Affinity Separation of Lectins Using Porous Membranes Immobilized with Glycopolymer Brushes Containing Mannose or N-Acetyl-D-Glucosamine


Abstract
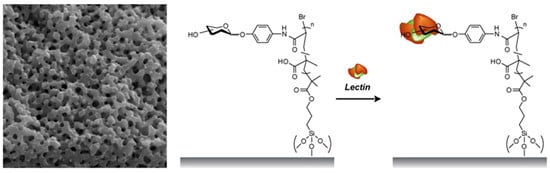
:
1. Introduction
2. Results and Discussion
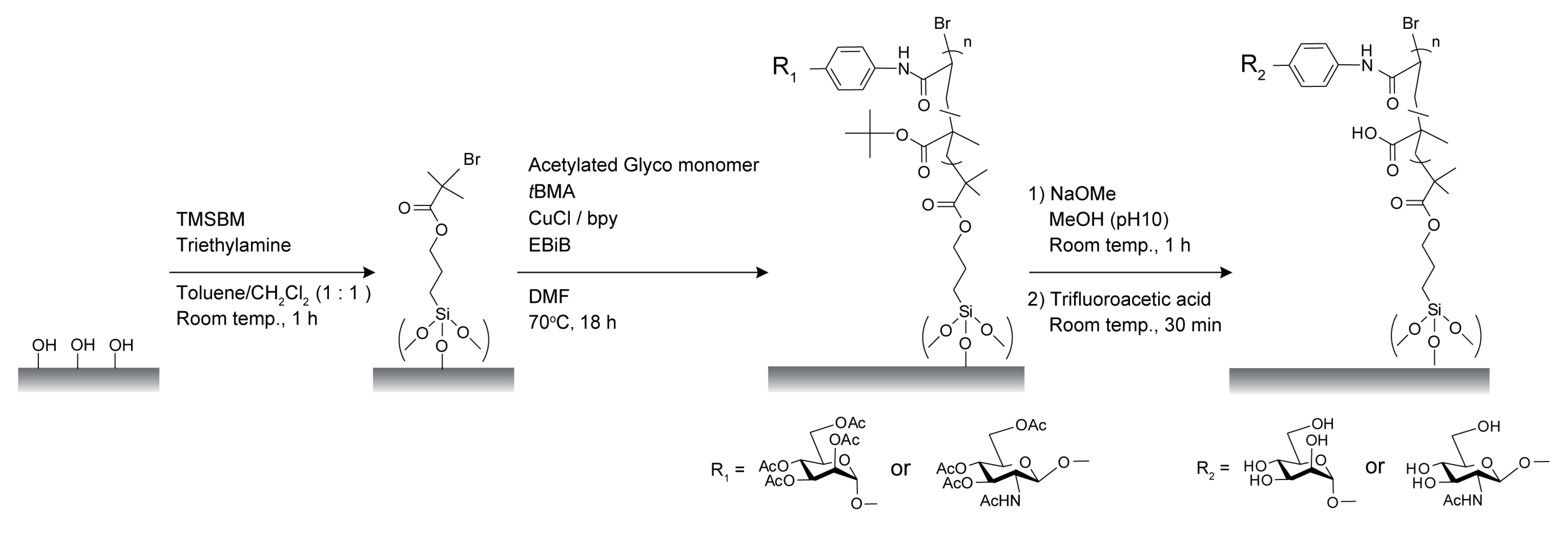
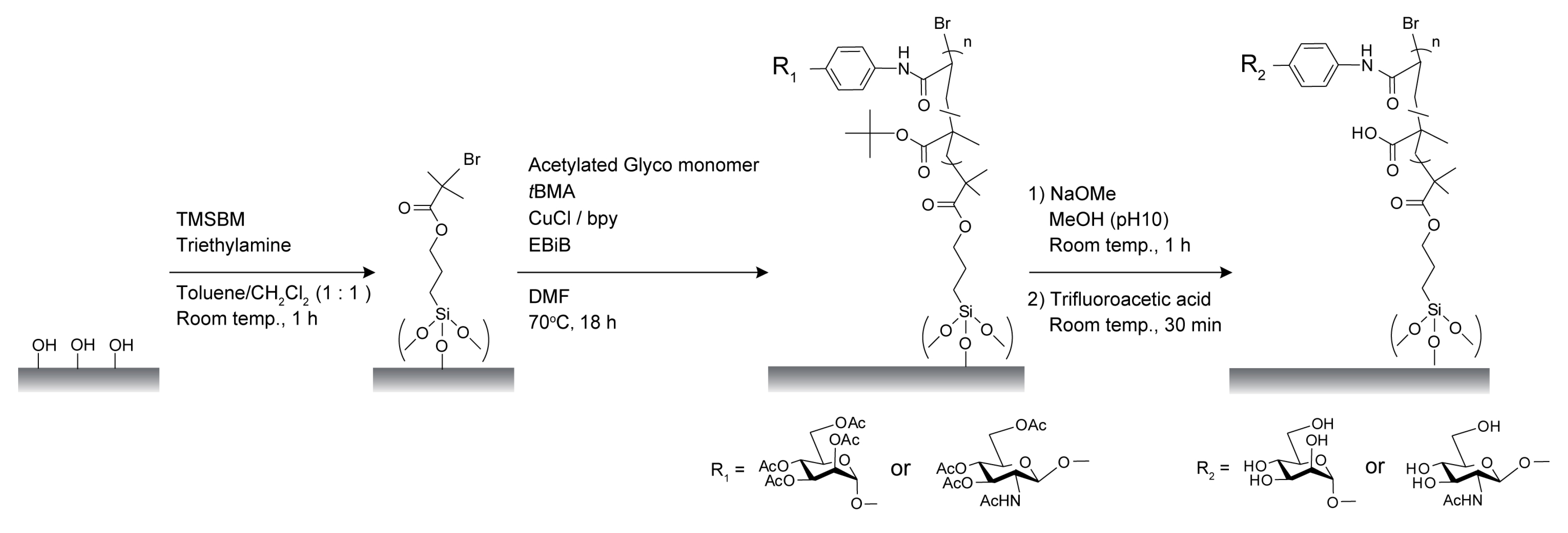
2.1. Characterization of Poly(glyco-MA) and Poly(glyco-MA)-Immobilized Surfaces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mw | Mn | ĐM |
|---|---|---|---|
| PolytBMA | 24,800 | 17,100 | 1.4 |
| Poly(acetylated Man-tBMA) | 27,000 | 12,300 | 2.2 |
| Poly(acetylated GlcNAc-tBMA) | 14,700 | 9,500 | 1.5 |
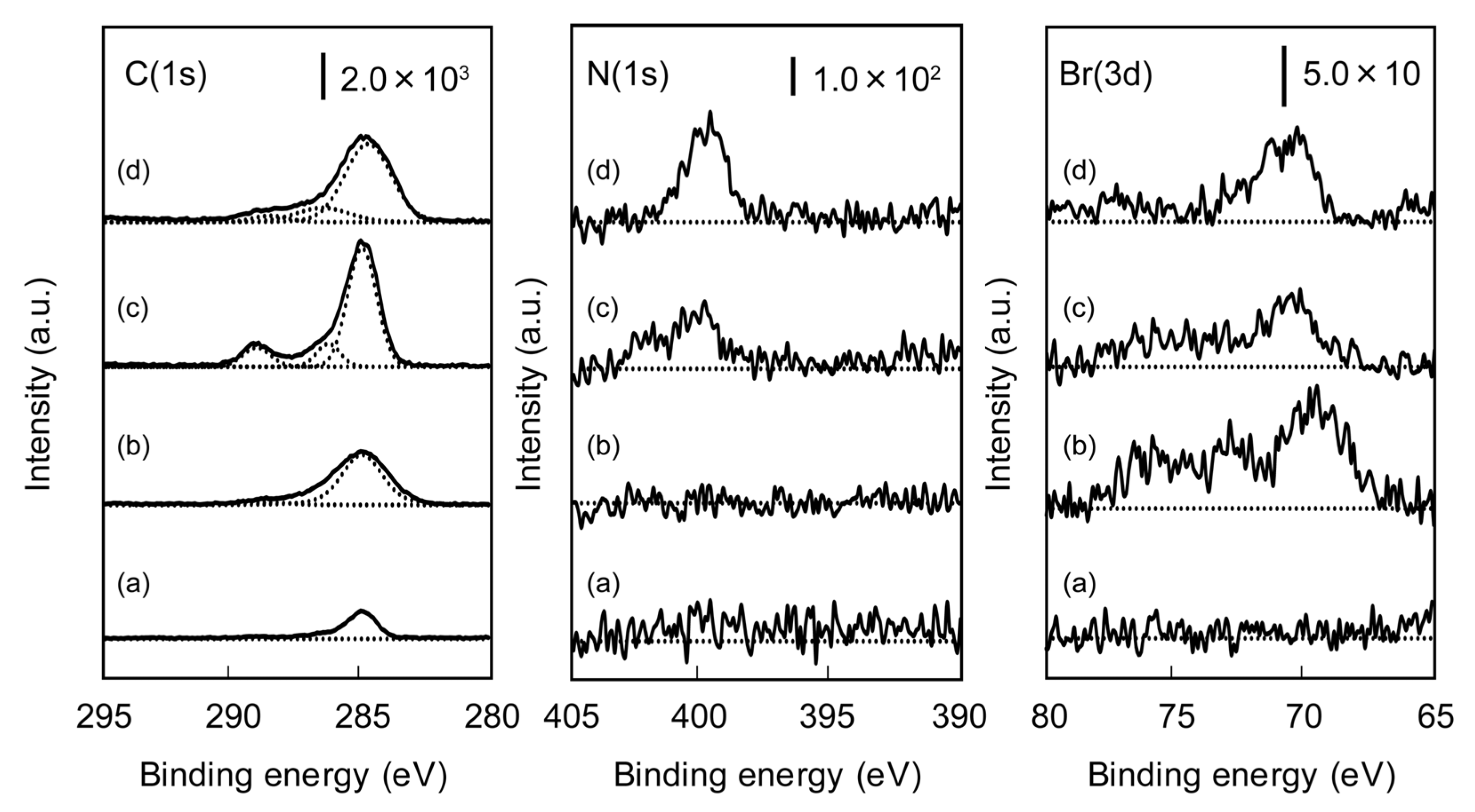
| Surface | Contact angle (°) | Thickness (nm) |
|---|---|---|
| Unmodified | 7.5 ± 0.8 | – |
| TMSBM-immobilized | 76.5 ± 1.2 | 4.2 |
| Poly(acetylated Man-tBMA)-immobilized | 87.6 ± 5.0 | 14.1 |
| Poly(Man-MA)-immobilized | 60.4 ± 2.6 | 13.9 |
| Poly(acetylated GlcNAc-tBMA)-immobilized | 83.7 ± 3.0 | 12.3 |
| Poly(GlcNAc-MA)-immobilized | 69.3 ± 1.4 | 11.8 |
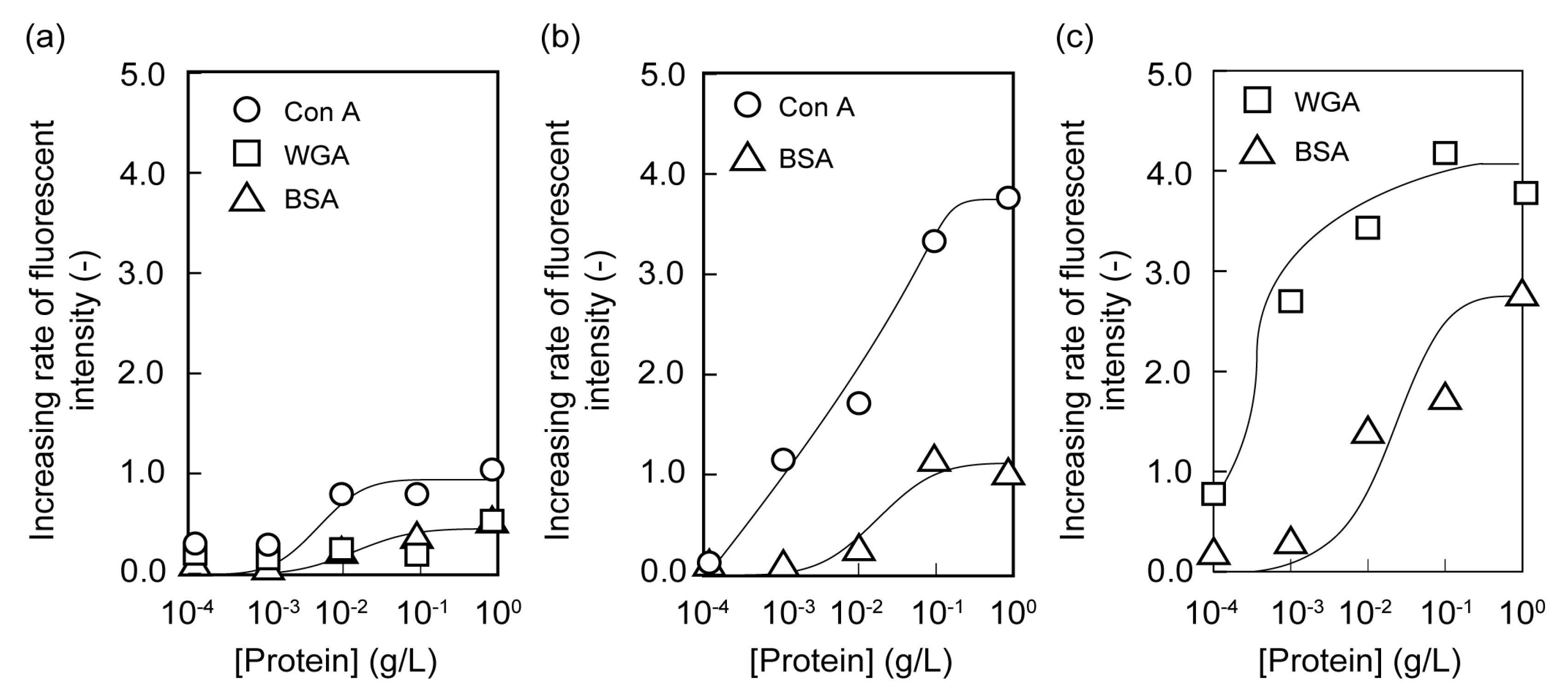
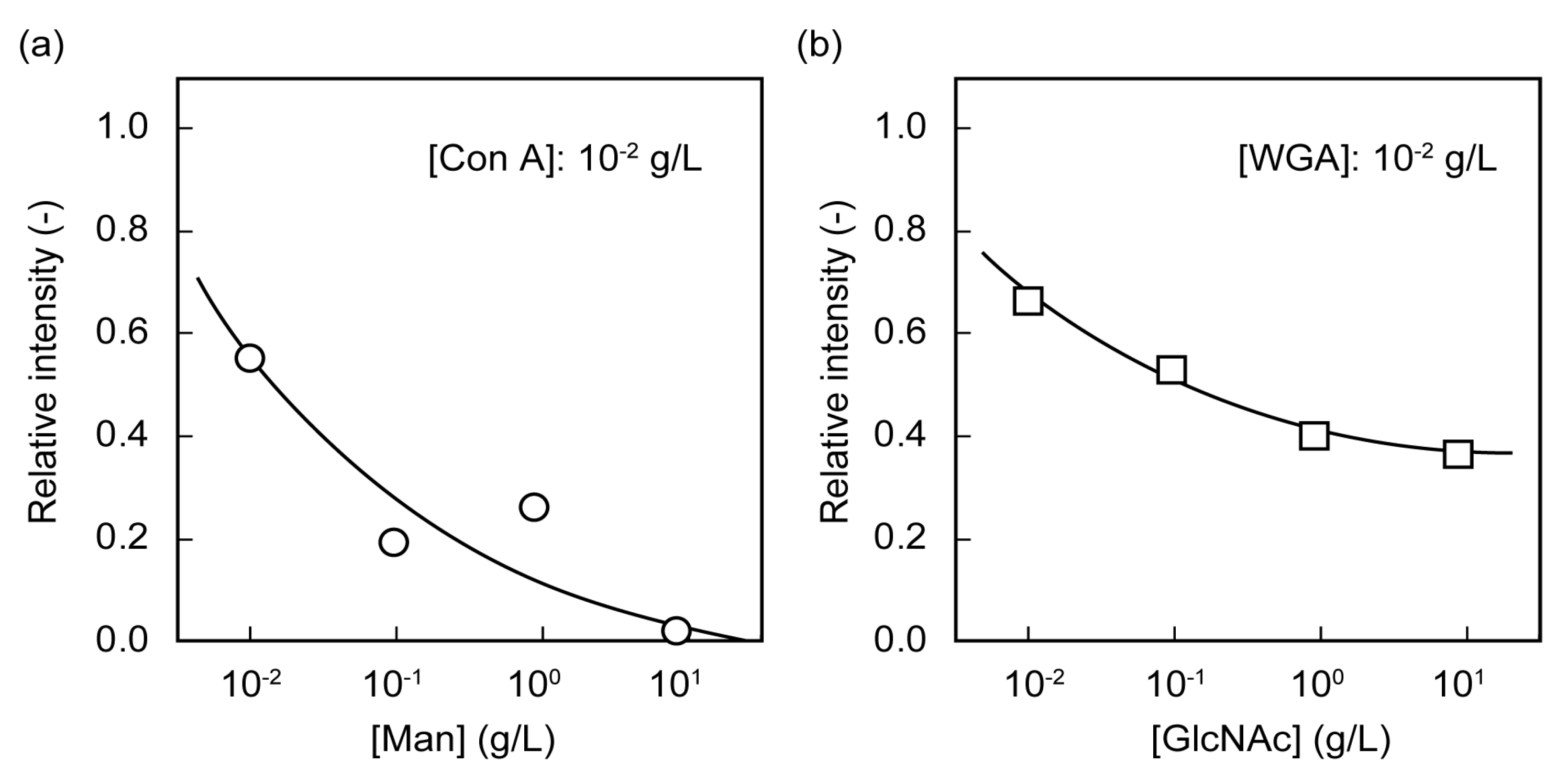
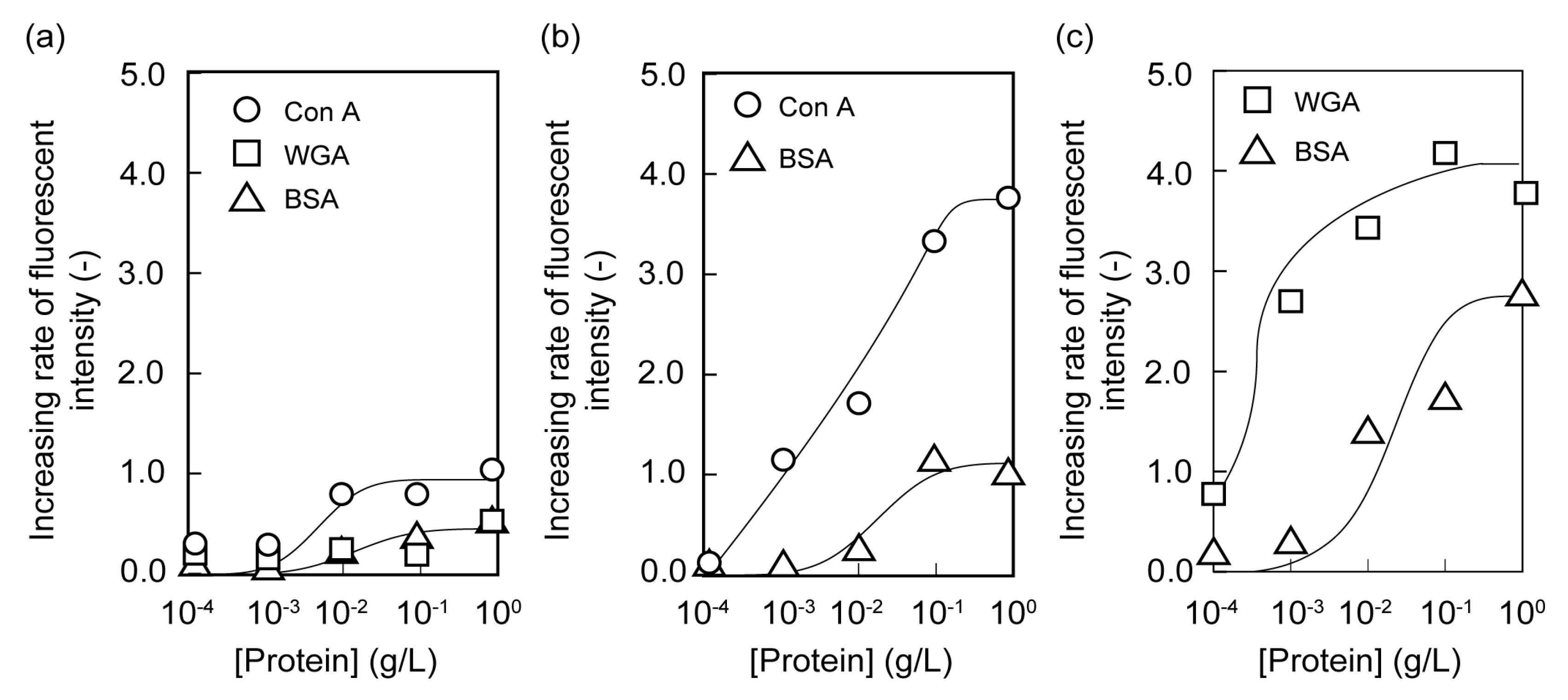
2.2. Microarrays of FITC-Protein Using Poly(glyco-MA)-Immobilized Glass Slides
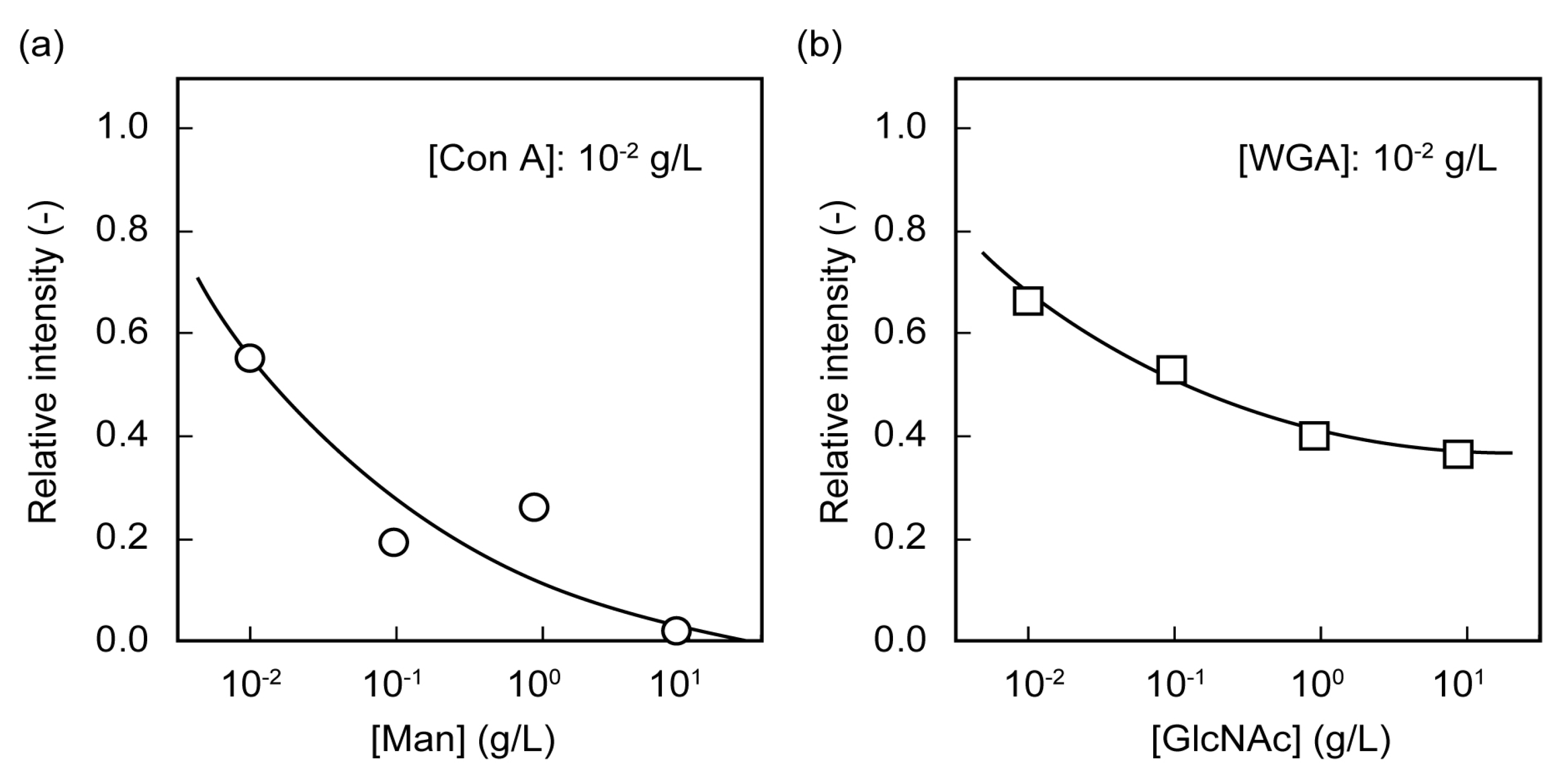
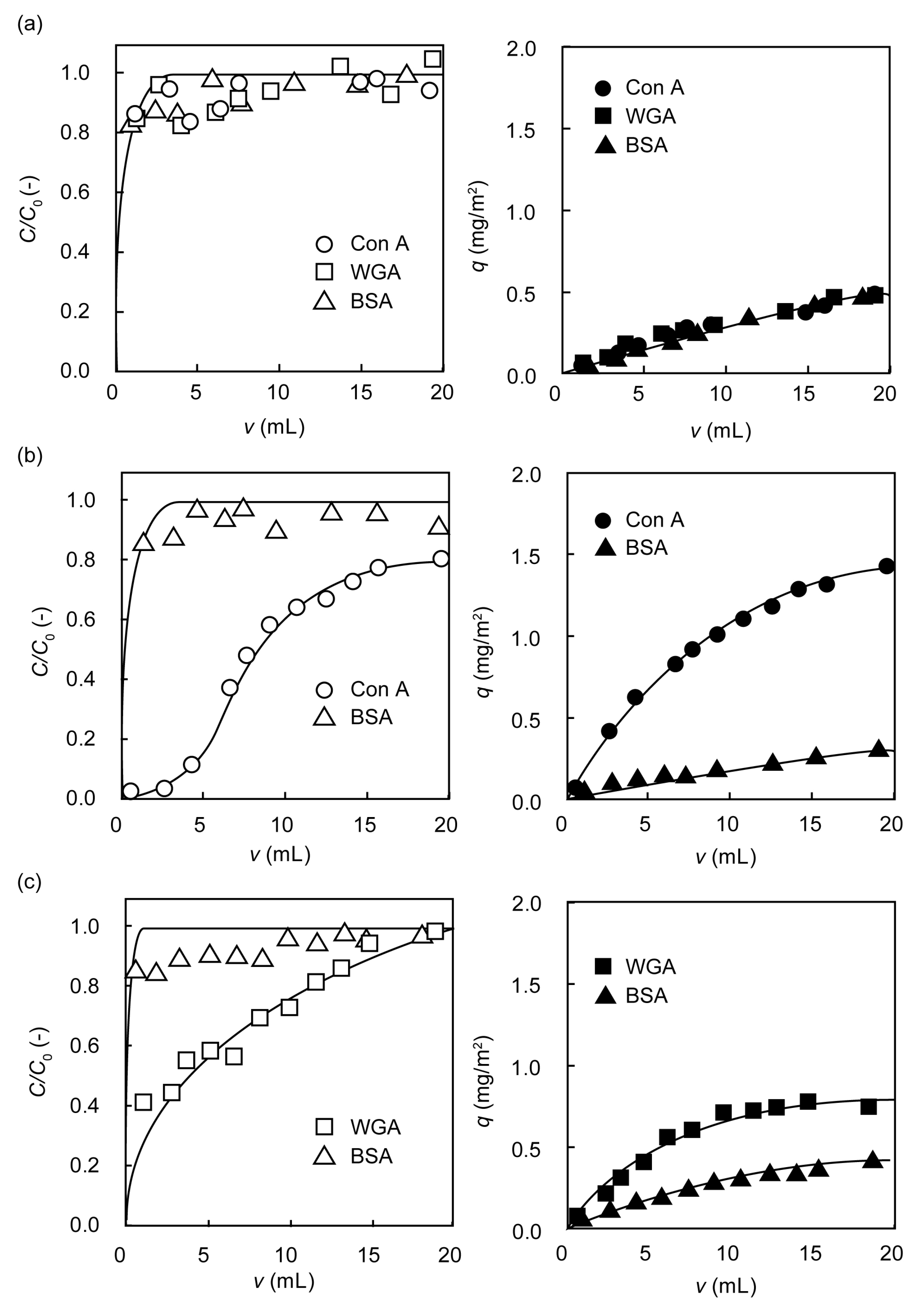
2.3. Separation of Lectin Using Poly(glyco-MA)-Immobilized Membranes

3. Experimental Section
3.1. Reagents and Materials
3.2. Synthesis
3.2.1. 3-(Trimethoxysilyl)propyl 2-Bromo-2-methyl Propanoate (TMSBM)
3.2.2. Glyco Monomers
3.2.3. FITC-BSA
3.3. Preparation of Initiator-Immobilized Glass Slides and Membranes for SI-ATRP
3.4. Surface Modification of Glass Slides and Membranes with Glyco-Polymer Brushes via SI-ATRP
3.5. SEC Determination
3.6. Adsorption of Fluorescent-Labeled Lectin on the Surface of Poly(glyco-MA)-Immobilized Glass Slide

3.7. Rejection of Lectin on the Surface of Poly(glyco-MA)-Immobilized Membrane
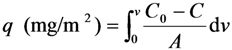
4. Conclusions
Supplementary Files
Acknowledgments
Conflict of Interest
References
- Taylor, M.E.; Drickamer, K. Introduction to Glycobiology; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Kato, K.; Kamiya, Y. Structural views of glycoprotein-fate determination in cells. Glycobiology 2007, 17, 1031–1044. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lee, R.T. Carbohydrate-protein interactions: Basis of glycobiology. Acc. Chem. Res. 1995, 28, 321–327. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Miura, Y. Design and synthesis of well-defined glycopolymers for the control of biological functionalities. Polym. J. 2012, 44, 679–689. [Google Scholar] [CrossRef]
- Ambrosi, M.; Cameron, N.R.; Davis, B.G.; Stolnik, S. Investigation of the interaction between peanut agglutinin and synthetic glycopolymeric multivalent ligands. Org. Biomol. Chem. 2005, 3, 1476–1480. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Chen, G.; Stenzel, M.H. Synthesis of glycopolymers and their multivalent recognitions with lectins. Polym. Chem. 2010, 1, 1392–1412. [Google Scholar] [CrossRef]
- Nishimura, S.; Furuike, T.; Matsuoka, K.; Maruyama, K.; Nagata, K.; Kurita, K.; Nishi, N.; Tokura, S. Synthetic glycoconjugates. 4. Use of ω-(acrylamido)alkyl glycosides for the preparation of cluster glycopolymers. Macromolecules 1994, 27, 4876–4880. [Google Scholar] [CrossRef]
- Gruner, S.A.W.; Locardi, E.; Lohof, E.; Kessler, H. Carbohydrate-based mimetics in drug design: Sugar amino acids and carbohydrate scaffolds. Chem. Rev. 2002, 102, 491–514. [Google Scholar] [CrossRef]
- Jelinek, R.; Kolusheva, S. Carbohydrate biosensor. Chem. Rev. 2004, 104, 5987–6015. [Google Scholar] [CrossRef]
- Costantino, L.; Gandolfi, F.; Bossy-Nobs, L.; Tosi, G.; Gurny, R.; Rivasi, F.; Vandelli, M.A.; Forni, F. Nanoparticulate drug carriers based on hybridpoly(D,L-lactide-co-glycolide)-dendron structures. Biomaterials 2006, 27, 4635–4645. [Google Scholar] [CrossRef]
- Cho, C.S.; Seo, S.J.; Park, I.K.; Kim, S.H.; Kim, T.H.; Hoshiba, T.; Harada, I.; Akaike, T. Galactose-carrying polymers as extracellular matrices for liver tissue engineering. Biomaterials 2006, 27, 576–585. [Google Scholar] [CrossRef]
- Park, S.; Lee, K.B.; Choi, I.S.; Langer, R.; Jon, S. Dual functional, polymeric self-assembled monolayers as a facile platform for construction of patterns of biomolecules. Langmuir 2007, 23, 10902–10905. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Hirase, T.; Madsen, J.; Armes, S.P.; Nagasaki, Y. Non-fouling character of poly[2-(methacryloyloxy)ethyl phosphorylcholine]-modified gold surfaces fabricated by the “grafting to” method: Comparison of its protein resistance with poly(ethylene glycol)-modified gold surfaces. Macromol. Rapid Commun. 2009, 30, 2136–2140. [Google Scholar] [CrossRef]
- Sung, D.; Park, S.; Jon, S. Facile method for selective immobilization of biomolecules on plastic surfaces. Langmuir 2009, 25, 11289–11294. [Google Scholar] [CrossRef]
- Zdyrko, B.; Iyer, K.S.; Luzinov, I. Macromolecular anchoring layers for polymer grafting: Comparative study. Polymer 2006, 47, 272–279. [Google Scholar] [CrossRef]
- Ayres, N.; Boyes, S.G.; Brittain, W.J. Stimuli-responsive polyelectrolyte polymer brushes prepared via atom-transfer radical polymerization. Langmuir 2007, 23, 182–189. [Google Scholar] [CrossRef]
- Shah, R.R.; Merreceyes, D.; Husemann, M.; Rees, I.; Abbott, N.L.; Hawker, C.J.; Hedrick, J.L. Using atom transfer radical polymerization to amplify monolayers of initiators patterned by microcontact printing into polymer brushes for pattern transfer. Macromolecules 2000, 33, 597–605. [Google Scholar] [CrossRef]
- Ejaz, M.; Ohno, K.; Tsujii, Y.; Fukuda, T. Controlled grafting of a well-defined glycopolymer on a solid surface by surface-initiated atom transfer radical polymerization. Macromolecules 2000, 33, 2870–2874. [Google Scholar] [CrossRef]
- Mittal, V.; Matsko, N.B.; Butte, A.; Morbidelli, M. Functionalized polystyrene latex particles as substrates for ATRP: Surface and colloidal characterization. Polymer 2007, 48, 2806–2817. [Google Scholar] [CrossRef]
- Matsugi, T.; Saito, J.; Kawahara, N.; Matsuo, S.; Kaneko, H.; Kashiwa, N.; Kobayashi, M.; Takahara, A. Surface modification of polypropylene molded sheets by means of surface-initiated ATRP of methacrylates. Polym. J. 2009, 41, 547–554. [Google Scholar] [CrossRef]
- Ghadban, A.; Albertin, L. Synthesis of glycopolymer architectures by reversible-deactivation radical polymerization. Polymers 2013, 5, 431–526. [Google Scholar] [CrossRef]
- Lis, H.; Sharon, N. Lectins: Carbohydrate-specific proteins that mediate cellular recognition. Chem. Rev. 1998, 98, 637–674. [Google Scholar] [CrossRef]
- Merritt, E.A.; Sarfaty, S.; van den Akker, F.; L’Hoir, C.; Martial, J.A.; Hol, W.G.J. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci. 1994, 3, 166–175. [Google Scholar] [PubMed]
- Hammache, D.; Pieroni, G.; Yahi, N.; Delezay, O.; Koch, N.; Lafont, H.; Tamalet, C.; Fantini, J. Specific interaction of HIV-1 and HIV-2 surface envelope glycoproteins with monolayers of galactosylceramide and ganglioside GM3. J. Biol. Chem. 1998, 27, 7967–7971. [Google Scholar]
- Schilling, J.D.; Mulvey, M.A.; Hultgren, S.J. Structure and function of Escherichia coli Type 1 Pili: New insight into the pathogenesis of urinary tract infections. J. Infect. Dis. 2001, 183, S36–S40. [Google Scholar] [CrossRef]
- Ejaz, M.; Yamamoto, S.; Ohno, K.; Tsujii, Y.; Fukuda, T. Controlled graft polymerization of methyl methacrylate on silicon substrate by the combined use of the Langmuir-Blodgett and atom transfer radical polymerization techniques. Macromolecules 1998, 31, 5934–5936. [Google Scholar] [CrossRef]
- Hoshino, Y.; Nakamoto, M.; Miura, Y. Control of protein-binding kinetics on synthetic polymer nanoparticles by tuning flexibility and inducing conformation changes of polymer chains. J. Am. Chem. Sci. 2012, 134, 15209–15212. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ogata, Y.; Seto, H.; Murakami, T.; Hoshino, Y.; Miura, Y. Affinity Separation of Lectins Using Porous Membranes Immobilized with Glycopolymer Brushes Containing Mannose or N-Acetyl-D-Glucosamine. Membranes 2013, 3, 169-181. https://doi.org/10.3390/membranes3030169
Ogata Y, Seto H, Murakami T, Hoshino Y, Miura Y. Affinity Separation of Lectins Using Porous Membranes Immobilized with Glycopolymer Brushes Containing Mannose or N-Acetyl-D-Glucosamine. Membranes. 2013; 3(3):169-181. https://doi.org/10.3390/membranes3030169
Chicago/Turabian StyleOgata, Yutaro, Hirokazu Seto, Tatsuya Murakami, Yu Hoshino, and Yoshiko Miura. 2013. "Affinity Separation of Lectins Using Porous Membranes Immobilized with Glycopolymer Brushes Containing Mannose or N-Acetyl-D-Glucosamine" Membranes 3, no. 3: 169-181. https://doi.org/10.3390/membranes3030169