
Computational Investigation of the Effect of Lipid Membranes on Ion Permeation in Gramicidin A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
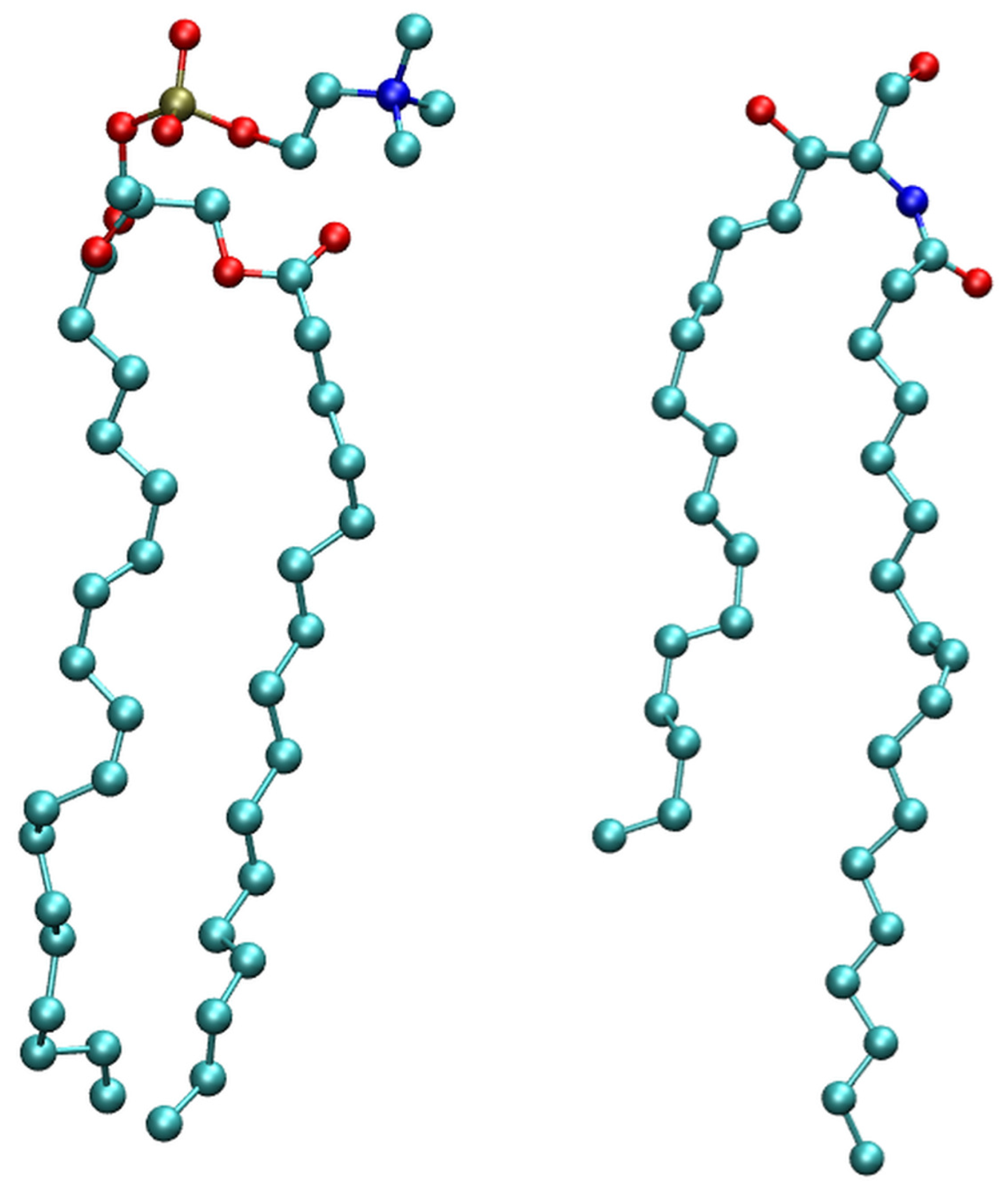
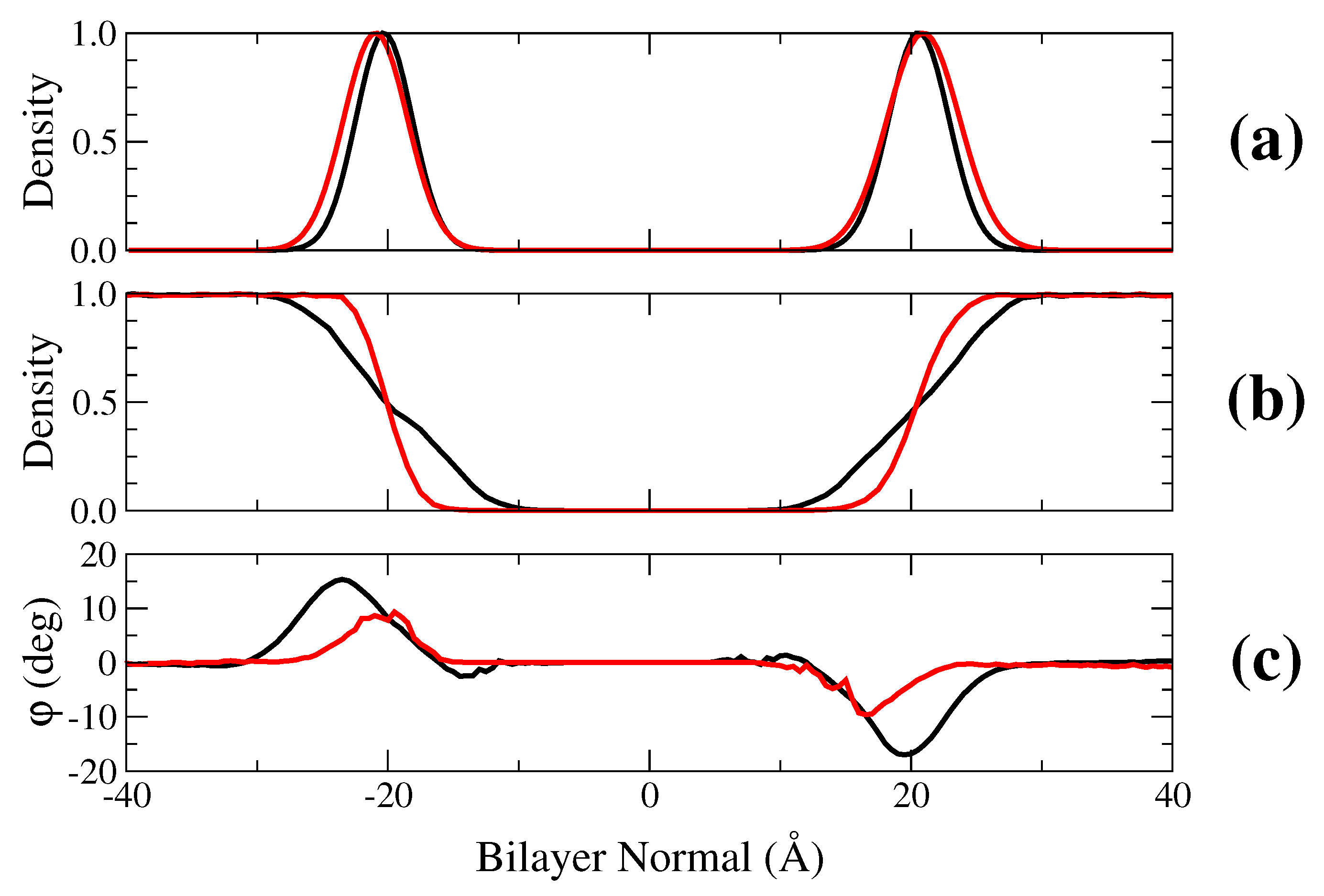
2.1. Membrane Bilayer
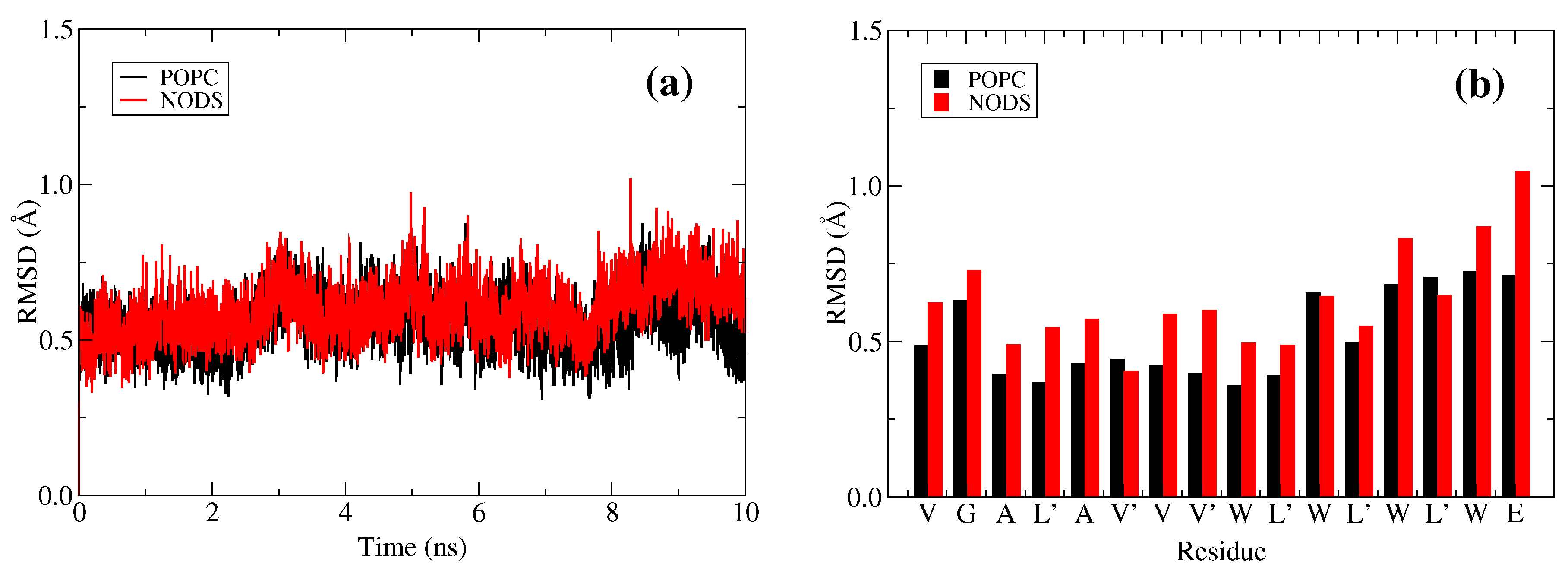
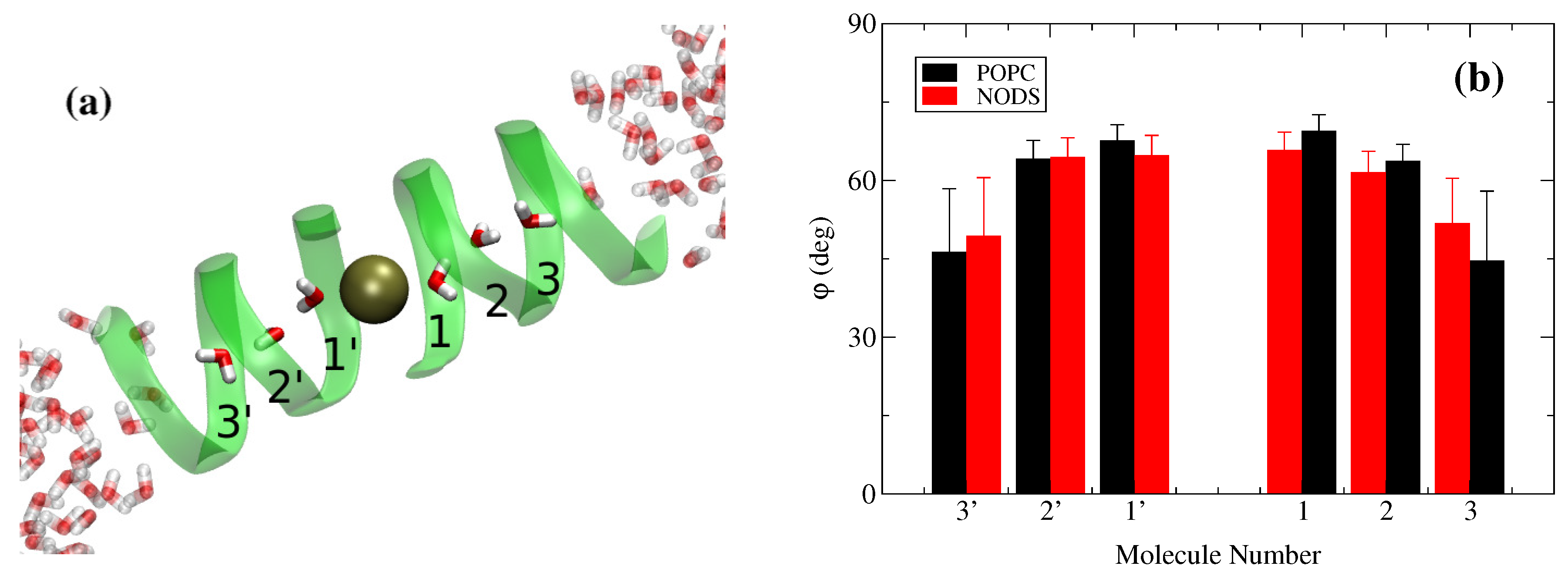
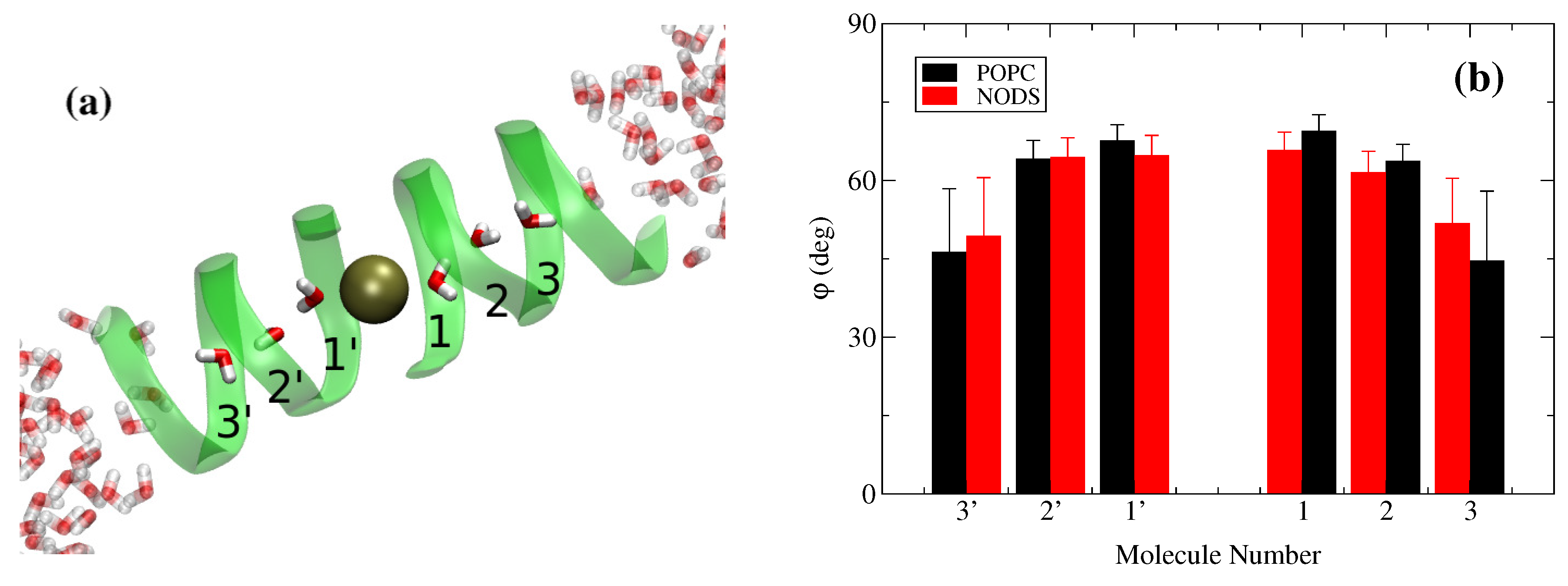
2.2. gA Embedded in Membrane Bilayer
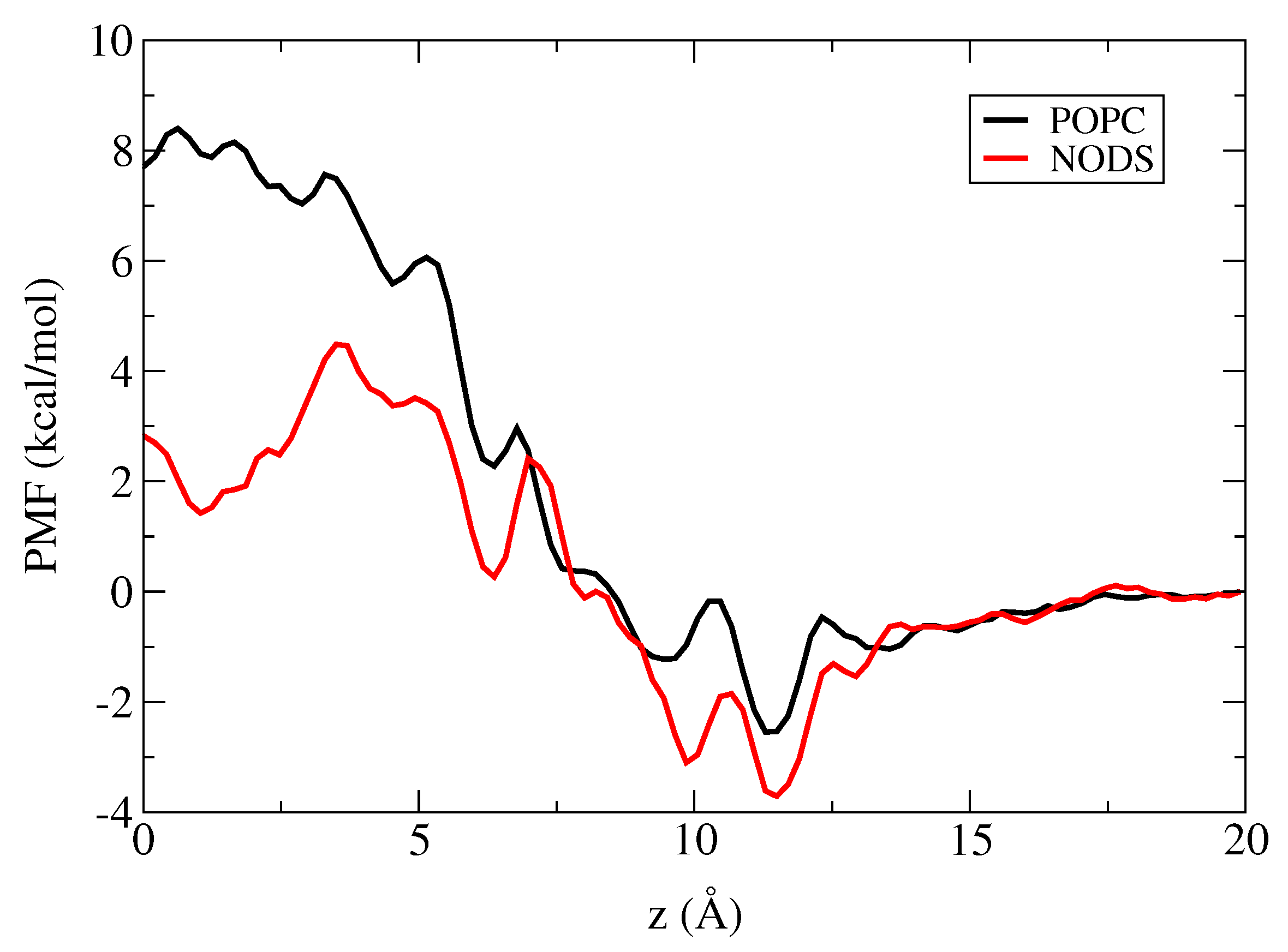
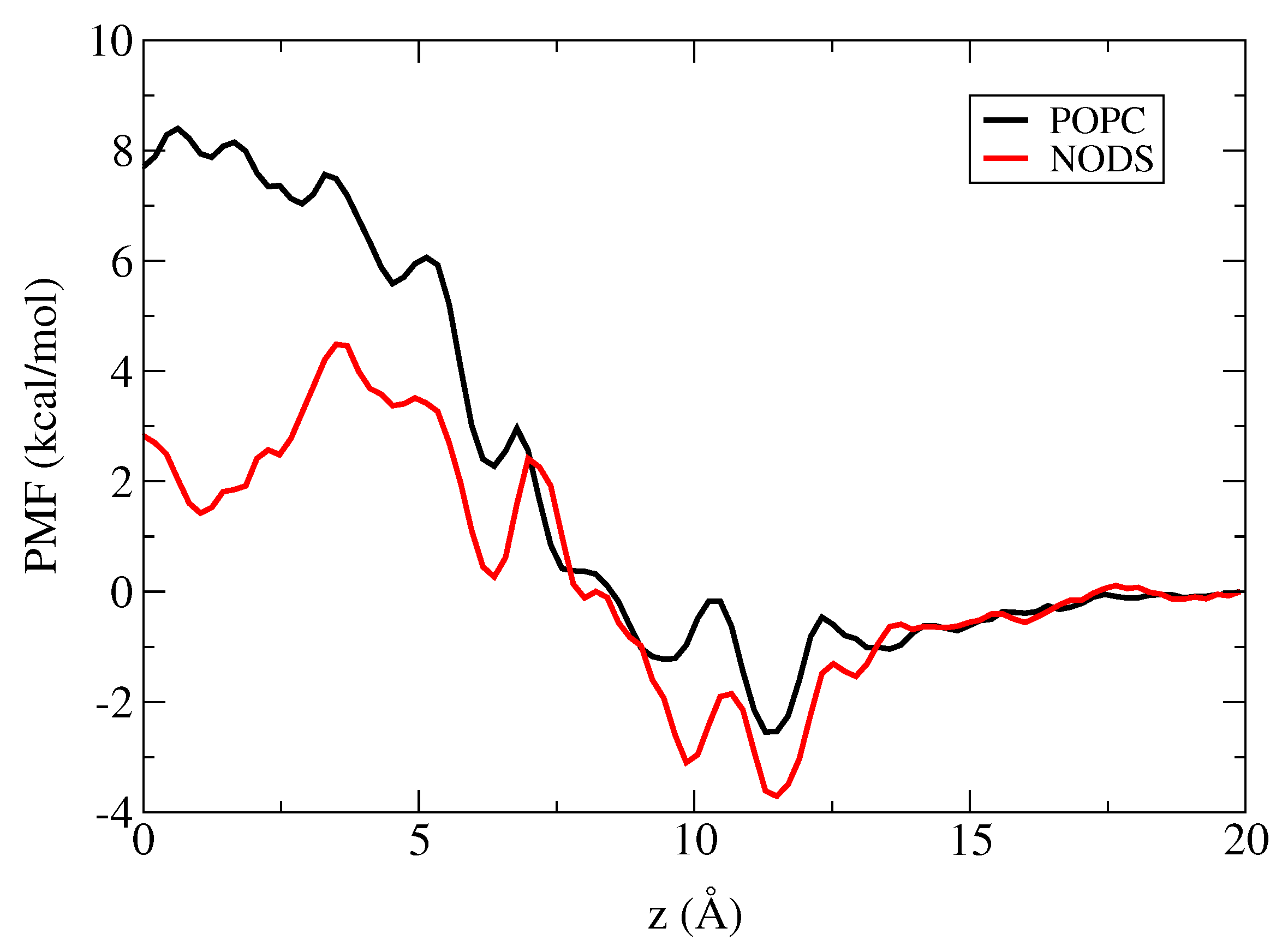
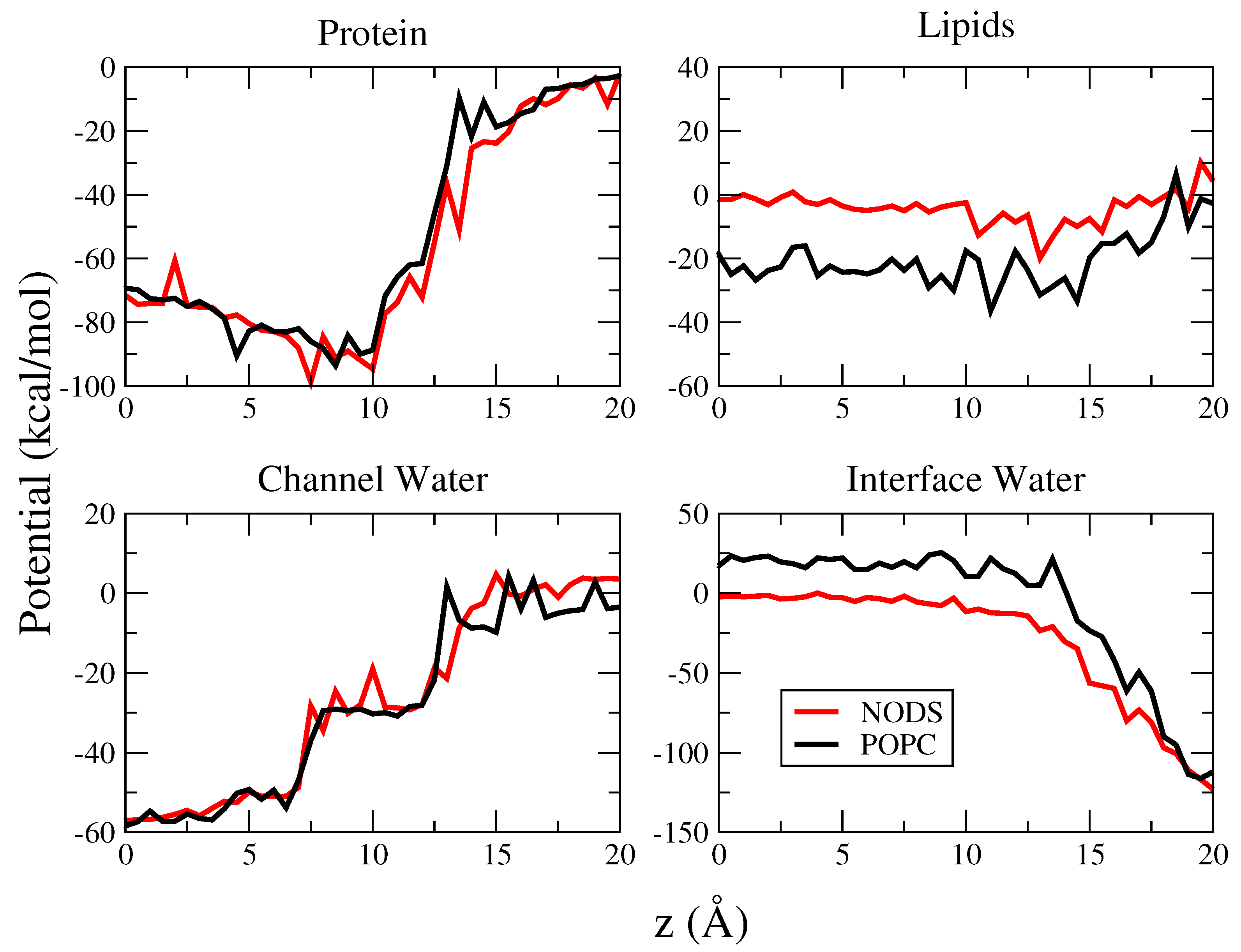
2.3. Potential of Mean Force of K in gA
3. Materials and Methods
3.1. Model System
3.2. PMF Calculations
3.3. MD Simulations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| gA | gramicidin A |
| MD | molecular dynamics |
| NODS | ceramide C18, N-oleoyl-D-erythro-sphingosine |
| PC | phosphatidylcholine |
| POPC | phospholipid, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine |
| PMF | potential of mean force |
References
- Arseniev, A.S.; Barsukov, I.L.; Bystrov, V.F.; Lomize, A.L.; Ovchinnikov, Y.A. H-NMR study of gramicidin A transmembrane channel single-stranded helices. FEBS Lett. 1985, 2, 168–174. [Google Scholar] [CrossRef]
- Ketchem, R.R.; Hu, W.; Cross, T.A. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 1993, 261, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Sarges, R.; Witkop, B. Gramicidin A.V. the structure of valine- and isoleucine-gramicidin A. J. Amer. Chem. Soc. 1965, 87, 2011–2019. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Chandrasekaran, R. Studies on dipeptide conformation and on peptides with sequences of alternating L and D residues with special reference to antibiotic and ion transport peptides. Prog. Peptides Res. 1972, 2, 195–215. [Google Scholar]
- Ketchem, R.R.; Lee, K.C.; Huo, S.; Cross, T.A. Macromolecular structural elucidation with solid-state NMR-derived orientational constraints. J. Biomol. NMR 1996, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Townsley, L.E.; Tucker, W.A.; Sham, S.; Hinton, J.F. Structures of gramicidin A, B and C incorporated into sodium dodecyl sulfate micelles. Biochem. J. 2001, 40, 11676–11686. [Google Scholar] [CrossRef]
- Allen, T.W.; Andersen, O.S.; Roux, B. Structure of gramicidin A in a lipid bilayer environment determined using molecular dynamics simulations and solid-state NMR data. J. Am. Chem. Soc. 2003, 125, 9868–9877. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.W.; Andersen, O.S.; Roux, B. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA 2004, 101, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.W.; Andersen, O.S.; Roux, B. Ion permeation through a narow channel: Using gramicidin to ascertain all-atom molecular dynamics potential of mean force methodology and biomolecular force fields. Biophys. J. 2006, 90, 3447–3460. [Google Scholar] [CrossRef] [PubMed]
- Bastug, T.; Kuyucak, S. Energetics of ion permeation, rejection, binding and block in gramicidin A from free energy simulations. Biophys. J. 2006, 90, 3941–3950. [Google Scholar] [CrossRef] [PubMed]
- Bastug, T.; Kuyucak, S. Free energy simulations of single and double ion occupancy in gramicidin A. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Partenskii, M.B.; Jordan, P.C. Theoretical perspectives on ion-channel electrostatics: Continuum and microscopic approaches. Q. Rev. Biophys. 1992, 25, 477–510. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Karplus, M. Molecular dynamics simulations of the gramicidin channel. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 731–761. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.; Corry, B.; Kuyucak, S.; Chung, S.H. Continuum electrostatics fails to describe ion permeation in the gramicidin channel. Biophys. J. 2000, 83, 1348–1360. [Google Scholar] [CrossRef]
- Allen, T.W.; Bastug, T.; Kuyucak, S.; Chung, S.H. Gramicidin A channel as a test ground for molecular dynamics force fields. Biophys. J. 2003, 84, 2159–2168. [Google Scholar] [CrossRef]
- Bastug, T.; Gray-Weale, A.; Patra, S.M.; Kuyucak, S. Role of protein flexibility in ion permeation: A case study in gramicidin A. Biophys. J. 2006, 90, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Bastug, T.; Kuyucak, S. Application of Jarzynski’s equality in simple versus complex systems. Chem. Phys. Lett. 2007, 436, 383–387. [Google Scholar] [CrossRef]
- Timko, J.; Kuyucak, S. Investigation of polarization effects in the gramicidin A channel from ab initio molecular dynamics simulations. J. Chem. Phys. 2012, 137. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Valiyaveetil, F.I.; Zhou, Y.; Mackinnon, R. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry 2002, 41, 10771–10777. [Google Scholar] [CrossRef] [PubMed]
- Elmore, D.E.; Dougherty, D.A. Investigating lipid composition effects on the mechanosensitive channel of large conductance (MscL) using molecular dynamics simulations. Biophys. J. 2003, 85, 1512–1524. [Google Scholar] [CrossRef]
- Jensen, M.O.; Mouritsen, O.G. Lipids do influence protein function-the hydrophobic matching hypothesis revisited. Biochim. Biophys. Acta 2004, 1666, 205–226. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, A.; Chen, J.; MacKinnon, R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 15441–15446. [Google Scholar] [CrossRef] [PubMed]
- De Godoy, C.M.; Cukierman, S. Membrane phosphate headgroups’ modulation of permeation of alkaline cations in gramicidin channels. J. Phys. Chem. B 2011, 115, 5026–5031. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, D.L.; de Godoy, C.M.; Cukierman, S. Enhancement of proton transfer in ion channels by membrane phosphate headgroups. J. Phys. Chem. B 2009, 113, 6725–6731. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Tepper, H.L.; Voth, G.A. Effect of membrane environment on proton permeation through gramicidin A channels. J. Phys. Chem. B 2007, 111, 9931–9939. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, S. Proton mobilities in water and in different stereoisomers of covalently linked gramicidin A channels. Biophys. J. 2000, 78, 1825–1834. [Google Scholar] [CrossRef]
- Dreyer, J.; Zhang, C.; Ippoliti, E.; Carloni, P. Role of the membrane dipole potential for proton transport in gramicidin A embedded in a DMPC bilayer. J. Chem. Theory Comput. 2013, 9, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Kucerka, N.; Nieh, M.P.; Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta 2011, 1808, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Dutagaci, B.; Becker-Baldus, J.; Faraldo-GÃşmez, J.D.; Glaubitz, C. Ceramide-lipid interactions studied by MD simulations and solid-state NMR. Biochim. Biophys. Acta 2014, 1838, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Carrer, D.C.; Schreier, S.; Patrito, M.; Maggio, B. Effects of a short-chain ceramide on bilayer domain formation, thickness, and chain mobililty: DMPC and asymmetric ceramide mixtures. Biophys. J. 2006, 90, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Jurkiewicz, P.; Cwiklik, L.; Vojtiskova, A.; Jungwirth, P.; Hof, M. Structure, dynamics, and hydration of POPC/POPS bilayers suspended in NaCl, KCl, and CsCl solutions. Biochim. Biophys. Acta 2012, 1818, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Paloncyova, M.; Vavrova, K.; Sovova, Z.; DeVane, R.H.; Otyepka, M.; Berka, K. Structural changes in ceramide bilayers rationalize increased permeation through stratum Corneum models with shorter acyl tails. J. Phys. Chem. B 2015, 119, 9811–9819. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, T. Computing 1-D atomic densities in macromolecular simulations: The density profile tool for VMD. Comput. Phys. Commun. 2014, 185, 317–322. [Google Scholar] [CrossRef]
- Schmitt, U.W.; Voth, G.A. Multistate empirical valence bond model for proton transport in water. J. Phys. Chem. B 1998, 102, 5547–5551. [Google Scholar] [CrossRef]
- Kim, T.; Lee, K.I.; Morris, P.; Pastor, R.W.; Andersen, O.S.; Im, W. Influence of Hydrophobic Mismatch on Structures and Dynamics of Gramicidin A and Lipid Bilayers. Biophys. J. 2012, 7, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Li, Y.; Vostrikov, V.V.; Gu, H.; Hinton, J.F.; Koeppe, R.E.; Roux, B.; Andersen, O.S. Gramicidin A backbone and side chain dynamics evaluated by molecular dynamics simulations and nuclear magnetic resonance experiments. I: Nuclear magnetic resonance experiments. J. Phys. Chem. B 2011, 115, 7427–7432. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM General Force Field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force field. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Venable, R.M.; Sodt, A.J.; Rogaski, B.; Rui, H.; Hatcher, E.; MacKerell, A.D.; Pastor, R.W.; Klauda, J.B. CHARMM all-atom additive force field for sphingomyelin: Elucidation of hydrogen bonding and of positive curvature. Biophys. J. 2014, 107, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Mayne, C.G.; Saam, J.; Schulten, K.; Tajkhorshid, E.; Gumbart, J.C. Rapid parameterization of small molecules using the force field toolkit. J. Comput. Chem. 2013, 34, 2757–2770. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–28. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09; Revision D.01; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in monte carlo free energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kumar, S.; Bouzida, D.; Swensen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.; Zhang, Y.; Brooks, B. Constant pressure molecular dynamics: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setiadi, J.; Kuyucak, S. Computational Investigation of the Effect of Lipid Membranes on Ion Permeation in Gramicidin A. Membranes 2016, 6, 20. https://doi.org/10.3390/membranes6010020
Setiadi J, Kuyucak S. Computational Investigation of the Effect of Lipid Membranes on Ion Permeation in Gramicidin A. Membranes. 2016; 6(1):20. https://doi.org/10.3390/membranes6010020
Chicago/Turabian StyleSetiadi, Jeffry, and Serdar Kuyucak. 2016. "Computational Investigation of the Effect of Lipid Membranes on Ion Permeation in Gramicidin A" Membranes 6, no. 1: 20. https://doi.org/10.3390/membranes6010020