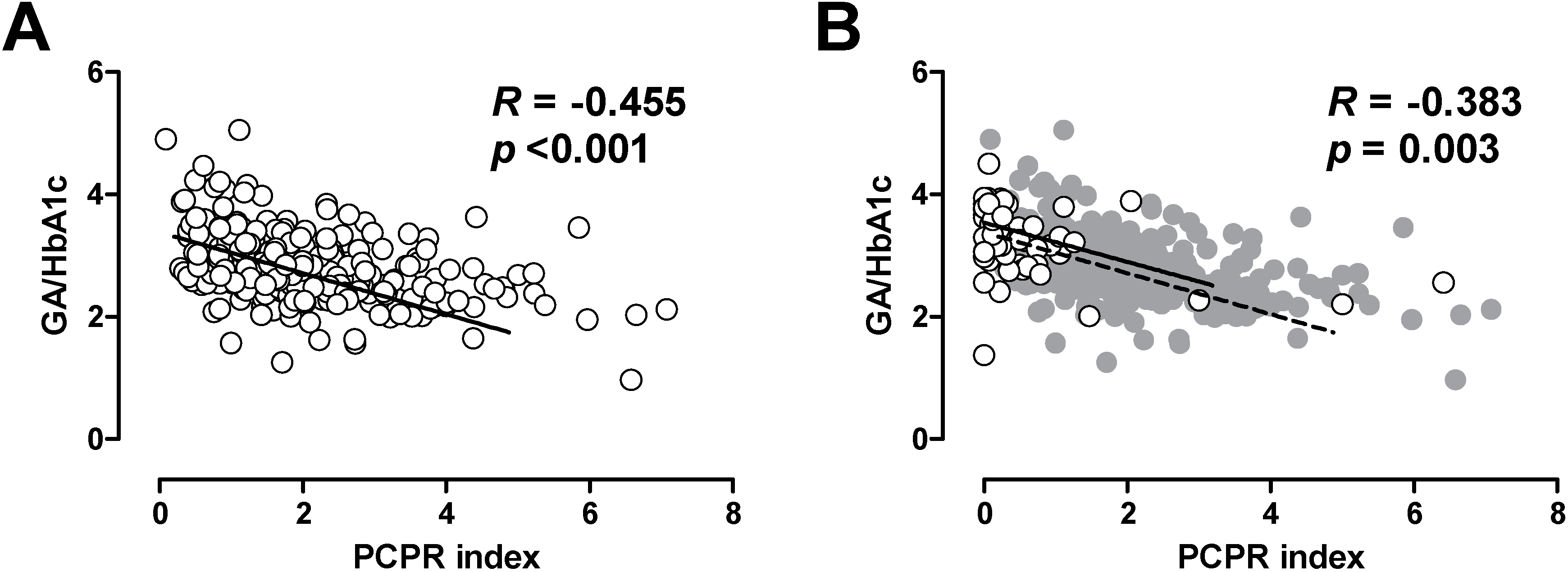
In patients with T1DM, it has been shown that lower residual C-peptide is associated with greater glycemic fluctuation [21,22,23]. Thus, we also evaluated the association between postprandial C-peptide index and glycemic excursion in patients with T2DM [24]. Since glycated albumin (GA) is more rapidly glycated than is HbA1c, GA or GA to HbA1c ratio more sensitively reflects postprandial glycemic excursions compared with HbA1c [24,25,26]. We, and others, have reported that postprandial C-peptide index is negatively correlated with GA to HbA1c ratio in patients with T2DM [24,27], indicating that lower beta cell function is associated with greater postprandial glycemic excursions in patients with T2DM. Interestingly, the relationship between postprandial C-peptide index and GA to HbA1c ratio was comparable between patients with T1DM and T2DM (Figure 2) [24,25], indicating that beta cell function similarly affects glycemic fluctuation in patients with T1DM and T2DM. These results further underpin the importance of beta cell function in both types of diabetes.
Figure 2.
Correlation between postprandial C-peptide index (PCPRI) and glycated albumin (GA) to HbA1c ratio in patients with type 2 diabetes (A) and type 1 diabetes (B). In Figure 2B, the data of patients with type 1 diabetes are superimposed on the data of those with type 2 diabetes (gray circles and dotted line). Reproduced with permission from [24].
Figure 2.
Correlation between postprandial C-peptide index (PCPRI) and glycated albumin (GA) to HbA1c ratio in patients with type 2 diabetes (A) and type 1 diabetes (B). In Figure 2B, the data of patients with type 1 diabetes are superimposed on the data of those with type 2 diabetes (gray circles and dotted line). Reproduced with permission from [24].
Thus, recent studies have shown that lower beta cell function is associated with a higher rate of treatment failure, poorer glycemic control and greater glycemic fluctuation in patients with T2DM, which are probably related to higher risk of diabetic complications in these patients, as suggested by several studies [28,29].
4. Current Issues in Treatment of Type 2 Diabetes: Inappropriate Insulin Supplementation
Current issues in the treatment of T2DM are summarized in Table 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Issue | Cause |
|---|---|
| Hypoglycemia | Excess insulin |
| Weight gain | Excess insulin |
| Concern of increased risk of malignancy and/or atherosclerosis | Excess insulin, especially peripheral hyperinsulinemia |
| Postprandial hyperglycemia | Insufficient insulin in postprandial state, especially in portal vein |
4.1. Hypoglycemia
T2DM is associated with a two- to four-fold increased risk of cardiovascular disease (CVD) [30]. As shown in the UKPDS, intensive glycemic control in patients with T2DM effectively suppressed the development or progression of diabetic microangiopathy; i.e., retinopathy, nephropathy and neuropathy, during the study; however, the effect of intensive glycemic control on CVD was less apparent [31], although a significant reduction in the rate of CVD was obtained after 10 years of study [32]. Recent randomized controlled trials (RCTs) aiming to control HbA1c to near normal levels (i.e., HbA1c < 6%–6.5%) failed to show a reduction of CVD [33,34,35], and in one trial, the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial, all-cause mortality was rather significantly increased in the intensive therapy group compared with the conventional therapy group. In the ACCORD trial, the intensive therapy group showed a higher incidence of hypoglycemia including severe hypoglycemia and greater weight gain compared with the conventional therapy group during the study [34].
Hypoglycemia is the most frequent and serious adverse event associated with the treatment of diabetes. Intensive glycemic control is inversely correlated with increased risk of hypoglycemia [31,33,34,35]. Hypoglycemia not only reduces patients’ quality of life (QOL) but also appears to induce inflammation, blood coagulation abnormality, sympathoadrenal response, and endothelial dysfunction, which could affect the onset of CVD [36,37], suggesting that hypoglycemia increases the risk of CVD.
4.2. Weight Gain
Weight gain associated with treatment of T2DM is also a major issue. Use of sulfonylureas (SU) and thiazolidinediones (TZD) is usually associated with weight gain [38]. Insulin therapy also often induces weight gain. Use of metformin is associated with less weight gain or even weight loss compared with SU or TZD treatment [13,39,40,41,42,43]. The effect of dipeptidyl peptidase-4 (DPP-4) inhibitors on body weight has been shown to be neutral, while glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1RA) have a favorable effect on body weight [44,45]. Weight gain not only may result in treatment failure but also may induce other obesity-related diseases and impair QOL [46].
4.3. Peripheral Hyperinsulinemia
The third issue is the concern that diabetes treatment may increase the risk of cancer and/or atherosclerosis. In 2009, a study reported by Hemkens et al. showed a significant increase in overall cancer risk by use of insulin glargine compared with human insulin [47]. However, papers reported by other groups did not find such a correlation [48,49,50]. A number of subsequent analyses including a subanalysis of randomized controlled trials also have not found any significant association between insulin glargine and risk of cancer [51,52,53,54], resulting in no warning label or restriction on the use of insulin glargine with regard to cancer to date. Nonetheless, the study by Hemkens et al. indeed showed a similar increase in risk of cancer or all-cause mortality between insulin glargine and human insulin in their Cox model adjusted for age and sex [47]. More recently, Currie et al. have examined the correlation between glycemic control and all-cause mortality [55]. They found a general U-shaped curve between HbA1c level and all-cause mortality. This relationship was observed in both patients treated with oral hypoglycemic agents (SU and/or metformin) and those treated with insulin, with the lowest hazard ratio (HR) at HbA1c of around 7.5%. HR for all-cause mortality was significantly higher in people given insulin-based regimens vs. those given combination oral agents (1.49, 95% confidence interval (CI) 1.39–1.59). Although the higher mortality in insulin-treated patients might simply reflect the severity of the disease, these findings indicate that the relationship between insulin therapy and mortality remains controversial. When insulin is subcutaneously injected, the distribution of insulin is not physiological, and systemic, rather than portal, hyperinsulinemia occurs. Although it has been reported that insulin has anti-inflammatory and anti-oxidative effects [56], insulin also has the potential to induce cell growth [57]. Thus, it has been suggested that peripheral hyperinsulinemia caused by subcutaneous insulin injection may promote tumor growth [57]. Peripheral hyperinsulinemia may also induce proliferation of vascular endothelial cells and increase plaque vulnerability within atherosclerotic plaques [58], which may increase the risk of CVD in insulin-treated patients [59].
The Outcome Reduction with Initial Glargine Intervention (ORIGIN) trial was designed to examine the effect of early basal insulin supplementation on the development of CVD in patients with IGT or T2DM [60]. A total of 12,537 patients participated, and the study showed that the effect of basal insulin supplementation was neutral in terms of development of CVD. In this study, no increase in cancer incidence in patients treated with insulin was also noted.
4.4. Postprandial Glycemic Excursion
Hyperglycemia consists of two components; fasting hyperglycemia and postprandial glycemic excursion. Epidemiological studies have shown that postprandial hyperglycemia rather than fasting hyperglycemia is more strongly correlated with CVD and all-cause mortality [62,63,64]. It has also been shown that intermittent exposure to high glucose more strongly induces apoptosis in endothelial cells compared with chronic high glucose exposure in an in vitro study [65]. The recent development of continuous glucose monitoring systems (CGMS) has allowed more precise evaluation of the daily glycemic profile. Using this system, Monnier et al. [66] have reported a strong positive association between the mean amplitude of glycemic excursions (MAGE), a measure of daily glycemic variability, and urinary 8-iso-prostaglandin (PG) F2alpha excretion rate in patients with T2DM, indicating that greater glycemic variability results in greater production of oxidative stress in patients with T2DM, which may cause vascular damage and diabetic complications. Thus, reducing postprandial glycemic excursion and glycemic variability in patients with T2DM may improve CVD outcomes, although the role of glycemic variability in CVD outcome remains controversial [67,68,69,70].
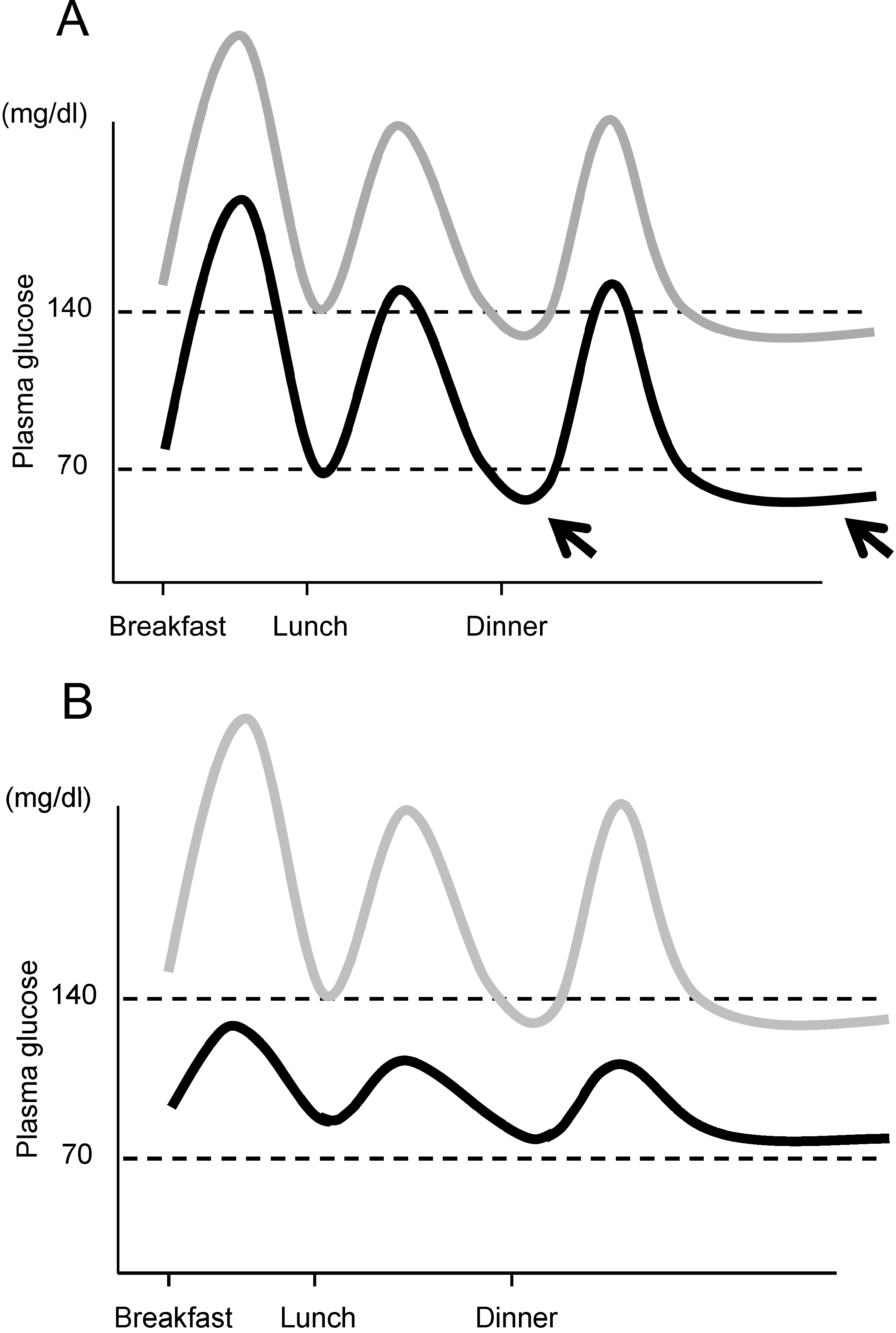
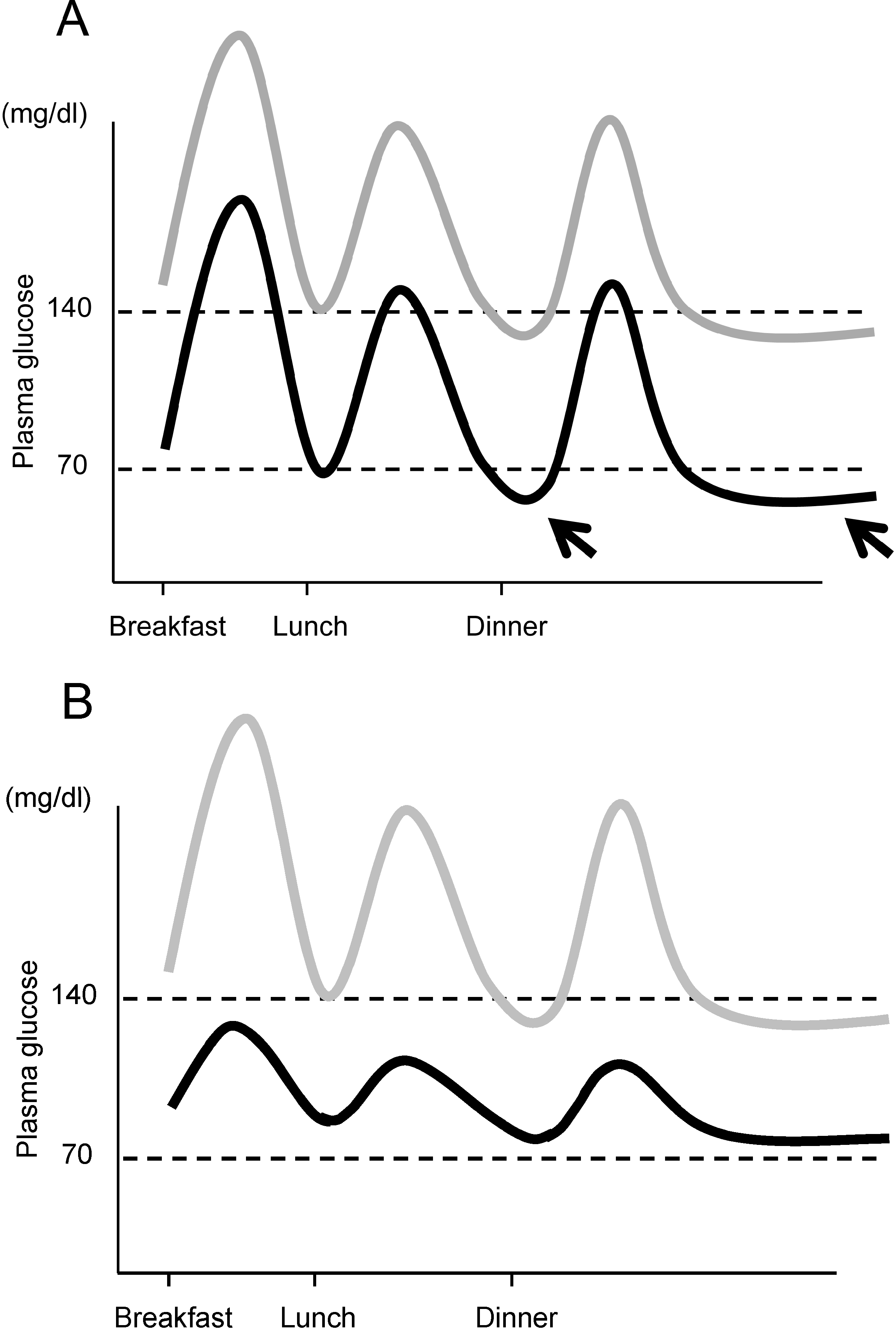
Moreover, another important aspect of managing postprandial glycemic excursion and glycemic variability is the prevention of hypoglycemia. The importance of reducing glycemic variability for prevention of hypoglycemia is illustrated in Figure 3. HbA1c reflects the average glucose. When lowering HbA1c, if glycemic variability persists, the risk of hypoglycemia increases (Figure 3A). We and others have reported that postprandial glycemic excursion and glycemic variability are increased in older patients with T2DM [25,71], and a greater risk of hypoglycemia has been reported in elderly patients with T2DM [72]. Thus, controlling postprandial glucose excursion and reducing glycemic variability are necessary to achieve good glycemic control without an increase in incidence of hypoglycemia, especially in the elderly.
Figure 3.
Importance of controlling postprandial glycemic excursion. (A) If mean plasma glucose level is lowered without controlling postprandial glycemic excursion (gray line → black line), the risk of hypoglycemia between meals and during the night increases (arrows); (B) Lowering the mean glucose level with control of postprandial glycemic excursion (gray line → black line) results in a low risk of hypoglycemia. Note that mean plasma glucose levels are similar in both cases, indicating similar HbA1c in both cases.
Figure 3.
Importance of controlling postprandial glycemic excursion. (A) If mean plasma glucose level is lowered without controlling postprandial glycemic excursion (gray line → black line), the risk of hypoglycemia between meals and during the night increases (arrows); (B) Lowering the mean glucose level with control of postprandial glycemic excursion (gray line → black line) results in a low risk of hypoglycemia. Note that mean plasma glucose levels are similar in both cases, indicating similar HbA1c in both cases.
5. Therapeutic Strategy in Relation to Beta Cell Function
The current issues in the treatment of T2DM described above are, simply said, all related to beta cell function; i.e., excess or insufficiency of insulin supply (Table 1). Therefore, to overcome these issues of current therapy of T2DM, it is inevitably important to preserve or recover endogenous beta cell function and physiological insulin secretion in patients with T2DM.
To date, the most effective therapeutic strategy to preserve or recover beta cell function is likely to be to reduce beta cell workload or induce beta cell rest. These include lifestyle modification and/or weight reduction, and use of metformin or TZD. Lifestyle modification, i.e., nutritional therapy and increase in physical activity, and weight reduction improve insulin sensitivity and thereby reduce beta cell workload. Intensive lifestyle modification with more than 7% weight loss suppressed the progression to T2DM by ~58% in patients with IGT [73]. In the same study, metformin therapy also reduced the progression to T2DM by ~31% [73]. Metformin improves insulin sensitivity mainly through suppressing hepatic glucose production, which results in reduced beta cell workload. TZDs have also been shown to effectively suppress the progression from IGT to T2DM [74,75,76]. TZDs improve insulin sensitivity in adipose tissue, thereby reducing beta cell workload. Insulin therapy is initiated to reproduce the physiological daily insulin profile. Insulin therapy has been shown to improve beta cell function [77,78], probably through inducing beta cell rest. In the ORIGIN study, adding basal insulin has been shown to suppress progression from IGT to T2DM [60]. On the other hand, nateglinide, a short-acting insulin secretagogue, failed to show a reduction in progression to T2DM in patients with IGT [79], suggesting that the therapeutic strategy to increase beta cell workload may not be effective to prevent deterioration of glucose metabolism.
Drugs to reduce beta cell workload have also been shown to be more effective to maintain optimal glycemic control compared with drugs to increase beta cell workload in patients with recent-onset T2DM. In the ADOPT study, treatment with metformin or rosiglitazone monotherapy resulted in a lower rate of treatment failure compared with SU monotherapy [13].
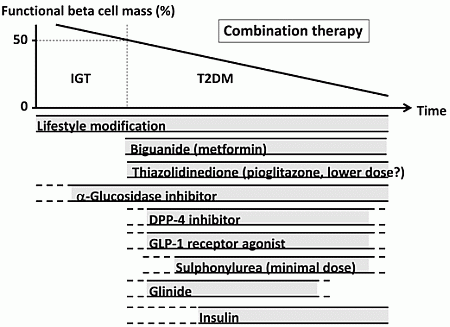
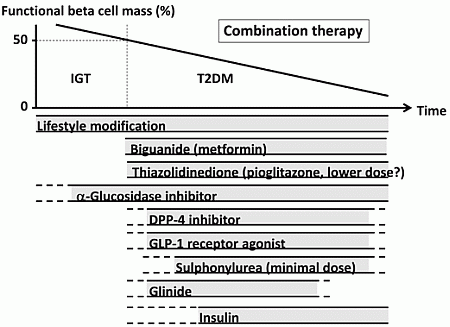
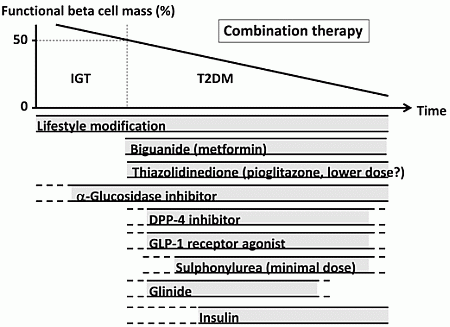
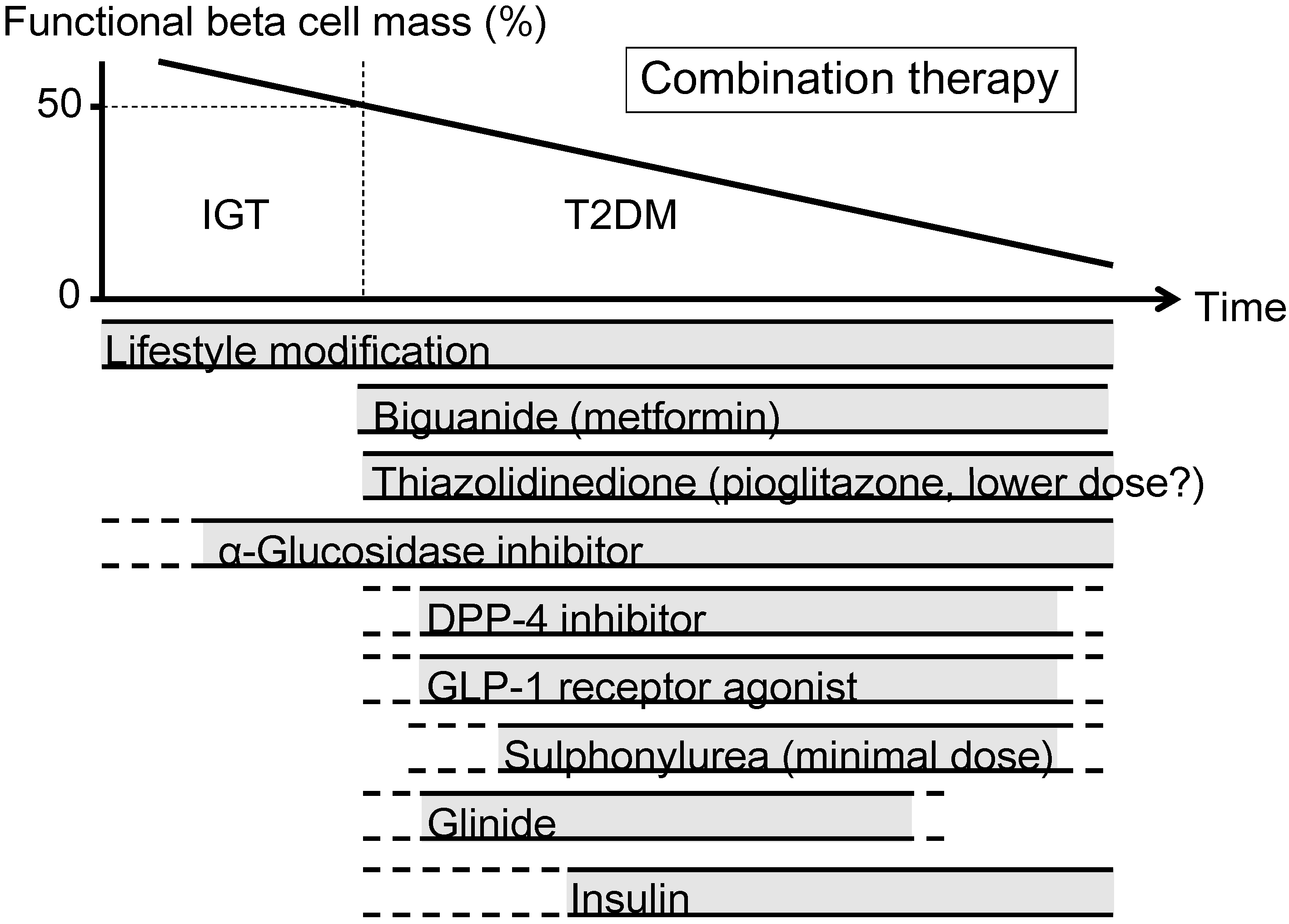
The currently proposed therapeutic strategy for T2DM aiming at preservation and/or recovery of beta cell function is shown in Figure 4. It is emphasized that, to reduce beta cell workload, lifestyle modification and weight reduction remain the most important therapy at any stage of T2DM. Although lifestyle modification failed to reduce the incidence of CVD in the Look AHEAD (Action for Health in Diabetes) trial [80], it has been reported that lifestyle modification improved cardiovascular risk factors, reduced the need for and cost of medication, reduced the rate of sleep apnea and urinary incontinence, improved well-being, and increased the rate of diabetes remission [81,82,83,84,85].
Metformin is positioned as first-line therapy in most guidelines for the treatment of T2DM [38,86]. Since metformin is effective in lean patients as well as obese patients with T2DM [87,88], it should be used in both lean and obese individuals unless contraindicated. Its efficacy in reducing HbA1c (~1.5%), low risk of hypoglycemia, favorable effect on body weight and low cost also support metformin as a first-line drug.
TZDs have been also shown to reduce beta cell workload and maintain glycemic control in the long term [13,15]. Rosiglitazone has been shown to increase low-density lipoprotein (LDL) cholesterol and the risk of coronary heart disease in patients with T2DM [89], and its use has been suspended or strictly restricted in Europe and USA [90,91], although recently the U.S. Food and Drug Administration (FDA) has lifted most of its restrictions [92]. On the other hand, pioglitazone has been shown to suppress the progression of atherosclerosis and reduce the risk of cardiovascular disease [93,94,95,96]. However, TZDs often induce weight gain and edema due to fluid retention, and are contraindicated in patients with heart failure [38]. Recent studies have also shown an increase in risk of bone fracture in women [97] and risk of bladder cancer [98,99,100] in patients treated with pioglitazone. The risk of bladder cancer may be dose dependent. In addition, since low-dose pioglitazone also reduces the risk of weight gain and edema, it may be preferable to use pioglitazone at lower doses, especially in women. Pioglitazone also should be used with caution in postmenopausal women with osteoporosis due to the increased fracture risk.
Figure 4.
Proposed concept of treatment strategy for type 2 diabetes (T2DM) in relation to functional beta cell mass. An α-glucosidase inhibitor is partly approved for use in patients with impaired glucose tolerance (IGT) in Japan. Medications not approved or marketed in Japan are not included in the figure. Reproduced with permission from [101]. Since currently no single therapy or agent can cure and even manage T2DM, an effective combination of current medications in addition to lifestyle modification aiming at reduction in beta cell workload is important to preserve or recover beta cell function.
Figure 4.
Proposed concept of treatment strategy for type 2 diabetes (T2DM) in relation to functional beta cell mass. An α-glucosidase inhibitor is partly approved for use in patients with impaired glucose tolerance (IGT) in Japan. Medications not approved or marketed in Japan are not included in the figure. Reproduced with permission from [101]. Since currently no single therapy or agent can cure and even manage T2DM, an effective combination of current medications in addition to lifestyle modification aiming at reduction in beta cell workload is important to preserve or recover beta cell function.
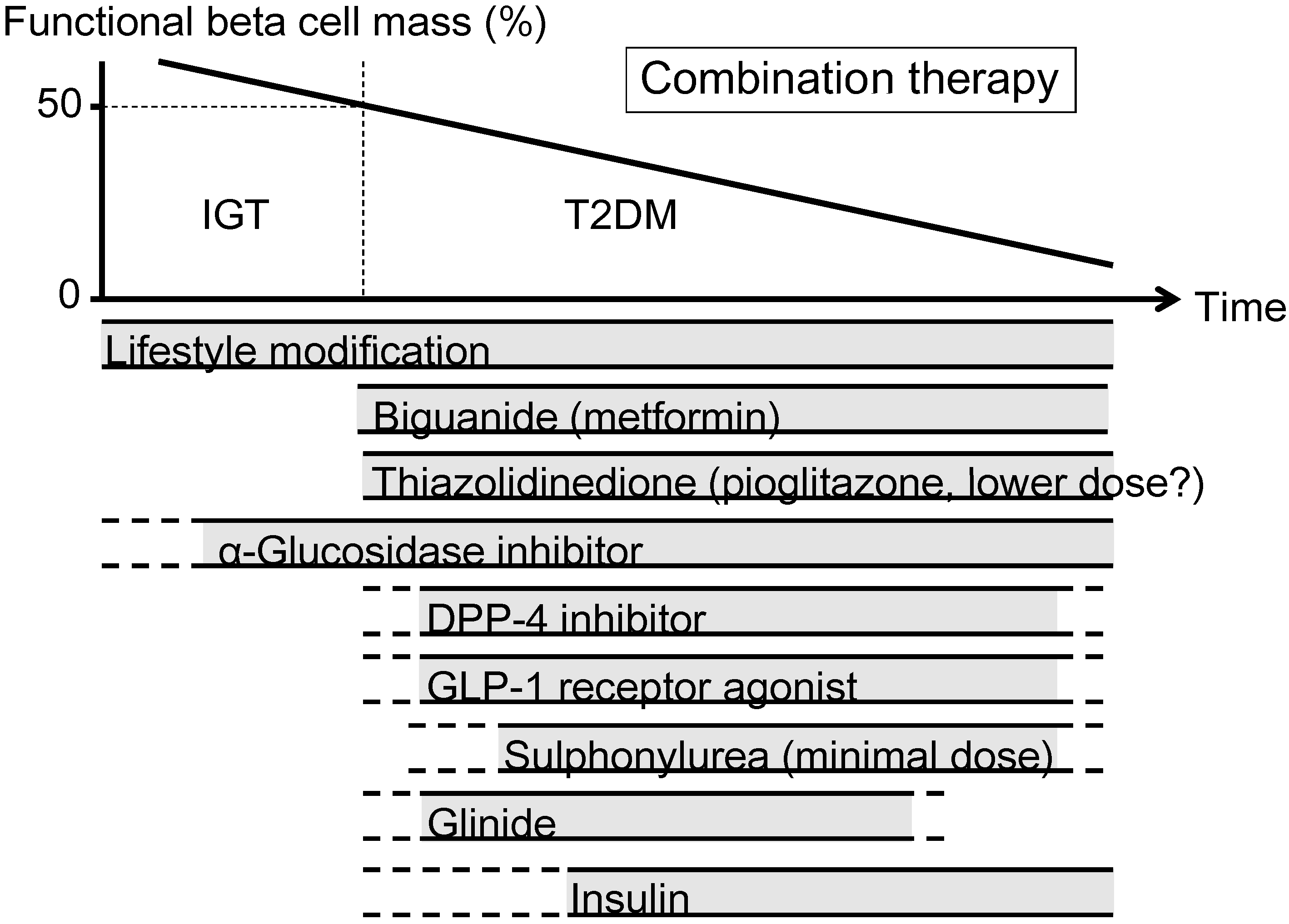
α-Glucosidase inhibitors (AGIs) delay the absorption of carbohydrate from the small intestine, and thereby reduce postprandial hyperglycemia, resulting in reduced beta cell workload in a postprandial state. AGIs have also been reported to reduce the progression to T2DM in patients with IGT [102,103]. Improving postprandial hyperglycemia by AGIs may also improve the cardiovascular outcome [104,105,106]. Therefore, although the reduction in HbA1c by AGIs is relatively small (~0.5%), their use is also considered in patients with T2DM, especially those with postprandial hyperglycemia. The major side effect of AGIs is gastrointestinal disturbance such as flatulence, diarrhea and abdominal pain. In Japan, AGIs are the only medication indicated for patients with IGT. Thus, AGIs are also considered for the treatment of T2DM at the early stage of the disease, if tolerated.
On the other hand, the use of insulin secretagogues, which increase beta cell workload, may be somewhat limited. SUs, while they remain among the most prescribed drugs for the treatment of T2DM, increase the risk of hypoglycemia and weight gain, resulting in a high rate of treatment failure [13]. These issues of SUs may be derived from their non-physiological augmentation of insulin secretion from beta cells.
Incretin drugs include DPP-4 inhibitors and GLP-1RAs. Both drug types reduce HbA1c mainly through an increase in insulin secretion, but also through suppression of glucagon secretion [107]. GLP-1RAs also slow gastric emptying and reduce appetite, resulting in weight loss. The most important characteristic of incretin drugs is probably that the enhancement of insulin secretion occurs in a glucose-dependent manner. Thus, the action of incretin drugs as insulin secretagogues is more physiological than that of SUs, thereby resulting in a low risk of hypoglycemia and weight gain with incretin therapy [44,108,109]. Whether this physiological enhancement of insulin secretion results in long-term maintenance of glycemic control remains to be elucidated. Although an increase in beta cell mass with incretin therapy has been reported in rodent studies [110,111], this effect has not been confirmed in humans [112,113,114]. As incretin therapy is usually well tolerated without serious adverse effects, the use of incretin drugs is rapidly increasing [115].
Glinides, short-acting insulin secretagogues, enhance early-phase insulin secretion, thereby reducing postprandial hyperglycemia [116]. As a defect in early-phase insulin secretion is a hallmark of glucose intolerance [117], enhancement of early-phase insulin secretion but not prolonged hyperinsulinemia by glinides is more physiological, unlike SUs, and is assumed to increase beta cell workload as well as risk of hypoglycemia to a lesser degree compared with SUs.
Thus, the use of insulin secretagogues may be limited because of an increase in beta cell workload as well as increased risk of hypoglycemia. As incretin enhances insulin secretion in a more physiological fashion, and is also expected to improve beta cell function and/or mass, incretin drugs could be used at any stage of T2DM. On the other hand, SUs may be used rather to enhance incretin action at only a minimal dose. To recover physiological insulin secretion, a combination of an incretin drug and a glinide may also be useful.
Insulin has been shown to improve beta cell function in patients with IGT and T2DM [60,77,78]. Since initial intensive insulin therapy has been shown to preserve beta cell function thereafter [76], insulin therapy should be considered as early as possible in patients with T2DM. Insulin therapy is also the most effective medication to reduce HbA1c [38]. However, increased risk of hypoglycemia, weight gain and non-physiological insulin delivery (i.e., systemic vs. portal), in addition to fear of injections, limit its use. Insulin therapy to overpower insulin resistance without eliminating excess calories may worsen ectopic lipid overload [118].
A sodium-glucose cotransporter 2 (SGLT2) inhibitor has been recently approved in several countries including USA, EU, and Japan. SGLT2 inhibitors suppress reabsorption of glucose by SGLT2 in the proximal renal tubule and increase glucose excretion in urine (~60–80 g glucose/day) [119]. As a result, SGLT2 inhibitors not only decrease HbA1c, but also reduce body weight and blood pressure and improve the lipid profile. The action of SGLT2 inhibitors is independent of insulin. Thus, the efficacy of SGLT2 inhibitors seems to be regardless of beta cell function. SGLT2 inhibitors show a low risk of hypoglycemia but increase the incidence of bacterial urinary tract infections and fungal genital infections especially in women. Higher risk of hypotension has also been reported [120]. SGLT2 inhibitors may be suitable for obese patients with T2DM and metabolic syndrome; however, their longer-term safety including cardiovascular and cancer risk and efficacy remain unknown [120,121].
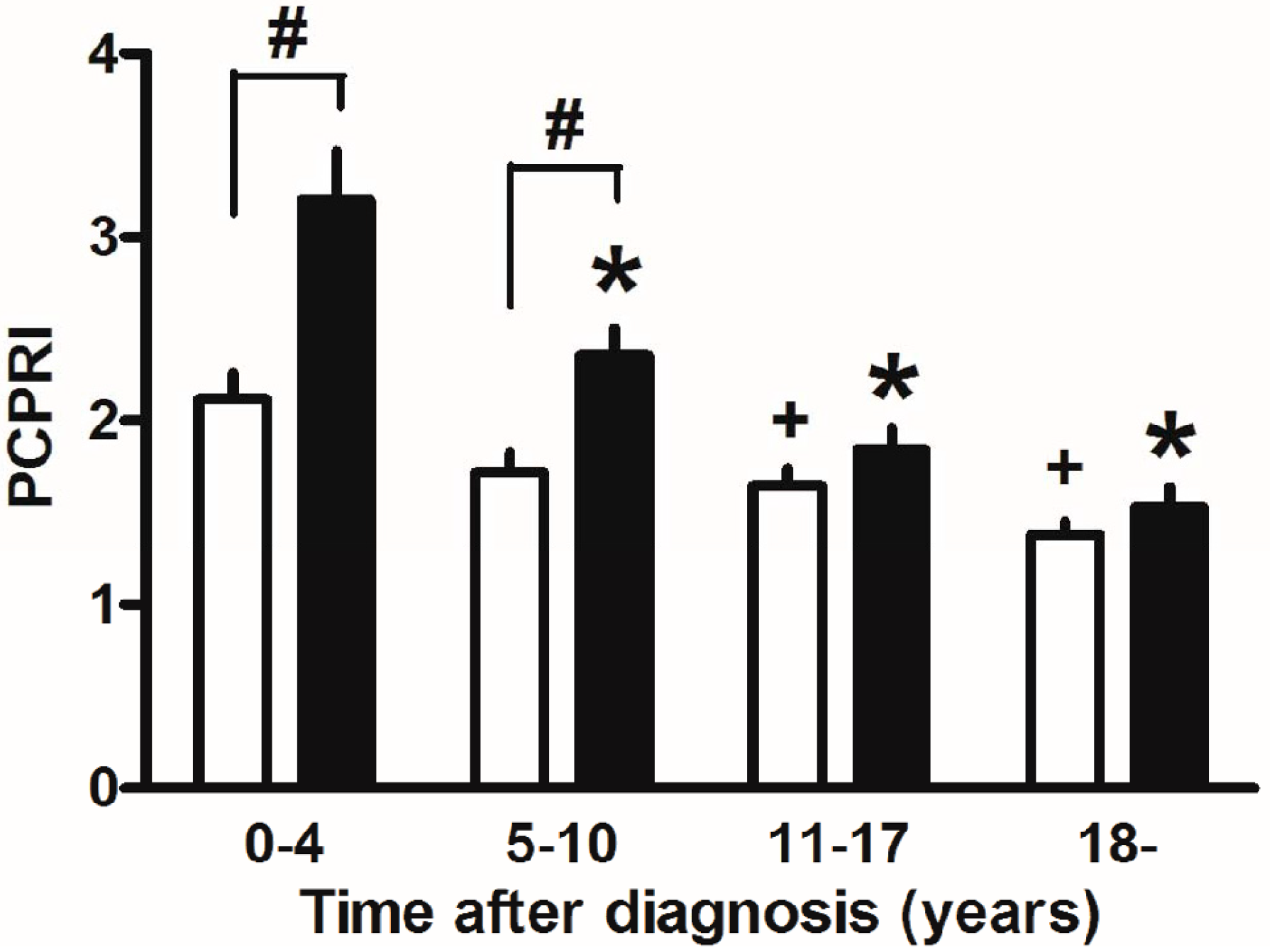
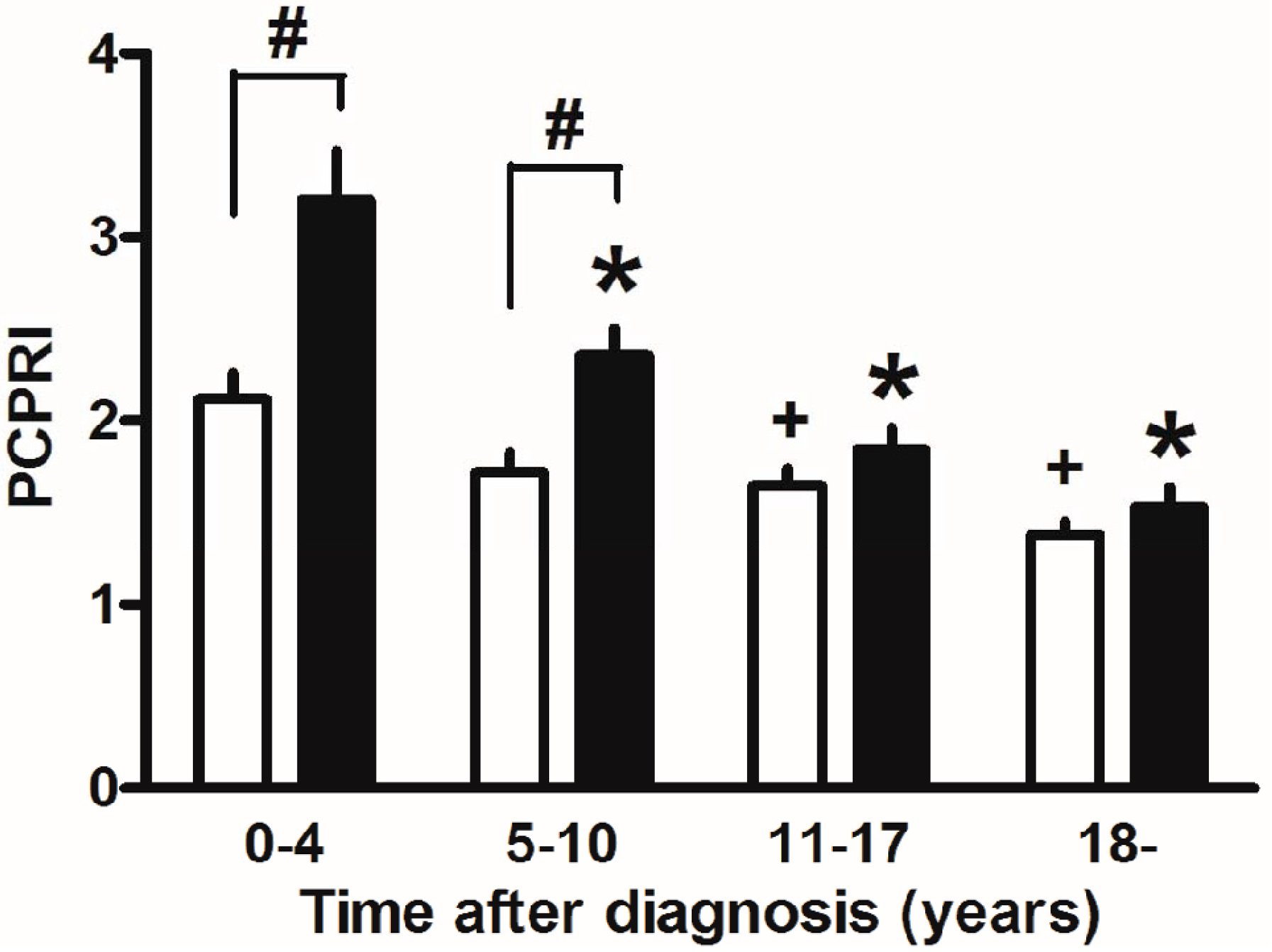
Finally, marked weight reduction by bariatric surgery such as gastric bypass and sleeve gastrectomy has been reported to markedly improve glycemic control and even achieve remission of T2DM in severely obese T2DM patients [122]. This also suggests the importance of reducing beta cell workload, although change in incretin secretion has also been proposed as another mechanism by which glucose metabolism is improved after gastric bypass. In our retrospective cohort, the progressive decline in beta cell function seemed exaggerated in the presence of obesity in Japanese patients with T2DM (Figure 5) [123]. The remission of T2DM after bariatric surgery is associated with residual beta cell function [124,125], indicating the importance of residual beta cell function to manage and/or cure T2DM.
Figure 5.
Postprandial C-peptide index (PCPRI) in subjects according to obesity and time after diagnosis (0–4, 5–10, 11–17 and ≥18 years). There were significant differences in PCPRI between lean (open bars) and obese subjects (solid bars) in the first and second quartiles of time after diagnosis, but no significant differences were observed in the third and fourth quartiles. * p < 0.05 vs. obese subjects ≤4 years after diagnosis, + p < 0.05 vs. lean subjects ≤4 years after diagnosis, # p < 0.05 vs. lean subjects. Reproduced with permission from [123].
Figure 5.
Postprandial C-peptide index (PCPRI) in subjects according to obesity and time after diagnosis (0–4, 5–10, 11–17 and ≥18 years). There were significant differences in PCPRI between lean (open bars) and obese subjects (solid bars) in the first and second quartiles of time after diagnosis, but no significant differences were observed in the third and fourth quartiles. * p < 0.05 vs. obese subjects ≤4 years after diagnosis, + p < 0.05 vs. lean subjects ≤4 years after diagnosis, # p < 0.05 vs. lean subjects. Reproduced with permission from [123].
6. Conclusions
This review summarizes the current knowledge on beta cell dysfunction in T2DM and discusses the treatment strategy for T2DM in relation to beta cell function. The hypothetical change in beta cell function, beta cell mass and resulting beta cell workload is shown in Figure 6. Beta cell dysfunction resulting from increased beta cell workload due to genetic and/or environmental factors such as excess caloric intake and physical inactivity is central in the pathogenesis of T2DM, and beta cell function progressively declines with disease duration. Beta cell function relates to glycemic control, and thereby possibly relates to complications and mortality in patients with T2DM. Although a reduction in beta cell workload seems an effective therapeutic strategy to prevent or manage T2DM, currently, no single therapy or agent can cure and even manage T2DM. Thus, an effective combination of current medications in addition to lifestyle modification aiming at reduction in beta cell workload is important to preserve or recover beta cell function and achieve life-long optimal glycemic control in patients with T2DM. Furthermore, progressive decline in beta cell functional mass even prior to the development of diabetes emphasizes the importance of lifestyle modification for prevention of the disease. Such efforts may lead to a further reduction in the incidence of diabetic complications and increase longevity in patients with T2DM.
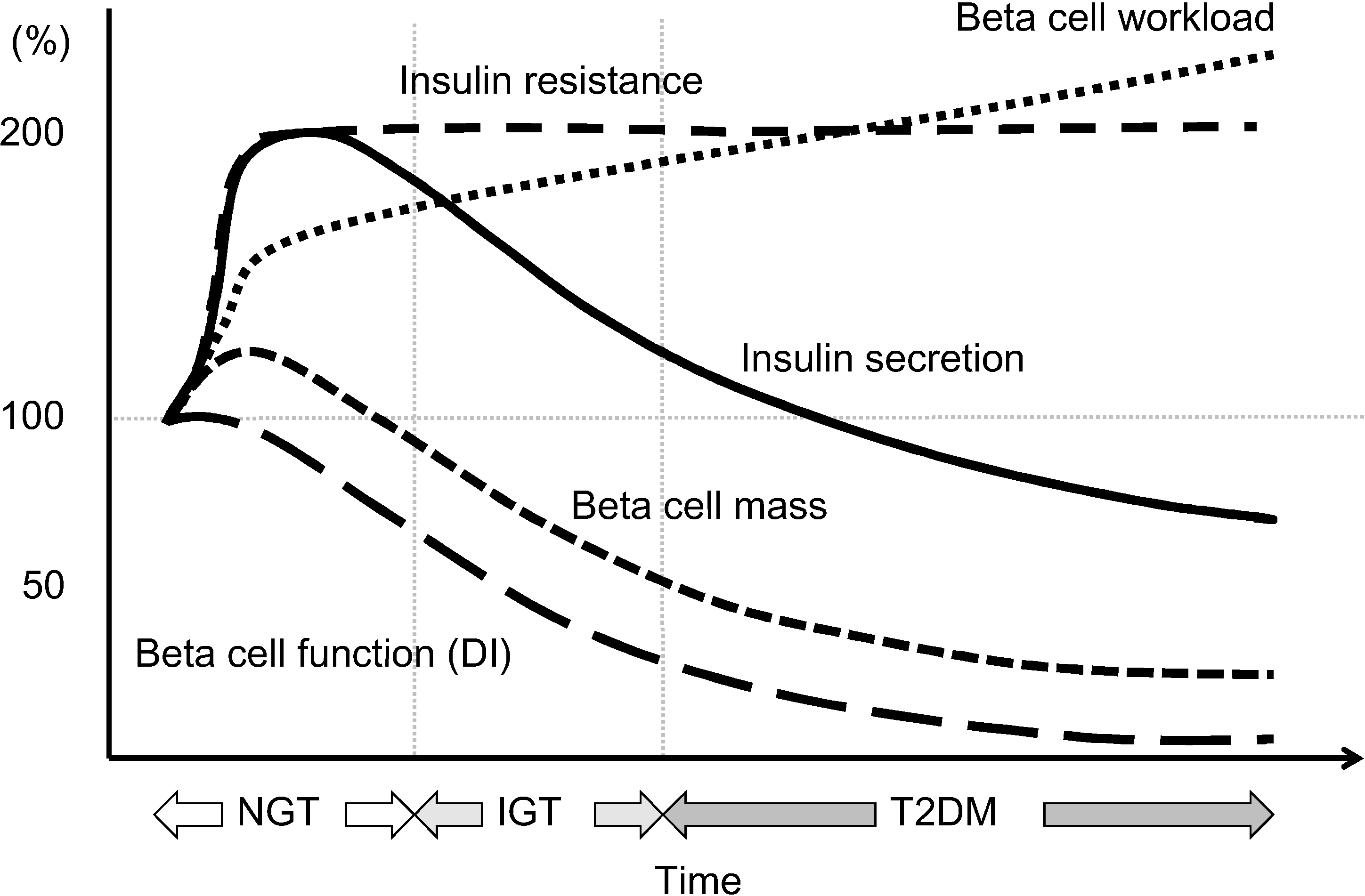
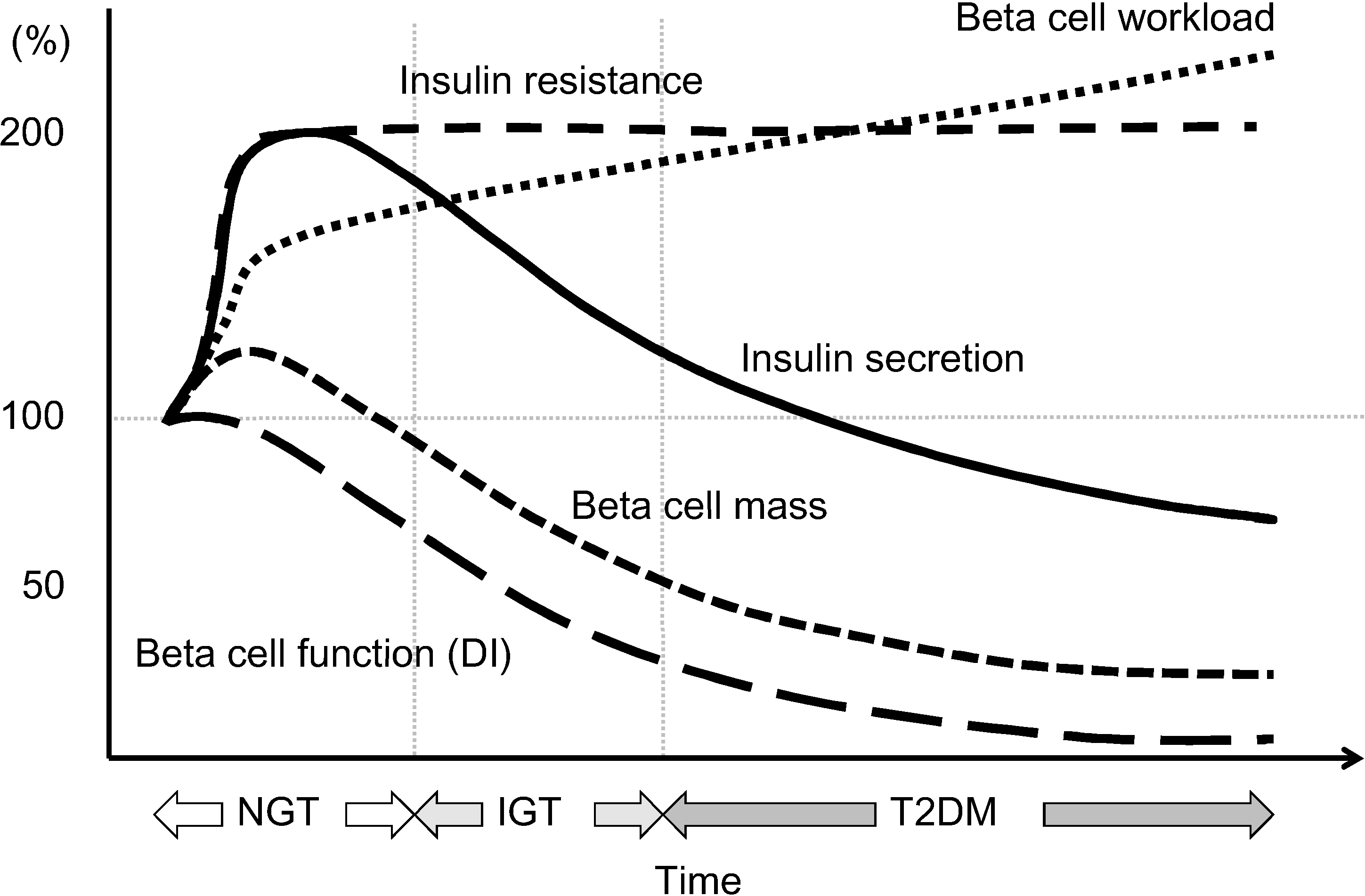
Figure 6.
Hypothesis of change in beta cell function and mass during development of abnormal glucose tolerance. The magnitude of the increased demand for insulin due to insulin resistance caused by excess caloric intake and physical inactivity exceeds the magnitude of beta cell mass expansion, resulting in an increase in beta cell workload. In individuals who are susceptible to type 2 diabetes (T2DM), increased beta cell workload may lead to beta cell failure and the development of T2DM. Reproduced with permission from [101].
Figure 6.
Hypothesis of change in beta cell function and mass during development of abnormal glucose tolerance. The magnitude of the increased demand for insulin due to insulin resistance caused by excess caloric intake and physical inactivity exceeds the magnitude of beta cell mass expansion, resulting in an increase in beta cell workload. In individuals who are susceptible to type 2 diabetes (T2DM), increased beta cell workload may lead to beta cell failure and the development of T2DM. Reproduced with permission from [101].
Acknowledgments
The author is grateful to Wendy Gray for editing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- International Diabetes Federation (2013) IDF Diabetes Atlas, 6th ed. Available online: http://www.idf.org/diabetesatlas (accessed on 1 March 2014).
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef]
- Defronzo, R.A.; Eldor, R.; Abdul-Ghani, M. Pathophysiologic approach to therapy in patients with newly diagnosed type 2 diabetes. Diabetes Care 2013, 36, S127–S138. [Google Scholar] [CrossRef]
- Defronzo, R.A. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef]
- Bergman, R.N.; Ader, M.; Huecking, K.; van Citters, G. Accurate assessment of beta-cell function: The hyperbolic correction. Diabetes 2002, 51, S212–S220. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef]
- Yoon, K.H.; Ko, S.H.; Cho, J.H.; Lee, J.M.; Ahn, Y.B.; Song, K.H.; Yoo, S.J.; Kang, M.I.; Cha, B.Y.; Lee, K.Y.; et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J. Clin. Endocrinol. Metab. 2003, 88, 2300–2308. [Google Scholar] [CrossRef]
- Sakuraba, H.; Mizukami, H.; Yagihashi, N.; Wada, R.; Hanyu, C.; Yagihashi, S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002, 45, 85–96. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Abdul-Ghani, M.A. Preservation of beta-cell function: The key to diabetes prevention. J. Clin. Endocrinol. Metab. 2011, 96, 2354–2366. [Google Scholar] [CrossRef]
- U.K. Prospective Diabetes Study Group. U.K. prospective diabetes study 16. Overview of 6 years therapy of type II diabetes: A progressive disease. Diabetes 1995, 44, 1249–1258. [Google Scholar] [CrossRef]
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443. [Google Scholar] [CrossRef]
- Matthews, D.R.; Cull, C.A.; Stratton, I.M.; Holman, R.R.; Turner, R.C. UKPDS 26: Sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabet. Med. 1998, 15, 297–303. [Google Scholar] [CrossRef]
- Kahn, S.E.; Lachin, J.M.; Zinman, B.; Haffner, S.M.; Aftring, R.P.; Paul, G.; Kravitz, B.G.; Herman, W.H.; Viberti, G.; Holman, R.R. Effects of rosiglitazone, glyburide, and metformin on beta-cell function and insulin sensitivity in ADOPT. Diabetes 2011, 60, 1552–1560. [Google Scholar] [CrossRef]
- TODAY Study Group. Effects of metformin, metformin plus rosiglitazone, and metformin plus lifestyle on insulin sensitivity and beta-cell function in TODAY. Diabetes Care 2013, 36, 1749–1757. [Google Scholar] [CrossRef]
- Saisho, Y.; Kou, K.; Tanaka, K.; Abe, T.; Kurosawa, H.; Shimada, A.; Meguro, S.; Kawai, T.; Itoh, H. Postprandial serum C-peptide to plasma glucose ratio as a predictor of subsequent insulin treatment in patients with type 2 diabetes. Endocr. J. 2011, 58, 315–322. [Google Scholar] [CrossRef]
- Saisho, Y.; Kou, K.; Tanaka, K.; Abe, T.; Shimada, A.; Kawai, T.; Itoh, H. Association between beta cell function and future glycemic control in patients with type 2 diabetes. Endocr. J. 2013, 60, 517–523. [Google Scholar]
- Saisho, Y.; Kou, K.; Tanaka, K.; Abe, T.; Shimada, A.; Kawai, T.; Itoh, H. Postprandial serum C-peptide to plasma glucose ratio predicts future insulin therapy in Japanese patients with type 2 diabetes. Acta Diabetol. 2013, 50, 987–988. [Google Scholar] [CrossRef]
- Saisho, Y.; Tanaka, K.; Abe, T.; Kawai, T.; Itoh, H. Lower beta cell function relates to sustained higher glycated albumin to glycated hemoglobin ratio in Japanese patients with type 2 diabetes. Endocr. J. 2014, 61, 149–157. [Google Scholar] [CrossRef]
- Cha, T.; Tahara, Y.; Ikegami, H.; Fukuda, M.; Yoneda, H.; Yamato, E.; Yamamoto, Y.; Noma, Y.; Shima, K.; Ogihara, T. Urinary C-peptide as an index of unstable glycemic control in insulin-dependent diabetes mellitus (IDDM). Diabetes Res. Clin. Pract. 1991, 13, 181–187. [Google Scholar] [CrossRef]
- Nakanishi, K.; Kobayashi, T.; Inoko, H.; Tsuji, K.; Murase, T.; Kosaka, K. Residual beta-cell function and HLA-A24 in IDDM. Markers of glycemic control and subsequent development of diabetic retinopathy. Diabetes 1995, 44, 1334–1339. [Google Scholar] [CrossRef]
- Fukuda, M.; Tanaka, A.; Tahara, Y.; Ikegami, H.; Yamamoto, Y.; Kumahara, Y.; Shima, K. Correlation between minimal secretory capacity of pancreatic beta-cells and stability of diabetic control. Diabetes 1988, 37, 81–88. [Google Scholar] [CrossRef]
- Saisho, Y.; Tanaka, K.; Abe, T.; Shimada, A.; Kawai, T.; Itoh, H. Glycated albumin to glycated hemoglobin ratio reflects postprandial glucose excursion and relates to beta cell function in both type 1 and type 2 diabetes. Diabetol. Int. 2011, 2, 146–153. [Google Scholar] [CrossRef]
- Tanaka, C.; Saisho, Y.; Tanaka, K.; Kou, K.; Tanaka, M.; Meguro, S.; Irie, J.; Jo, R.; Kawai, T.; Itoh, H. Factors associated with glycemic variability in Japanese patients with diabetes. Diabetol. Int. 2014, 5, 36–42. [Google Scholar] [CrossRef]
- Koga, M.; Kasayama, S. Clinical impact of glycated albumin as another glycemic control marker. Endocr. J. 2010, 57, 751–762. [Google Scholar] [CrossRef]
- Koga, M.; Murai, J.; Saito, H.; Kasayama, S. Glycated albumin and glycated hemoglobin are influenced differently by endogenous insulin secretion in patients with type 2 diabetes. Diabetes Care 2010, 33, 270–272. [Google Scholar] [CrossRef]
- Bo, S.; Gentile, L.; Castiglione, A.; Prandi, V.; Canil, S.; Ghigo, E.; Ciccone, G. C-Peptide and the risk for incident complications and mortality in type 2 diabetic patients: A retrospective cohort study after a 14-year follow-up. Eur. J. Endocrinol. 2012, 167, 173–180. [Google Scholar]
- Kim, B.Y.; Jung, C.H.; Mok, J.O.; Kang, S.K.; Kim, C.H. Association between serum C-peptide levels and chronic microvascular complications in Korean type 2 diabetic patients. Acta Diabetol. 2012, 49, 9–15. [Google Scholar] [CrossRef]
- Seshasai, S.R.; Kaptoge, S.; Thompson, A.; di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef] [Green Version]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-Year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef]
- Frier, B.M.; Schernthaner, G.; Heller, S.R. Hypoglycemia and cardiovascular risks. Diabetes Care 2011, 34, S132–S137. [Google Scholar] [CrossRef]
- Desouza, C.V.; Bolli, G.B.; Fonseca, V. Hypoglycemia, diabetes, and cardiovascular events. Diabetes Care 2010, 33, 1389–1394. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycemia in type 2 diabetes: A patient-centered approach: Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012, 35, 1364–1379. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar]
- Saenz, A.; Fernandez-Esteban, I.; Mataix, A.; Ausejo, M.; Roque, M.; Moher, D. Metformin monotherapy for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2005, 3. [Google Scholar] [CrossRef]
- McDonagh, M.S.; Selph, S.; Ozpinar, A.; Foley, C. Systematic review of the benefits and risks of metformin in treating obesity in children aged 18 years and younger. JAMA Pediatr. 2014, 168, 178–184. [Google Scholar] [CrossRef]
- Wang, H.; Ni, Y.; Yang, S.; Li, H.; Li, X.; Feng, B. The effects of gliclazide, metformin, and acarbose on body composition in patients with newly diagnosed type 2 diabetes mellitus. Curr. Ther. Res. Clin. Exp. 2013, 75, 88–92. [Google Scholar] [CrossRef]
- Kostev, K.; Rex, J.; Rockel, T.; Heilmaier, C. Effects of selected antidiabetics on weight loss—A retrospective database analysis. Prim. Care Diabetes 2014. [Google Scholar] [CrossRef]
- Karagiannis, T.; Paschos, P.; Paletas, K.; Matthews, D.R.; Tsapas, A. Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: Systematic review and meta-analysis. BMJ 2012, 344. [Google Scholar] [CrossRef]
- Russell-Jones, D.; Cuddihy, R.M.; Hanefeld, M.; Kumar, A.; Gonzalez, J.G.; Chan, M.; Wolka, A.M.; Boardman, M.K. Efficacy and safety of exenatide once weekly vs. metformin, pioglitazone, and sitagliptin used as monotherapy in drug-naive patients with type 2 diabetes (DURATION-4): A 26-week double-blind study. Diabetes Care 2012, 35, 252–258. [Google Scholar] [CrossRef]
- Apovian, C.M. The clinical and economic consequences of obesity. Am. J. Manag. Care 2013, 19, S219–S228. [Google Scholar]
- Hemkens, L.G.; Grouven, U.; Bender, R.; Gunster, C.; Gutschmidt, S.; Selke, G.W.; Sawicki, P.T. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: A cohort study. Diabetologia 2009, 52, 1732–1744. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef]
- Jonasson, J.M.; Ljung, R.; Talback, M.; Haglund, B.; Gudbjornsdottir, S.; Steineck, G. Insulin glargine use and short-term incidence of malignancies—A population-based follow-up study in Sweden. Diabetologia 2009, 52, 1745–1754. [Google Scholar] [CrossRef]
- Colhoun, H.M. Use of insulin glargine and cancer incidence in Scotland: A study from the Scottish Diabetes Research Network Epidemiology Group. Diabetologia 2009, 52, 1755–1765. [Google Scholar] [CrossRef]
- Rosenstock, J.; Fonseca, V.; McGill, J.B.; Riddle, M.; Halle, J.P.; Hramiak, I.; Johnston, P.; Davis, M. Similar risk of malignancy with insulin glargine and neutral protamine Hagedorn (NPH) insulin in patients with type 2 diabetes: Findings from a 5 year randomised, open-label study. Diabetologia 2009, 52, 1971–1973. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; He, Z.; Liu, J. Insulin glargine and cancer risk in patients with diabetes: A meta-analysis. PLoS One 2012, 7, e51814. [Google Scholar] [CrossRef]
- Sturmer, T.; Marquis, M.A.; Zhou, H.; Meigs, J.B.; Lim, S.; Blonde, L.; Macdonald, E.; Wang, R.; Lavange, L.M.; Pate, V.; et al. Cancer incidence among those initiating insulin therapy with glargine vs. human NPH insulin. Diabetes Care 2013, 36, 3517–3525. [Google Scholar] [CrossRef]
- Habel, L.A.; Danforth, K.N.; Quesenberry, C.P.; Capra, A.; van den Eeden, S.K.; Weiss, N.S.; Ferrara, A. Cohort study of insulin glargine and risk of breast, prostate, and colorectal cancer among patients with diabetes. Diabetes Care 2013, 36, 3953–3960. [Google Scholar] [CrossRef]
- Currie, C.J.; Peters, J.R.; Tynan, A.; Evans, M.; Heine, R.J.; Bracco, O.L.; Zagar, T.; Poole, C.D. Survival as a function of HbA(1c) in people with type 2 diabetes: A retrospective cohort study. Lancet 2010, 375, 481–489. [Google Scholar] [CrossRef]
- Dandona, P.; Chaudhuri, A.; Ghanim, H.; Mohanty, P. Insulin as an anti-inflammatory and antiatherogenic modulator. J. Am. Coll. Cardiol. 2009, 53, S14–S20. [Google Scholar] [CrossRef]
- Smith, U.; Gale, E.A. Does diabetes therapy influence the risk of cancer? Diabetologia 2009, 52, 1699–1708. [Google Scholar] [CrossRef]
- Rensing, K.L.; von der Thusen, J.H.; Weijers, E.M.; Houttuijn Bloemendaal, F.M.; van Lammeren, G.W.; Vink, A.; van der Wal, A.C.; van Hinsbergh, V.W.; van der Loos, C.M.; Stroes, E.S.; et al. Endothelial insulin receptor expression in human atherosclerotic plaques: Linking micro- and macrovascular disease in diabetes? Atherosclerosis 2012, 222, 208–215. [Google Scholar] [CrossRef]
- Gamble, J.M.; Simpson, S.H.; Eurich, D.T.; Majumdar, S.R.; Johnson, J.A. Insulin use and increased risk of mortality in type 2 diabetes: A cohort study. Diabetes Obes. Metab. 2010, 12, 47–53. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Bosch, J.; Dagenais, G.R.; Diaz, R.; Jung, H.; Maggioni, A.P.; Pogue, J.; Probstfield, J.; Ramachandran, A.; Riddle, M.C.; et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N. Engl. J. Med. 2012, 367, 319–328. [Google Scholar] [CrossRef]
- Monnier, L.; Hanefeld, M.; Schnell, O.; Colette, C.; Owens, D. Insulin and atherosclerosis: How are they related? Diabetes Metab. 2013, 39, 111–117. [Google Scholar] [CrossRef]
- The DECODE Study Group on behalf of the European Diabetes Epidemiology Group. Glucose tolerance and cardiovascular mortality: Comparison of fasting and 2-hour diagnostic criteria. Arch. Intern. Med. 2001, 161, 397–405. [Google Scholar] [CrossRef]
- Nakagami, T. Hyperglycaemia and mortality from all causes and from cardiovascular disease in five populations of Asian origin. Diabetologia 2004, 47, 385–394. [Google Scholar] [CrossRef]
- Tominaga, M.; Eguchi, H.; Manaka, H.; Igarashi, K.; Kato, T.; Sekikawa, A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The Funagata Diabetes Study. Diabetes Care 1999, 22, 920–924. [Google Scholar] [CrossRef]
- Risso, A.; Mercuri, F.; Quagliaro, L.; Damante, G.; Ceriello, A. Intermittent high glucose enhances apoptosis in human umbilical vein endothelial cells in culture. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E924–E930. [Google Scholar]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef]
- Raz, I.; Wilson, P.W.; Strojek, K.; Kowalska, I.; Bozikov, V.; Gitt, A.K.; Jermendy, G.; Campaigne, B.N.; Kerr, L.; Milicevic, Z.; et al. Effects of prandial vs. fasting glycemia on cardiovascular outcomes in type 2 diabetes: The HEART2D trial. Diabetes Care 2009, 32, 381–386. [Google Scholar] [CrossRef]
- Raz, I.; Ceriello, A.; Wilson, P.W.; Battioui, C.; Su, E.W.; Kerr, L.; Jones, C.A.; Milicevic, Z.; Jacober, S.J. Post hoc subgroup analysis of the HEART2D trial demonstrates lower cardiovascular risk in older patients targeting postprandial vs. fasting/premeal glycemia. Diabetes Care 2011, 34, 1511–1513. [Google Scholar] [CrossRef]
- Kilpatrick, E.S.; Rigby, A.S.; Atkin, S.L. Effect of glucose variability on the long-term risk of microvascular complications in type 1 diabetes. Diabetes Care 2009, 32, 1901–1903. [Google Scholar] [CrossRef]
- Cavalot, F.; Pagliarino, A.; Valle, M.; di Martino, L.; Bonomo, K.; Massucco, P.; Anfossi, G.; Trovati, M. Postprandial blood glucose predicts cardiovascular events and all-cause mortality in type 2 diabetes in a 14-year follow-up: Lessons from the San Luigi Gonzaga Diabetes Study. Diabetes Care 2011, 34, 2237–2243. [Google Scholar] [CrossRef]
- Munshi, M.N.; Pandya, N.; Umpierrez, G.E.; DiGenio, A.; Zhou, R.; Riddle, M.C. Contributions of basal and prandial hyperglycemia to total hyperglycemia in older and younger adults with type 2 diabetes mellitus. J. Am. Geriatr. Soc. 2013, 61, 535–541. [Google Scholar] [CrossRef]
- Miller, M.E.; Williamson, J.D.; Gerstein, H.C.; Byington, R.P.; Cushman, W.C.; Ginsberg, H.N.; Ambrosius, W.T.; Lovato, L.; Applegate, W.B. Effects of randomization to intensive glucose control on adverse events, cardiovascular disease, and mortality in older vs. younger adults in the ACCORD Trial. Diabetes Care 2014, 37, 634–643. [Google Scholar] [CrossRef]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D.; Schwenke, D.C.; Banerji, M.; Bray, G.A.; Buchanan, T.A.; Clement, S.C.; Henry, R.R.; Hodis, H.N.; Kitabchi, A.E. Pioglitazone for diabetes prevention in impaired glucose tolerance. N. Engl. J. Med. 2011, 364, 1104–1115. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Yusuf, S.; Bosch, J.; Pogue, J.; Sheridan, P.; Dinccag, N.; Hanefeld, M.; Hoogwerf, B.; Laakso, M.; Mohan, V.; et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: A randomised controlled trial. Lancet 2006, 368, 1096–1105. [Google Scholar] [CrossRef]
- Knowler, W.C.; Hamman, R.F.; Edelstein, S.L.; Barrett-Connor, E.; Ehrmann, D.A.; Walker, E.A.; Fowler, S.E.; Nathan, D.M.; Kahn, S.E. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes 2005, 54, 1150–1156. [Google Scholar] [CrossRef]
- Weng, J.; Li, Y.; Xu, W.; Shi, L.; Zhang, Q.; Zhu, D.; Hu, Y.; Zhou, Z; Yan, X; Tian, H.; et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: A multicentre randomised parallel-group trial. Lancet 2008, 371, 1753–1760. [Google Scholar] [CrossRef]
- Pennartz, C.; Schenker, N.; Menge, B.A.; Schmidt, W.E.; Nauck, M.A.; Meier, J.J. Chronic reduction of fasting glycemia with insulin glargine improves first- and second-phase insulin secretion in patients with type 2 diabetes. Diabetes Care 2011, 34, 2048–2053. [Google Scholar] [CrossRef]
- Holman, R.R.; Haffner, S.M.; McMurray, J.J.; Bethel, M.A.; Holzhauer, B.; Hua, T.A.; Belenkov, Y.; Boolell, M.; Buse, J.B.; Buckley, B.M.; et al. Effect of nateglinide on the incidence of diabetes and cardiovascular events. N. Engl. J. Med. 2010, 362, 1463–1476. [Google Scholar] [CrossRef]
- Wing, R.R.; Bolin, P.; Brancati, F.L.; Bray, G.A.; Clark, J.M.; Coday, M.; Crow, R.S.; Curtis, J.M.; Egan, C.M.; Espeland, M.A.; et al. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N. Engl. J. Med. 2013, 369, 145–154. [Google Scholar] [CrossRef]
- Williamson, D.A.; Rejeski, J.; Lang, W.; van Dorsten, B.; Fabricatore, A.N.; Toledo, K. Impact of a weight management program on health-related quality of life in overweight adults with type 2 diabetes. Arch. Intern. Med. 2009, 169, 163–171. [Google Scholar] [CrossRef]
- Wing, R.R.; Lang, W.; Wadden, T.A.; Safford, M.; Knowler, W.C.; Bertoni, A.G.; Hill, J.O.; Brancati, F.L.; Peters, A.; Wagenknecht, L. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care 2011, 34, 1481–1486. [Google Scholar] [CrossRef]
- Gregg, E.W.; Chen, H.; Wagenknecht, L.E.; Clark, J.M.; Delahanty, L.M.; Bantle, J.; Pownall, H.J.; Johnson, K.C.; Safford, M.M.; Kitabchi, A.E.; et al. Association of an intensive lifestyle intervention with remission of type 2 diabetes. JAMA 2012, 308, 2489–2496. [Google Scholar] [CrossRef]
- Breyer, B.N.; Phelan, S.; Hogan, P.E.; Rosen, R.C.; Kitabchi, A.E.; Wing, R.R.; Brown, J.S. Intensive lifestyle intervention reduces urinary incontinence in overweight/obese men with type 2 diabetes: Results from the Look AHEAD trial. J. Urol. 2014. [Google Scholar] [CrossRef]
- Gerstein, H.C. Do lifestyle changes reduce serious outcomes in diabetes? N. Engl. J. Med. 2013, 369, 189–190. [Google Scholar] [CrossRef]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; Davidson, M.B.; Einhorn, D.; Garvey, W.T.; et al. AACE comprehensive diabetes management algorithm 2013. Endocr. Pract. 2013, 19, 327–336. [Google Scholar]
- DeFronzo, R.A.; Barzilai, N.; Simonson, D.C. Mechanism of metformin action in obese and lean noninsulin-dependent diabetic subjects. J. Clin. Endocrinol. Metab. 1991, 73, 1294–1301. [Google Scholar] [CrossRef]
- Kim, C.H.; Han, K.A.; Oh, H.J.; Tan, K.E.; Sothiratnam, R.; Tjokroprawiro, A.; Klein, M. Safety, tolerability, and efficacy of metformin extended-release oral antidiabetic therapy in patients with type 2 diabetes: An observational trial in Asia. J. Diabetes 2012, 4, 395–406. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- FDA places greater restrictions on access to rosiglitazone. BMJ 2010, 341. [CrossRef]
- Blind, E.; Dunder, K.; de Graeff, P.A.; Abadie, E. Rosiglitazone: A European regulatory perspective. Diabetologia 2011, 54, 213–218. [Google Scholar] [CrossRef]
- McCarthy, M. US regulators relax restrictions on rosiglitazone. BMJ 2013, 347. [Google Scholar] [CrossRef]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefebvre, P.J.; Murray, G.D.; et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial in macroVascular Events): A randomised controlled trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Tzoulaki, I.; Molokhia, M.; Curcin, V.; Little, M.P.; Millett, C.J.; Ng, A.; Hughes, R.I.; Khunti, K.; Wilkins, M.R.; Majeed, A.; et al. Risk of cardiovascular disease and all cause mortality among patients with type 2 diabetes prescribed oral antidiabetes drugs: Retrospective cohort study using UK general practice research database. BMJ 2009, 339. [Google Scholar] [CrossRef]
- Loke, Y.K.; Kwok, C.S.; Singh, S. Comparative cardiovascular effects of thiazolidinediones: Systematic review and meta-analysis of observational studies. BMJ 2011, 342. [Google Scholar] [CrossRef]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A.; Jure, H.; De Larochelliere, R.; Staniloae, C.S.; Mavromatis, K.; et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef]
- Meier, C.; Kraenzlin, M.E.; Bodmer, M.; Jick, S.S.; Jick, H.; Meier, C.R. Use of thiazolidinediones and fracture risk. Arch. Intern. Med. 2008, 168, 820–825. [Google Scholar] [CrossRef]
- Lewis, J.D.; Ferrara, A.; Peng, T.; Hedderson, M.; Bilker, W.B.; Quesenberry, C.P., Jr.; Vaughn, D.J.; Nessel, L.; Selby, J.; Strom, B.L. Risk of bladder cancer among diabetic patients treated with pioglitazone: Interim report of a longitudinal cohort study. Diabetes Care 2011, 34, 916–922. [Google Scholar] [CrossRef]
- Neumann, A.; Weill, A.; Ricordeau, P.; Fagot, J.P.; Alla, F.; Allemand, H. Pioglitazone and risk of bladder cancer among diabetic patients in France: A population-based cohort study. Diabetologia 2012, 55, 1953–1962. [Google Scholar] [CrossRef]
- Colmers, I.N.; Bowker, S.L.; Majumdar, S.R.; Johnson, J.A. Use of thiazolidinediones and the risk of bladder cancer among people with type 2 diabetes: A meta-analysis. CMAJ 2012, 184. [Google Scholar] [CrossRef]
- Saisho, Y. Obesity, type 2 diabetes and beta cell failure: An Asian perspective. J. Mol. Genet. Med. 2014. [Google Scholar] [CrossRef]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Kawamori, R.; Tajima, N.; Iwamoto, Y.; Kashiwagi, A.; Shimamoto, K.; Kaku, K. Voglibose for prevention of type 2 diabetes mellitus: A randomised, double-blind trial in Japanese individuals with impaired glucose tolerance. Lancet 2009, 373, 1607–1614. [Google Scholar] [CrossRef]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: The STOP-NIDDM trial. JAMA 2003, 290, 486–494. [Google Scholar] [CrossRef]
- Hanefeld, M.; Chiasson, J.L.; Koehler, C.; Henkel, E.; Schaper, F.; Temelkova-Kurktschiev, T. Acarbose slows progression of intima-media thickness of the carotid arteries in subjects with impaired glucose tolerance. Stroke 2004, 35, 1073–1078. [Google Scholar] [CrossRef]
- Hanefeld, M.; Cagatay, M.; Petrowitsch, T.; Neuser, D.; Petzinna, D.; Rupp, M. Acarbose reduces the risk for myocardial infarction in type 2 diabetic patients: Meta-analysis of seven long-term studies. Eur. Heart J. 2004, 25, 10–16. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Kendall, D.M.; Cuddihy, R.M.; Bergenstal, R.M. Clinical application of incretin-based therapy: Therapeutic potential, patient selection and clinical use. Am. J. Med. 2009, 122, S37–S50. [Google Scholar] [CrossRef]
- Aroda, V.R.; Henry, R.R.; Han, J.; Huang, W.; DeYoung, M.B.; Darsow, T.; Hoogwerf, B.J. Efficacy of GLP-1 receptor agonists and DPP-4 inhibitors: Meta-analysis and systematic review. Clin. Ther. 2012, 34. [Google Scholar] [CrossRef]
- Rolin, B.; Larsen, M.O.; Gotfredsen, C.F.; Deacon, C.F.; Carr, R.D.; Wilken, M.; Knudsen, L.B. The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E745–E752. [Google Scholar]
- Lamont, B.J.; Li, Y.; Kwan, E.; Brown, T.J.; Gaisano, H.; Drucker, D.J. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. J. Clin. Investig. 2012, 122, 388–402. [Google Scholar] [CrossRef]
- Farilla, L.; Bulotta, A.; Hirshberg, B.; Li Calzi, S.; Khoury, N.; Noushmehr, H.; Bertolotto, C.; Di Mario, U.; Harlan, D.M.; Perfetti, R. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003, 144, 5149–5158. [Google Scholar] [CrossRef]
- Bunck, M.C.; Corner, A.; Eliasson, B.; Heine, R.J.; Shaginian, R.M.; Taskinen, M.R.; Smith, U.; Yki-Jarvinen, H.; Diamant, M. Effects of exenatide on measures of beta-cell function after 3 years in metformin-treated patients with type 2 diabetes. Diabetes Care 2011, 34, 2041–2047. [Google Scholar] [CrossRef]
- Foley, J.E.; Bunck, M.C.; Moller-Goede, D.L.; Poelma, M.; Nijpels, G.; Eekhoff, E.M.; Schweizer, A.; Heine, R.J.; Diamant, M. Beta cell function following 1 year vildagliptin or placebo treatment and after 12 week washout in drug-naive patients with type 2 diabetes and mild hyperglycaemia: A randomised controlled trial. Diabetologia 2011, 54, 1985–1991. [Google Scholar] [CrossRef]
- Kohro, T.; Yamazaki, T.; Sato, H.; Harada, K.; Ohe, K.; Komuro, I.; Nagai, R. Trends in antidiabetic prescription patterns in Japan from 2005 to 2011. Int. Heart J. 2013, 54, 93–97. [Google Scholar] [CrossRef]
- Kodani, N.; Saisho, Y.; Tanaka, K.; Kawai, T.; Itoh, H. Effects of mitiglinide, a short-acting insulin secretagogue, on daily glycemic variability and oxidative stress markers in Japanese patients with type 2 diabetes mellitus. Clin. Drug Investig. 2013, 33, 563–570. [Google Scholar]
- Del Prato, S.; Tiengo, A. The importance of first-phase insulin secretion: Implications for the therapy of type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 2001, 17, 164–174. [Google Scholar] [CrossRef]
- Unger, R.H. Reinventing type 2 diabetes: Pathogenesis, treatment, and prevention. JAMA 2008, 299, 1185–1187. [Google Scholar] [CrossRef]
- Tahrani, A.A.; Barnett, A.H.; Bailey, C.J. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 2013, 1, 140–151. [Google Scholar] [CrossRef]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef]
- Riser Taylor, S.; Harris, K.B. The clinical efficacy and safety of sodium glucose cotransporter-2 inhibitors in adults with type 2 diabetes mellitus. Pharmacotherapy 2013, 33, 984–999. [Google Scholar] [CrossRef]
- Gloy, V.L.; Briel, M.; Bhatt, D.L.; Kashyap, S.R.; Schauer, P.R.; Mingrone, G.; Bucher, H.C.; Nordmann, A.J. Bariatric surgery vs. non-surgical treatment for obesity: A systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 347. [Google Scholar] [CrossRef]
- Saisho, Y.; Tanaka, K.; Abe, T.; Shimada, A.; Kawai, T.; Itoh, H. Effect of obesity on declining beta cell function after diagnosis of type 2 diabetes: A possible link suggested by cross-sectional analysis. Endocr. J. 2012, 59, 187–195. [Google Scholar] [CrossRef]
- Dixon, J.B.; Chuang, L.M.; Chong, K.; Chen, S.C.; Lambert, G.W.; Straznicky, N.E.; Lambert, E.A.; Lee, W.J. Predicting the glycemic response to gastric bypass surgery in patients with type 2 diabetes. Diabetes Care 2013, 36, 20–26. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lee, W.J.; Liew, P.L. Predictors of remission of type 2 diabetes mellitus in obese patients after gastrointestinal surgery. Obes. Res. Clin. Pract. 2013, 7. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Saisho, Y. Importance of Beta Cell Function for the Treatment of Type 2 Diabetes. J. Clin. Med. 2014, 3, 923-943. https://doi.org/10.3390/jcm3030923
AMA Style
Saisho Y. Importance of Beta Cell Function for the Treatment of Type 2 Diabetes. Journal of Clinical Medicine. 2014; 3(3):923-943. https://doi.org/10.3390/jcm3030923
Chicago/Turabian StyleSaisho, Yoshifumi. 2014. "Importance of Beta Cell Function for the Treatment of Type 2 Diabetes" Journal of Clinical Medicine 3, no. 3: 923-943. https://doi.org/10.3390/jcm3030923