
The Dichotomy of Tumor Exosomes (TEX) in Cancer Immunity: Is It All in the ConTEXt?

Abstract
:1. Introduction
Concerning Nomenclature
2. Exosomes
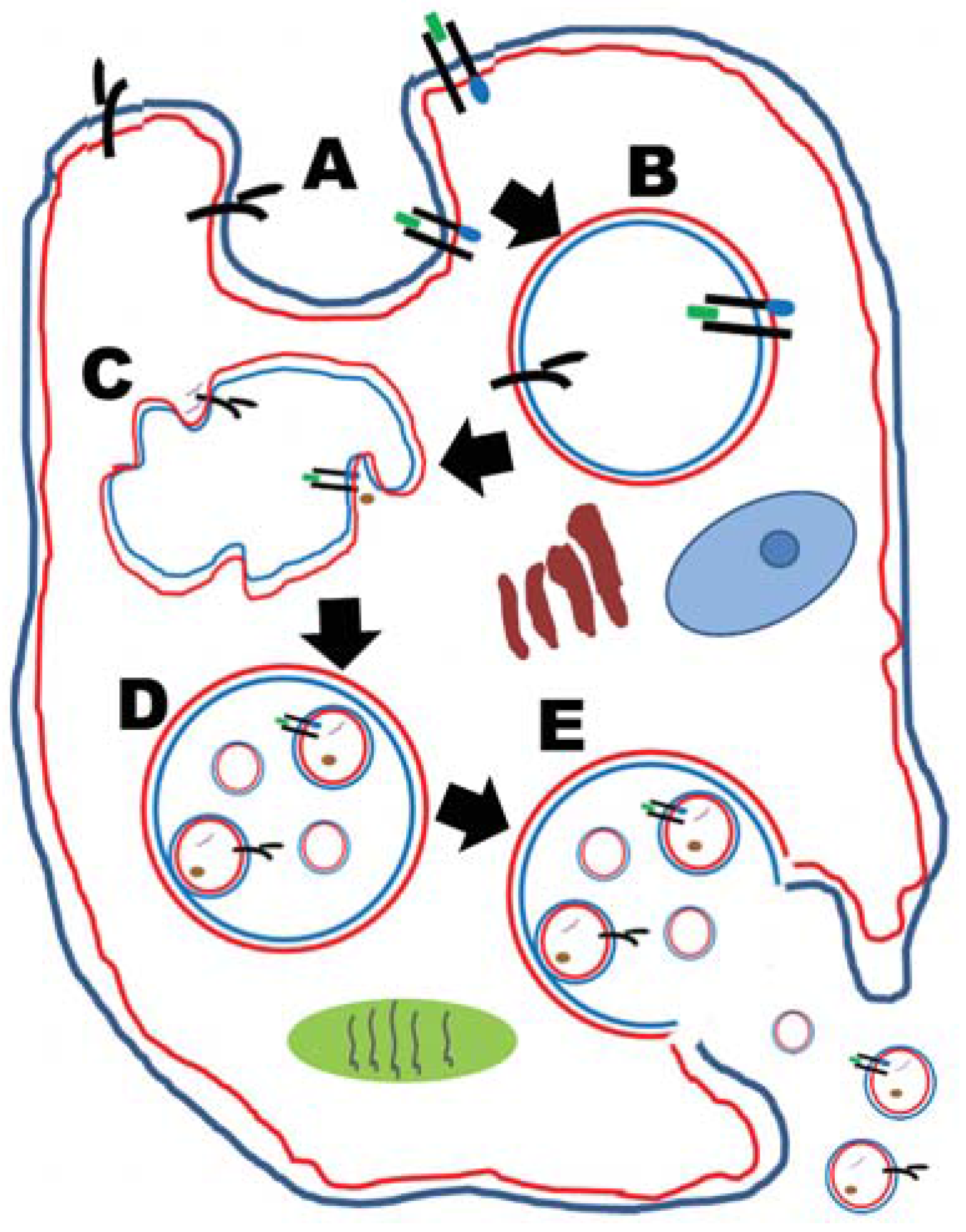
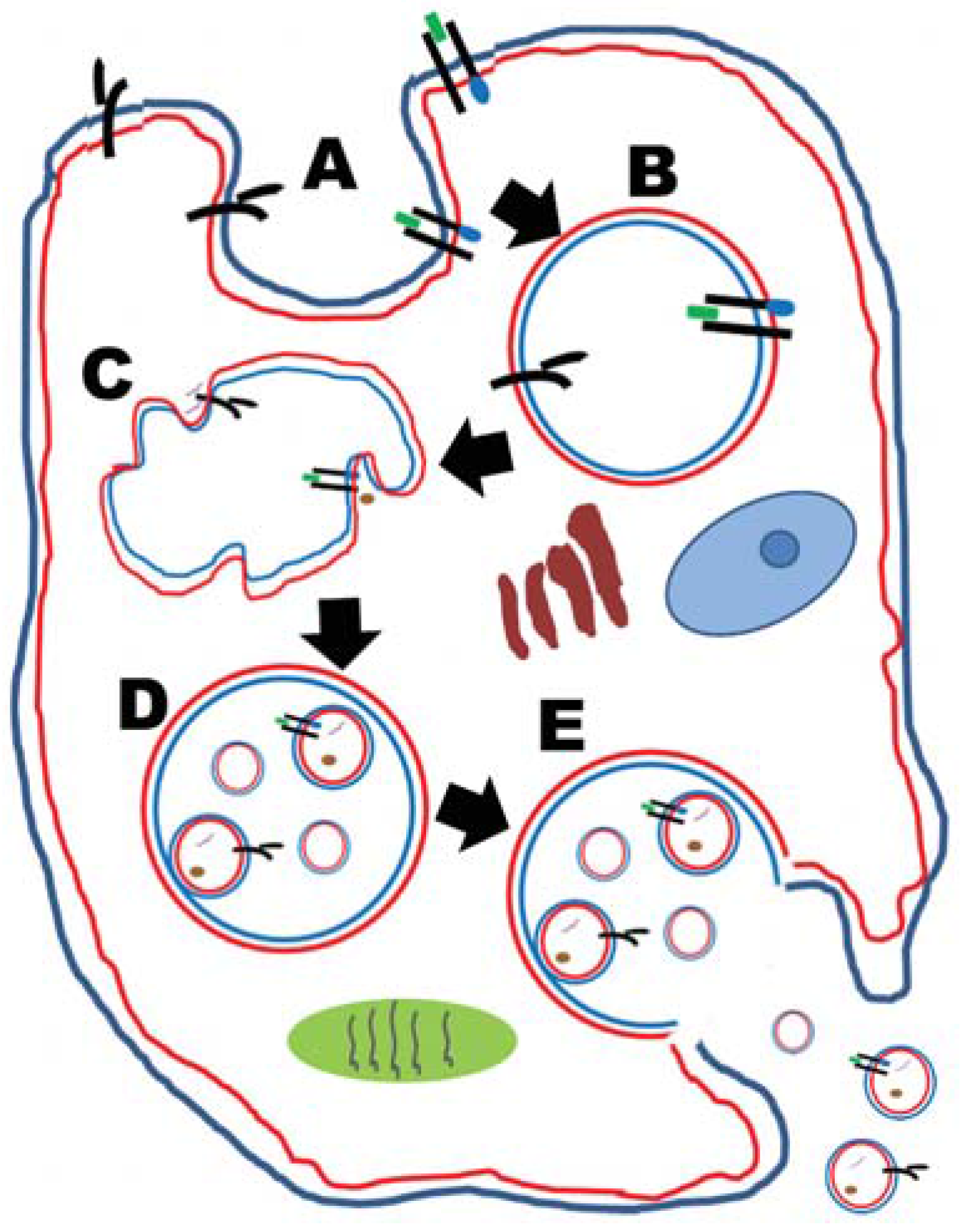
2.1. Tumor-Derived Exosomes (TEX)
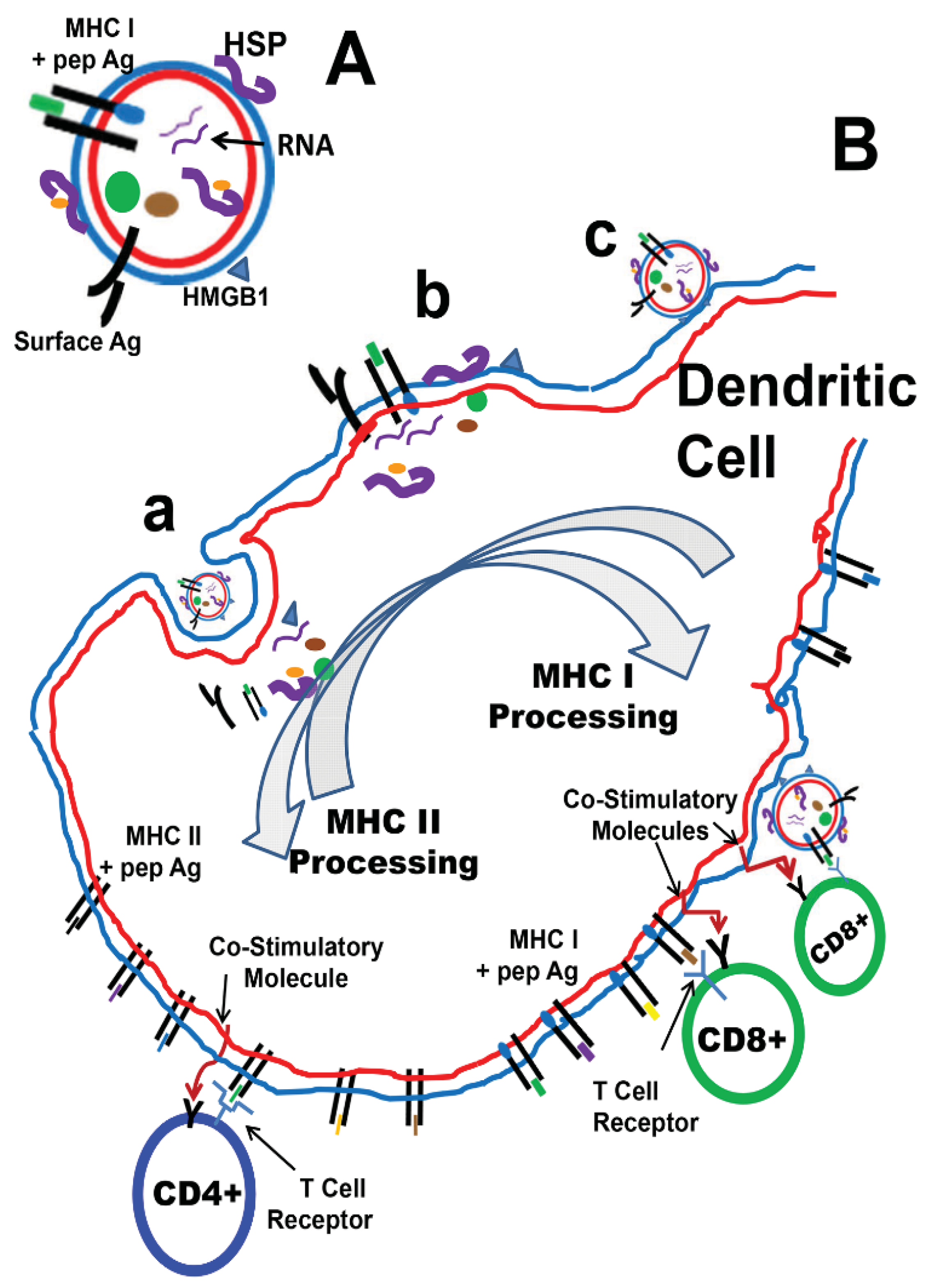
2.2. Dendritic Cell-Based Cancer Vaccines
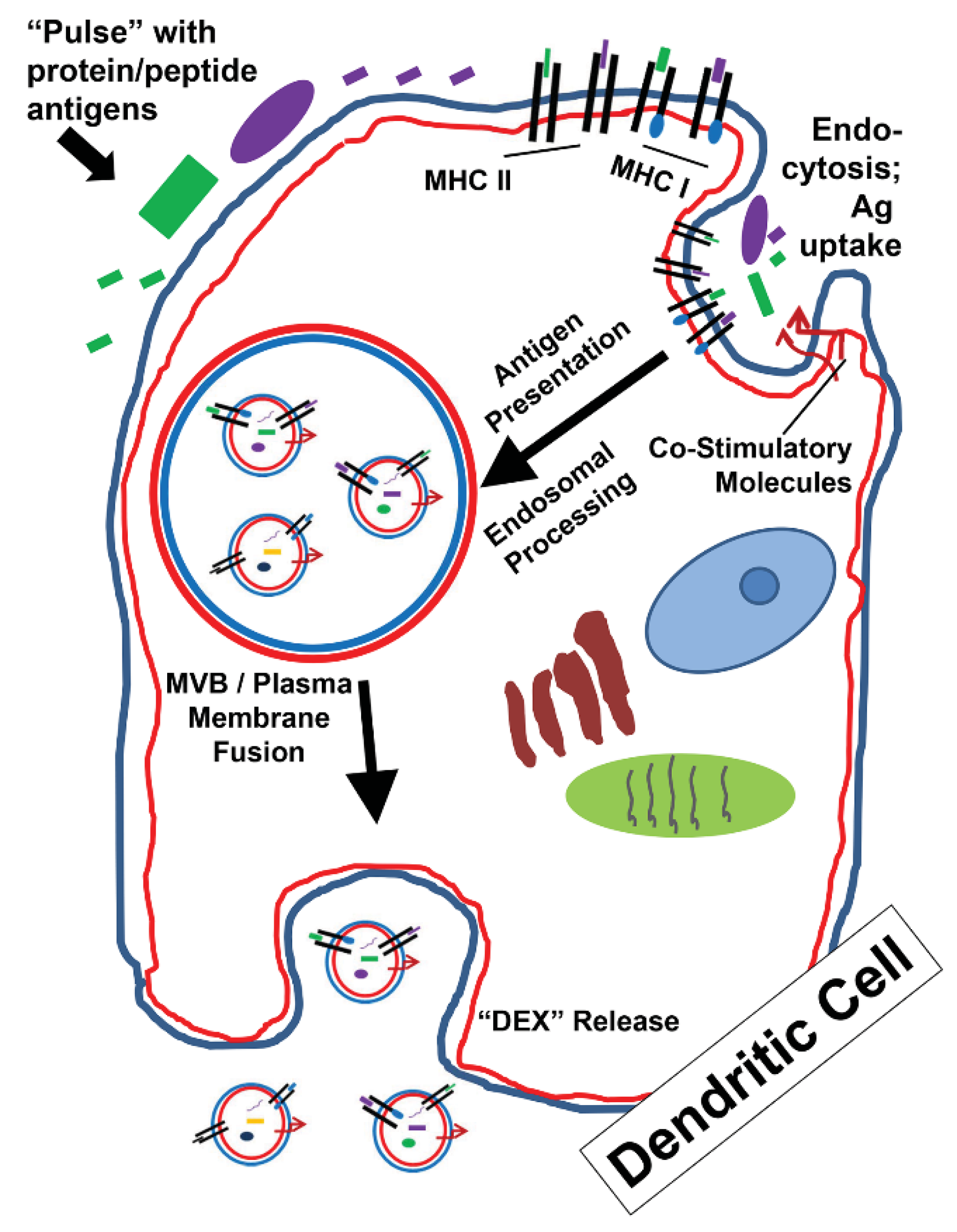
3. Exosomes as Potential Vaccine Candidates (All Hands on DEX)


4. TEX as Anti-Cancer Vaccines
4.1. Stand-Alone TEX

{kind=link}
{kind=link}
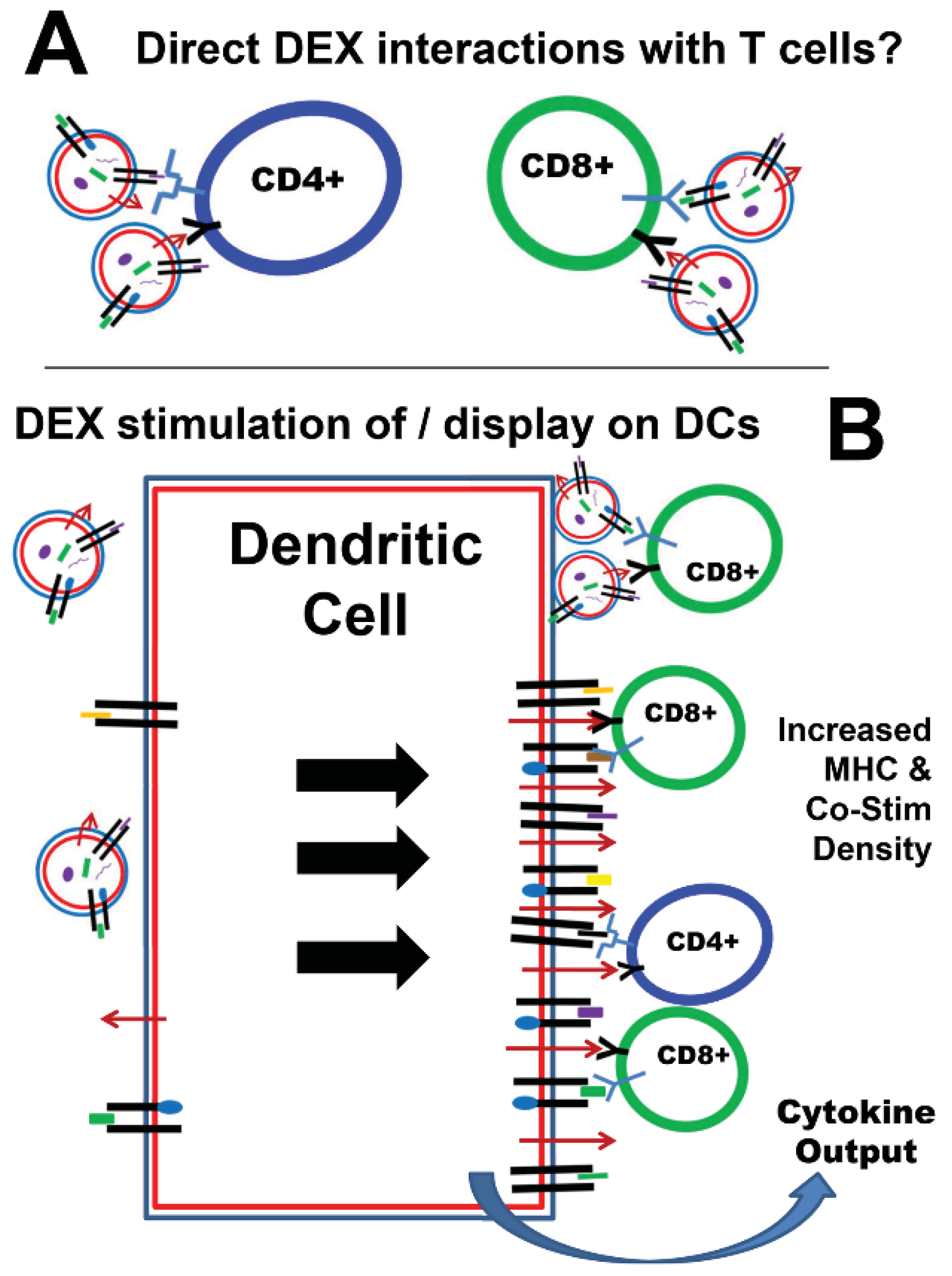{kind=link}
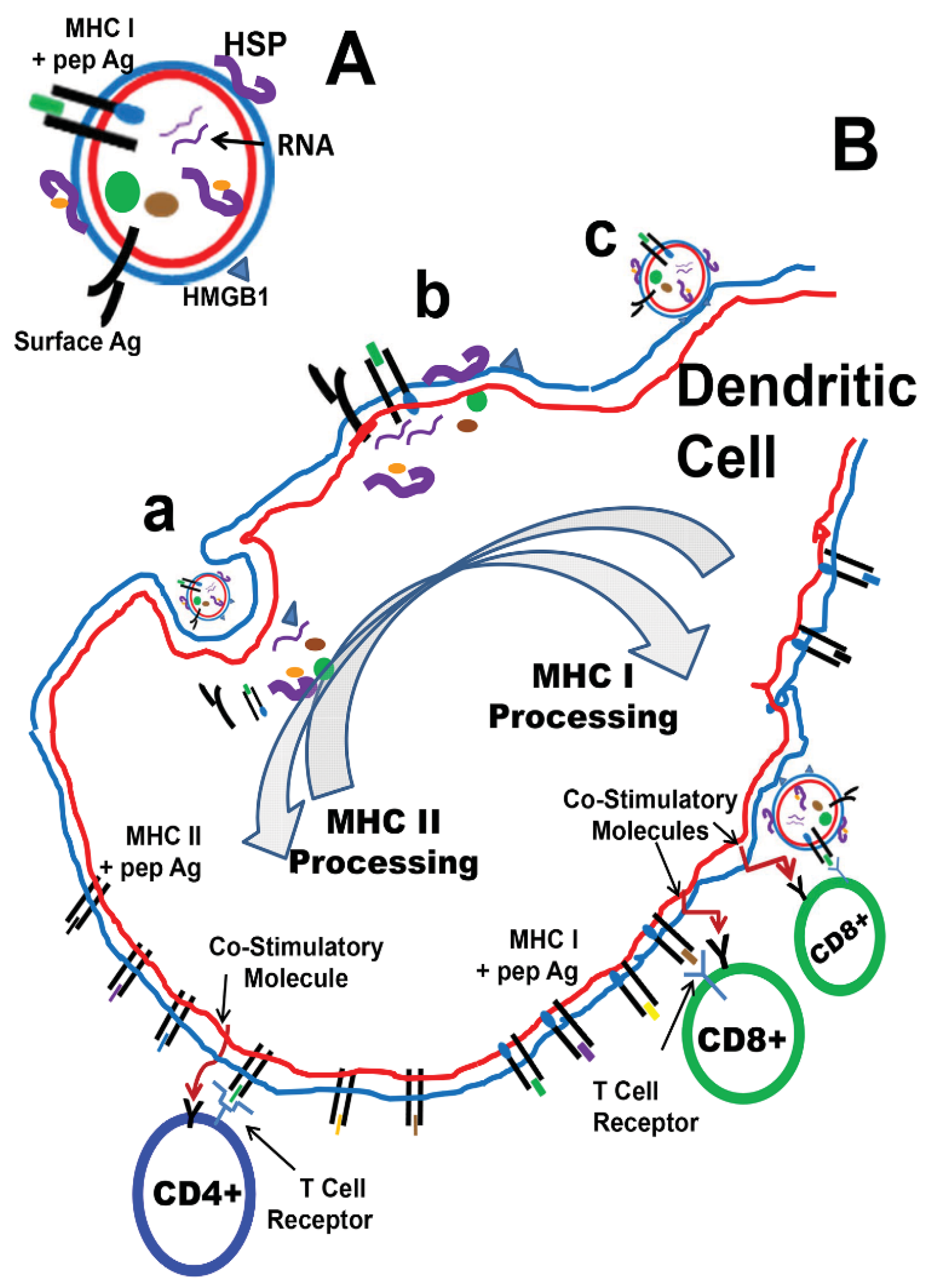{kind=link}
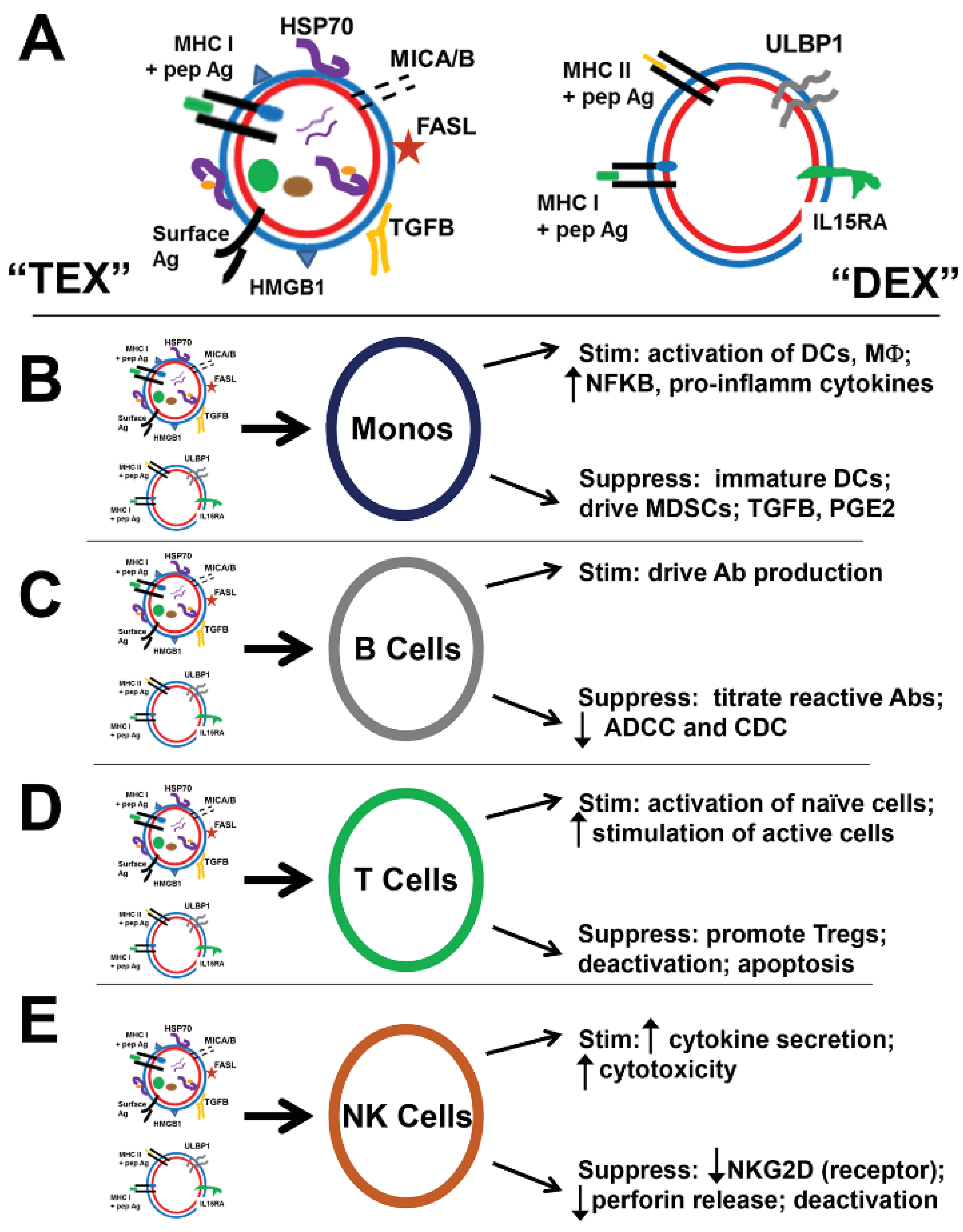{kind=link}
| Reference | Tumor Type | TEX (Mod/Engin) | Vax Route | Adjuvant/StiMulus | Outcome | Notes |
|---|---|---|---|---|---|---|
| Wolfers et al., 2001 [84] | TS/A; MC38 (mouse) | TEX only | SC/ID | None | Autologous and allogeneic cross protection DC presentaion of XO Ags | CD4+ and CD8+ dependent |
| L1210; AK7; Ba/F3; (mouse) | or TEX on DCs | |||||
| Fon; Mel-888 (human) | ||||||
| Altieri et al., 2004 [85] | J558; MPC11; Colon 26 (mouse) | TEX only | SC | None | Prophylactic and rechallenge protection | Tumor-specific |
| CTLs active | ||||||
| Bu et al., 2006 [88] | L1210 (mouse) | TEX only | SC | None | Prophylactic protection | CTLs active |
| Graner et al., 2009 [90] | SMA-560vIII (mouse) | Prophylactic and rechallenge protection | B, T cells active | |||
| Hao et al., 2006 [92] | EG7/OVA (mouse for Exos) | EG7 TEX; DEX fromOVA-pulsed DCs | IV | None | DEX > TEX vs. Metastatic disease (prophylaxis) | DEX > TEX CTL activity |
| B16-OVA (mouse tumor target) | ||||||
| Gu et al., 2015 [93] | WEHI3B; RENCA (mouse) | DEX pulsed with TEX as vax | SC | None | DEX/TEX > DEX/lys for preventing tumor growth/overall survival | Increase CTL and NK activity |
| Dai et al., 2005 [94] | LS-174T (CEA+, human) | TEX from cells +/− heat shock | SC | HS TEX from CEA+ cells > anti-tumor responses | Increase CD4+/CD8+ activity | |
| SW480 (CEA+, human) | ||||||
| LoVo (CEA+, human) | ||||||
| A549 (CEA−, human) | ||||||
| (mice txg for HLA-A2.1) | ||||||
| Cho et al., 2009 [95] | CT26-MUC1; B16-MUC1 (mouse) | TEX from cells +/− heat shock | ID | CPG (anti-tumor) IFA (for Abs) | HS TEX > auto/allogeneic anti-tumor responses | HS TEX > B and T cell responses |
| Xie et al., 2010 [96] | J558 (mouse) | TEX from cells expressing HSP70 on surfaces | SC | None | TEX70 > TEXhs > TEX in anti-tumor assays | IncreaseCD4+, CD8+, NK responses |
| Chen, et al., 2011 [97] | Lewis lung carcinoma (3LL, mouse) | TEX from cells +/− heat shock | IT, SC | none | HS-TEX > TEX in anti-tumor assays | HS-TEX contain chemokines, attract DCs and T cells |
| Yang, et al., 2007 [98] | EG7/OVA [IL2 tfxt for TEX] (mouse) | TEX w/IL2 | SC | none | TEX-IL2 > TEX + IL2, TEX in anti-tumor assays | CD8+ > CD4+ > NK effectors |
| Xie et al., 2010 [99] | J558 (mouse, P1A Ag) tfxt to express TNFA, IL2, IFNG | TEX from each transfectant | IV | none | TEX/TNFA > TEX.IL2 > TEX/INFG in anti-tumor assays | Same order for P1A-specific CTL |
| Lee et al., 2011 [100] | B16F1 (mouse)[CIITA tfxt for TEX] | TEX w/CIITA | ID | none | TEX/CITTA > TEX in in anti-tumor assays | TEX/CITTA > TEX for DC, T cells, B cells |
| Rountree et al., 2011 [101] | CT26-PAP; E6-PSA (mouse, expressing human Ags) | Immunize w/virus to drive PAP or PSA exo expression linked to C1C2 lactadherin domain | SC (virus) | none | Ag/C1C2 > untargeted Ag in anti-tumor assays | Similar responses in B cell and T cell assays; B cells were strain- dependent |
| Zeelenberg et al., 2011 [102] | MCA101-OVA (mouse) | cells tfxt for soluble OVA, membrane OVA, or TEX-OVA (via C1C2) | cyroablation | none | TEX-OVA > sOVA ≥ fcOVA tumors for immune activation and anti-tumor response | |
| Sedlik et al., 2014 [103] | MCA101-OVA, EL4-OVA, B16F-OVA (all mouse, expressing OVA) | cells tfxt for gag-OVA or C1C2-OVA (DNA vaccine) | ID, IM electroporation | none | both vax had ~ equal benefit in anti-tumor and cellular responses | |
| Hartman et al., 2011 [104] | 4T1--HER2 (mouse, human HER2) | C1C2-CEA, C1C2-HER2 (ECDs fused to C1C2) AdVir vaccine | ID | none | C1C2-HER2 > ECD-HER2 in anti-tumor responses | C1C2-Ag > ECD-Ag for B and T cell responses |
| Zeelenberg et al. 2008 [105] | MCA101-OVA (mouse) | C1C2-OVA, solb OVA (DNA vaccine) | IM | none | C1C2-OVA > sOVA in anti-tumor response | C1C2-OVA > sOVA in T cell responses |
| Xiu et al., 2007 [106] | EG7 (OVA) (mouse) | TEX with SEA or TM-SEA “transfer” | SC | none | TEX/TM-SEA > TEX/SEA > TEX > SEA in anti-tumor responses | TEX/TM-SEA > TEX/SEA > TEX > SEA in T cell assays |
| Dai et al., 2008 [107] | CRC with ascites Stage III−IV (human) CEA+ in sera | AEX (TEX from ascites) | SC | some GM-CSF; various chemos | 1 pt w/stable disease 1 pt w/minor response | AEX+GM-CSF > AEX for DTH and anti-CEA T cells |
4.2. TEXing while Driving Immune Responses: Manipulations of Cells to Produce TEX with Enhanced Immune Properties
4.2.1. Heat Shock, Cell Stress, HSPs, and TEX Effects
4.2.2. Engineering the Parent Cells—The TEX of New Immune Ideas
4.2.3. Exosome Display—Filling the TEX Box
4.2.4. Altering TEX Directly—New Immune Fonts
4.2.5. Clinical TEX: Use in a Clinical Cancer Vaccine Trial
5. TEX in Immune Suppression
5.1. “Bad” TEX: Vessels of Tumor-Induced Immune Suppression of Lymphocytes and Monocytes
5.1.1. T Lymphocytes
5.1.2. B Lymphocytes
5.1.3. NK Cells
5.1.4. Monocytes, Macrophage, Dendritic Cells
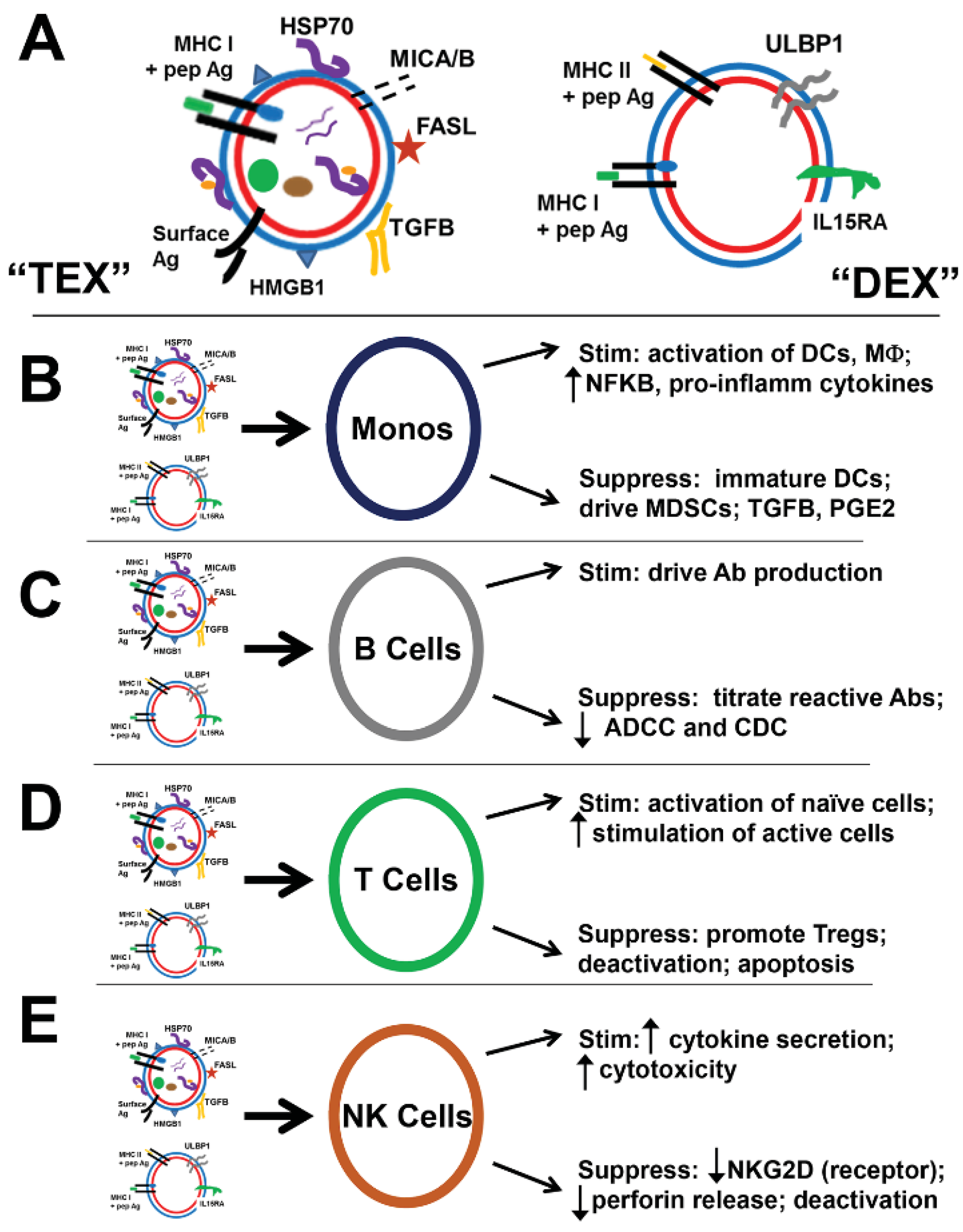
6. Immune Stimulation or Immune Suppression; Is It All in the ConTEXt?
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Toro, J.; Herschlik, L.; Waldner, C.; Mongini, C. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015. [Google Scholar] [CrossRef] [Green Version]
- Benito-Martin, A.; di Giannatale, A.; Ceder, S.; Peinado, H. The new deal: A potential role for secreted vesicles in innate immunity and tumor progression. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Hellwinkel, J.E.; Madsen, H.; Graner, M.W. Immune modulation by tumor-derived extracellular vesicles in glioblastoma. In Molecular Considerations and Evolving Surgical Management Issues in the Treatment of Patients with a Brain Tumor; Lichtor, T., Ed.; InTech: Rijeka, Croatia, 2015; pp. 329–354. [Google Scholar]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Gercel-Taylor, C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br. J. Cancer 2005, 92, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W. Brain tumor exosomes and microvesicles: Pleiotropic effects from tiny cellular surrogates. In Molecular Targets of Cns Tumors; Garami, M., Ed.; InTech Open Access: Rijeka, Croatia, 2012. [Google Scholar]
- Lin, J.; Li, J.; Huang, B.; Liu, J.; Chen, X.; Chen, X.M.; Xu, Y.M.; Huang, L.F.; Wang, X.Z. Exosomes: Novel biomarkers for clinical diagnosis. Sci. World J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, F.; Zhang, J.; Zhang, Q.; Lin, J. Exosome analysis: A promising biomarker system with special attention to saliva. J. Membr. Biol. 2014, 247, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Dieppa, D.R.; Steinberg, J.; Gonda, D.; Cheung, V.J.; Carter, B.S.; Chen, C.C. Extracellular vesicles as a platform for “liquid biopsy” in glioblastoma patients. Expert Rev. Mol. Diagn. 2014, 14, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabieres, C. Real-time liquid biopsy in cancer patients: Fact or fiction? Cancer Res. 2013, 73, 6384–6388. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Mallegol, J.; Bevilacqua, C.; Candalh, C.; Brugiere, S.; Tomaskovic-Crook, E.; Heath, J.K.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial exosomes carry mhc class ii/peptides able to inform the immune system in mice. Gut 2003, 52, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Thery, C. Exosomes: Immune properties and potential clinical implementations. Semin. Immunopathol. 2011, 33, 419–440. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.V.; Grunewald, T.G. Tumour-derived exosomes: Tiny envelopes for big stories. Biol. Cell 2015, 107, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Lasser, C. Exosomes in diagnostic and therapeutic applications: Biomarker, vaccine and RNA interference delivery vehicle. Expert Opin. Biol. Ther. 2015, 15, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Redzic, J.S.; Ung, T.H.; Graner, M.W. Glioblastoma extracellular vesicles: Reservoirs of potential biomarkers. Pharmacogen. Personal. Med. 2014, 7, 65–77. [Google Scholar]
- Whiteside, T.L. Immune suppression in cancer: Effects on immune cells, mechanisms and future therapeutic intervention. Semin. Cancer Biol. 2006, 16, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Hellwinkel, J.E.; Redzic, J.S.; Harland, T.A.; Gunaydin, D.; Anchordoquy, T.J.; Graner, M.W. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Vremec, D.; Shortman, K. What’s in a name? Some early and current issues in dendritic cell nomenclature. Front. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. Pillars article: The immunological synapse: A molecular machine controlling T cell activation. Science. 1999. 285: 221–227. J. Immunol. 2015, 194, 4066–4072. [Google Scholar] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Carballido, E.; Fishman, M. Sipuleucel-T: Prototype for development of anti-tumor vaccines. Curr. Oncol. Rep. 2011, 13, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Eyrich, M.; Schreiber, S.C.; Rachor, J.; Krauss, J.; Pauwels, F.; Hain, J.; Wolfl, M.; Lutz, M.B.; de Vleeschouwer, S.; Schlegel, P.G.; et al. Development and validation of a fully gmp-compliant production process of autologous, tumor-lysate-pulsed dendritic cells. Cytotherapy 2014, 16, 946–964. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Sprague, L.; McGinty, J.; Pate, M.; Muccioli, M. Dendritic cells the tumor microenvironment and the challenges for an effective antitumor vaccination. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, M.; Horiguchi, S.; Yamasaki, K.; Uchida, Y.; Motohashi, S.; Nakayama, T.; Sugimoto, A.; Okamoto, Y. Migration and immunological reaction after the administration of alphagalcer-pulsed antigen-presenting cells into the submucosa of patients with head and neck cancer. Cancer Immunol. Immunother. 2011, 60, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; de Vries, I.J.; Schreibelt, G.; Lambeck, A.J.; Aarntzen, E.H.; Jacobs, J.F.; Scharenborg, N.M.; van de Rakt, M.W.; de Boer, A.J.; Croockewit, S.; et al. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific t cells in advanced melanoma patients. Clin. Cancer Res. 2011, 17, 5725–5735. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.A.; Drake, V.; Huang, H.S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze t cells. Oncoimmunology 2015, 4, e1016700. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Watanabe, M. Myeloid-derived suppressor cells and therapeutic strategies in cancer. Mediat. inflamm. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Poschke, I.; Kiessling, R. Tumour-induced immune suppression: Role of inflammatory mediators released by myelomonocytic cells. J. Intern. Med. 2014, 276, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L.; Schuler, P.; Schilling, B. Induced and natural regulatory t cells in human cancer. Expert Opin. Biol. Ther. 2012, 12, 1383–1397. [Google Scholar] [CrossRef] [PubMed]
- Diener, K.R.; Need, E.F.; Buchanan, G.; Hayball, J.D. Tgf-beta signalling and immunity in prostate tumourigenesis. Expert Opin. Ther. Targets 2010, 14, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Waziri, A. Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurg. Clin. N. Am. 2010, 21, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.X. Dynamic behavior and function of Foxp3+ regulatory T cells in tumor bearing host. Cell Mol. Immunol. 2009, 6, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Vega, E.A.; Graner, M.W.; Sampson, J.H. Combating immunosuppression in glioma. Future Oncol. 2008, 4, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by texs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, D.M.; Lee, P.P. Tumor-derived exosomes contain micrornas with immunological function: Implications for a novel immunosuppression mechanism. Microrna 2014, 2, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Regnault, A.; Garin, J.; Wolfers, J.; Zitvogel, L.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 1999, 147, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Torigoe, T.; Kukita, K.; Saito, K.; Okuya, K.; Kutomi, G.; Hirata, K.; Sato, N. Heat-shock proteins as endogenous ligands building a bridge between innate and adaptive immunity. Immunotherapy 2012, 4, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, S.; Matzinger, P. Danger signals: Sos to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Seigneuric, R.; Mjahed, H.; Gobbo, J.; Joly, A.L.; Berthenet, K.; Shirley, S.; Garrido, C. Heat shock proteins as danger signals for cancer detection. Front. Oncol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Schneider, H.; Stumptner-Cuvelette, P.; Lankar, D.; Pain, S.; Raposo, G.; Benaroch, P.; Bonnerot, C. Exosomes bearing hla-dr1 molecules need dendritic cells to efficiently stimulate specific t cells. Int. Immunol. 2002, 14, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Denzer, K.; van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; de Groot, C.; Geuze, H.J. Follicular dendritic cells carry mhc class II-expressing microvesicles at their surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Duban, L.; Segura, E.; Veron, P.; Lantz, O.; Amigorena, S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Chaput, N.; Schartz, N.E.; Flament, C.; Aubert, N.; Bernard, J.; Lemonnier, F.; Raposo, G.; Escudier, B.; Hsu, D.H.; et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional mhc class I/peptide complexes to dendritic cells. J. Immunol. 2004, 172, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Utsugi-Kobukai, S.; Fujimaki, H.; Hotta, C.; Nakazawa, M.; Minami, M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol. Lett. 2003, 89, 125–131. [Google Scholar] [CrossRef]
- Sprent, J. Direct stimulation of naive t cells by antigen-presenting cell vesicles. Blood Cells Mol. Dis. 2005, 35, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Kovar, M.; Boyman, O.; Shen, X.; Hwang, I.; Kohler, R.; Sprent, J. Direct stimulation of T cells by membrane vesicles from antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11671–11676. [Google Scholar] [CrossRef] [PubMed]
- Amigorena, S. Anti-tumour immunotherapy using dendritic-cell-derived exosomes. Res. Immunol. 1998, 149, 661–662. [Google Scholar] [CrossRef]
- Zitvogel, L.; Fernandez, N.; Lozier, A.; Wolfers, J.; Regnault, A.; Raposo, G.; Amigorena, S. Dendritic cells or their exosomes are effective biotherapies of cancer. Eur. J. Cancer 1999, 35, S36–S38. [Google Scholar] [CrossRef]
- Quah, B.; O’Neill, H.C. Review: The application of dendritic cell-derived exosomes in tumour immunotherapy. Cancer Biother. Radiopharmcol. 2000, 15, 185–194. [Google Scholar] [CrossRef]
- Chaput, N.; Schartz, N.E.; Andre, F.; Zitvogel, L. Exosomes for immunotherapy of cancer. Adv. Exp. Med. Biol. 2003, 532, 215–221. [Google Scholar] [PubMed]
- Andre, F.; Escudier, B.; Angevin, E.; Tursz, T.; Zitvogel, L. Exosomes for cancer immunotherapy. Ann. Oncol. 2004, 15, iv141–iv144. [Google Scholar] [PubMed]
- Lamparski, H.G.; Metha-Damani, A.; Yao, J.Y.; Patel, S.; Hsu, D.H.; Ruegg, C.; le Pecq, J.B. Production and characterization of clinical grade exosomes derived from dendritic cells. J. Immunol. Methods 2002, 270, 211–226. [Google Scholar] [CrossRef]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; Andre, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Thery, C.; Ploix, S.; Tursz, T.; Lapierre, V.; Lantz, O.; Zitvogel, L.; Chaput, N. Dendritic cell-derived exosomes for cancer immunotherapy: What’s next? Cancer Res. 2010, 70, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Kanda, T.; Abe, T.; Uchiyama, T.; Iwafuchi, M.; Zheng, Z.; Liu, A.; Kaifu, T.; Kosugi, S.; Minagawa, M.; et al. Immune responses in patients with esophageal cancer treated with SART1 peptide-pulsed dendritic cell vaccine. Int. J. Oncol. 2015, 46, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Charrier, M.; Viaud, S.; Andre, F.; Besse, B.; Chaput, N.; Zitvogel, L. Dendritic cell-derived exosomes as immunotherapies in the fight against cancer. J. Immunol. 2014, 193, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Naslund, T.I.; Gehrmann, U.; Qazi, K.R.; Karlsson, M.C.; Gabrielsson, S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J. Immunol. 2013, 190, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, U.; Hiltbrunner, S.; Georgoudaki, A.M.; Karlsson, M.C.; Naslund, T.I.; Gabrielsson, S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with alpha-galactosylceramide on exosomes. Cancer Res. 2013, 73, 3865–3876. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, S.; Patel, J.M.; Bozeman, E.N.; Imasuen, I.E.; He, S.; Daniels, D.; Selvaraj, P. Allogeneic tumor cell vaccines: The promise and limitations in clinical trials. Hum. Vaccines Immunother. 2014, 10, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Li, Q.; Liu, P.; Xuan, X.; Du, Y. Umbilical cord blood-derived dendritic cells loaded with BGC823 tumor antigens and DC-derived exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumor immunity in vitro and in vivo. Cent. Eur. J. Immunol. 2014, 39, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Bu, N.; Wu, H.; Zhang, G.; Ma, X.; Zhao, P.; Zhai, N.; Xiang, L.; Cao, H.; Yang, X.; Liu, J. Exosome from chaperone-rich cell lysates-loaded dendritic cells produced by celline 1000 culture system exhibits potent immune activity. Biochem. Biophys. Res. Commun. 2015, 456, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.; Raymond, A.; Akporiaye, E.; Katsanis, E. Tumor-derived multiple chaperone enrichment by free-solution isoelectric focusing yields potent antitumor vaccines. Cancer Immunol. Immunother. 2000, 49, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Zeng, Y.; Feng, H.; Katsanis, E. Tumor-derived chaperone-rich cell lysates are effective therapeutic vaccines against a variety of cancers. Cancer Immunol. Immunother. 2003, 52, 226–234. [Google Scholar] [PubMed]
- Graner, M.W.; Romanoski, A.; Katsanis, E. The “peptidome” of tumour-derived chaperone-rich cell lysate anti-cancer vaccines reveals potential tumour antigens that stimulate tumour immunity. Int. J. Hyperth. 2013, 29, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Epple, L.M.; Bemis, L.T.; Cavanaugh, R.P.; Skope, A.; Mayer-Sonnenfeld, T.; Frank, C.; Olver, C.S.; Lencioni, A.M.; Dusto, N.L.; Tal, A.; et al. Prolonged remission of advanced bronchoalveolar adenocarcinoma in a dog treated with autologous, tumour-derived chaperone-rich cell lysate (CRCL) vaccine. Int. J. Hyperthermia 2013, 29, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Sonnenfeld, T.; Har-Noy, M.; Lillehei, K.O.; Graner, M.W. Proteomic analyses of different human tumour-derived chaperone-rich cell lysate (CRCL) anti-cancer vaccines reveal antigen content and strong similarities amongst the vaccines along with a basis for crcl’s unique structure: Crcl vaccine proteome leads to unique structure. Int. J. Hyperthermia 2013, 29, 520–527. [Google Scholar] [PubMed]
- Bu, N.; Wu, H.; Zhang, G.; Zhan, S.; Zhang, R.; Sun, H.; Du, Y.; Yao, L.; Wang, H. Exosomes from dendritic cells loaded with chaperone-rich cell lysates elicit a potent t cell immune response against intracranial glioma in mice. J. Mol. Neurosci. 2015, 56, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, G.G.; Zelante, B.B.; Toniolo, P.A.; Migliori, I.K.; Barbuto, J.A. Dendritic cell-derived exosomes may be a tool for cancer immunotherapy by converting tumor cells into immunogenic targets. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thery, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for ctl cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Altieri, S.L.; Khan, A.N.; Tomasi, T.B. Exosomes from plasmacytoma cells as a tumor vaccine. J. Immunother. 2004, 27, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Schartz, N.E.; Chaput, N.; Flament, C.; Raposo, G.; Amigorena, S.; Angevin, E.; Zitvogel, L. Tumor-derived exosomes: A new source of tumor rejection antigens. Vaccine 2002, 20, A28–A31. [Google Scholar] [CrossRef]
- Lee, E.Y.; Park, K.S.; Yoon, Y.J.; Lee, J.; Moon, H.G.; Jang, S.C.; Choi, K.H.; Kim, Y.K.; Gho, Y.S. Therapeutic effects of autologous tumor-derived nanovesicles on melanoma growth and metastasis. PLoS ONE 2012, 7, e33330. [Google Scholar] [CrossRef] [PubMed]
- Bu, N.; Li, Q.L.; Feng, Q.; Sun, B.Z. Immune protection effect of exosomes against attack of L1210 tumor cells. Leuk. Lymphoma 2006, 47, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Cumming, R.I.; Bigner, D.D. The heat shock response and chaperones/heat shock proteins in brain tumors: Surface expression, release, and possible immune consequences. J. Neurosci. 2007, 27, 11214–11227. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Alzate, O.; Dechkovskaia, A.M.; Keene, J.D.; Sampson, J.H.; Mitchell, D.A.; Bigner, D.D. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009, 23, 1541–1557. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Bai, O.; Yuan, J.; Qureshi, M.; Xiang, J. Dendritic cell-derived exosomes stimulate stronger CD8+ CTLresponses and antitumor immunity than tumor cell-derived exosomes. Cell Mol. Immunol. 2006, 3, 205–211. [Google Scholar] [PubMed]
- Gu, X.; Erb, U.; Buchler, M.W.; Zoller, M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wan, T.; Wang, B.; Zhou, X.; Xiu, F.; Chen, T.; Wu, Y.; Cao, X. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed cea-positive tumor cells. Clin. Cancer Res. 2005, 11, 7554–7563. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Lee, Y.S.; Kim, S.H.; Ko, J.K.; Kim, C.W. Mhc independent anti-tumor immune responses induced by hsp70-enriched exosomes generate tumor regression in murine models. Cancer Lett. 2009, 275, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bai, O.; Zhang, H.; Yuan, J.; Zong, S.; Chibbar, R.; Slattery, K.; Qureshi, M.; Wei, Y.; Deng, Y.; et al. Membrane-bound hsp70-engineered myeloma cell-derived exosomes stimulate more efficient CD8(+) ctl- and nk-mediated antitumour immunity than exosomes released from heat-shocked tumour cells expressing cytoplasmic hsp70. J. Cell. Mol. Med. 2010, 14, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guo, J.; Yang, M.; Zhu, X.; Cao, X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J. Immunol. 2011, 186, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiu, F.; Cai, Z.; Wang, J.; Wang, Q.; Fu, Y.; Cao, X. Increased induction of antitumor response by exosomes derived from interleukin-2 gene-modified tumor cells. J. Cancer Res. Clin. Oncol. 2007, 133, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bai, O.; Zhang, H.; Li, W.; Xiang, J. Tumor necrosis factor gene-engineered j558 tumor cell-released exosomes stimulate tumor antigen p1a-specific CD8+ ctl responses and antitumor immunity. Cancer Biother. Radiopharm. 2010, 25, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, S.H.; Cho, J.A.; Kim, C.W. Introduction of the ciita gene into tumor cells produces exosomes with enhanced anti-tumor effects. Exp. Mol. Med. 2011, 43, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Rountree, R.B.; Mandl, S.J.; Nachtwey, J.M.; Dalpozzo, K.; Do, L.; Lombardo, J.R.; Schoonmaker, P.L.; Brinkmann, K.; Dirmeier, U.; Laus, R.; et al. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011, 71, 5235–5244. [Google Scholar] [CrossRef] [PubMed]
- Zeelenberg, I.S.; van Maren, W.W.; Boissonnas, A.; van Hout-Kuijer, M.A.; den Brok, M.H.; Wagenaars, J.A.; van der Schaaf, A.; Jansen, E.J.; Amigorena, S.; Thery, C.; et al. Antigen localization controls T cell-mediated tumor immunity. J. Immunol. 2011, 187, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Sedlik, C.; Vigneron, J.; Torrieri-Dramard, L.; Pitoiset, F.; Denizeau, J.; Chesneau, C.; de la Rochere, P.; Lantz, O.; Thery, C.; Bellier, B. Different immunogenicity but similar antitumor efficacy of two DNA vaccines coding for an antigen secreted in different membrane vesicle-associated forms. J. Extracell. Vesicles 2014. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Wei, J.; Glass, O.K.; Guo, H.; Lei, G.; Yang, X.Y.; Osada, T.; Hobeika, A.; Delcayre, A.; le Pecq, J.B.; et al. Increasing vaccine potency through exosome antigen targeting. Vaccine 2011, 29, 9361–9367. [Google Scholar] [CrossRef] [PubMed]
- Zeelenberg, I.S.; Ostrowski, M.; Krumeich, S.; Bobrie, A.; Jancic, C.; Boissonnas, A.; Delcayre, A.; le Pecq, J.B.; Combadiere, B.; Amigorena, S.; et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008, 68, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Xiu, F.; Cai, Z.; Yang, Y.; Wang, X.; Wang, J.; Cao, X. Surface anchorage of superantigen sea promotes induction of specific antitumor immune response by tumor-derived exosomes. J. Mol. Med. 2007, 85, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Bigner, D.D. Chaperone proteins and brain tumors: Potential targets and possible therapeutics. Neuro Oncol. 2005, 7, 260–278. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Bigner, D.D. Therapeutic aspects of chaperones/heat-shock proteins in neuro-oncology. Expert Rev. Anticancer Ther. 2006, 6, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Toraya-Brown, S.; Fiering, S. Local tumour hyperthermia as immunotherapy for metastatic cancer. Int. J. Hyperth. 2014, 30, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Hazoume, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.H.; Wan, Y.L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.L.; Lin, H.M.; Shang, C.Z.; Chen, Y.J.; Min, J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Dong, W.; Zhang, C.; Saren, G.; Geng, P.; Zhao, H.; Li, Q.; Zhu, J.; Li, G.; Zhang, S.; et al. Effects of the epigenetic drug ms-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur. J. Med. Res. 2013, 18. [Google Scholar] [CrossRef] [PubMed]
- Mincheva-Nilsson, L.; Baranov, V. Cancer exosomes and NKG2D receptor-ligand interactions: Impairing NKG2D-mediated cytotoxicity and anti-tumour immune surveillance. Semin. Cancer Biol. 2014, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Parmiani, G.; Rodolfo, M.; Melani, C. Immunological gene therapy with ex vivo gene-modified tumor cells: A critique and a reappraisal. Hum. Gene Ther. 2000, 11, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Luo, C.; Xia, Y.; Chen, H.; Wu, X. Glycosyl-phosphatidylinositol-anchored interleukin-2 expressed on tumor-derived exosomes induces antitumor immune response in vitro. Tumori 2010, 96, 452–459. [Google Scholar] [PubMed]
- Napoletano, C.; Rughetti, A.; Landi, R.; Pinto, D.; Bellati, F.; Rahimi, H.; Spinelli, G.P.; Pauselli, S.; Sale, P.; Dolo, V.; et al. Immunogenicity of allo-vesicle carrying ErbB2 tumor antigen for dendritic cell-based anti-tumor immunotherapy. Int. J. Immunopathol. Pharmacol. 2009, 22, 647–658. [Google Scholar] [PubMed]
- Delcayre, A.; Estelles, A.; Sperinde, J.; Roulon, T.; Paz, P.; Aguilar, B.; Villanueva, J.; Khine, S.; le Pecq, J.B. Exosome display technology: Applications to the development of new diagnostics and therapeutics. Blood Cells Mol. Dis. 2005, 35, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Navabi, H.; Croston, D.; Coleman, S.; Tabi, Z.; Clayton, A.; Jasani, B.; Mason, M.D. The rationale for combined chemo/immunotherapy using a toll-like receptor 3 (TLR3) agonist and tumour-derived exosomes in advanced ovarian cancer. Vaccine 2005, 23, 2374–2378. [Google Scholar] [PubMed]
- Andre, F.; Schartz, N.E.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Navabi, H.; Croston, D.; Hobot, J.; Clayton, A.; Zitvogel, L.; Jasani, B.; Bailey-Wood, R.; Wilson, K.; Tabi, Z.; Mason, M.D.; et al. Preparation of human ovarian cancer ascites-derived exosomes for a clinical trial. Blood Cells Mol. Dis. 2005, 35, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, Y.; Cao, X. The exosomes in tumor immunity. Oncoimmunology 2015, 4, e1027472. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Zhuang, X.; Sun, D.; Liu, Y.; Xiang, X.; Grizzle, W.E. Exosomes and immune surveillance of neoplastic lesions: A review. Biotech. Histochem. 2012, 87, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Valenti, R.; Huber, V.; Iero, M.; Filipazzi, P.; Parmiani, G.; Rivoltini, L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res. 2007, 67, 2912–2915. [Google Scholar] [CrossRef] [PubMed]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of fasl-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Wieckowski, E.; Taylor, D.D.; Reichert, T.E.; Watkins, S.; Whiteside, T.L. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 2005, 11, 1010–1020. [Google Scholar] [PubMed]
- Abusamra, A.J.; Zhong, Z.; Zheng, X.; Li, M.; Ichim, T.E.; Chin, J.L.; Min, W.P. Tumor exosomes expressing fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis. 2005, 35, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ichim, T.E.; Zhong, Z.; Kaushal, S.; Zheng, X.; Ren, X.; Hao, X.; Joyce, J.A.; Hanley, H.H.; Riordan, N.H.; Koropatnick, J.; et al. Exosomes as a tumor immune escape mechanism: Possible therapeutic implications. J. Transl. Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress t cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [PubMed]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battke, C.; Ruiss, R.; Welsch, U.; Wimberger, P.; Lang, S.; Jochum, S.; Zeidler, R. Tumour exosomes inhibit binding of tumour-reactive antibodies to tumour cells and reduce ADCC. Cancer Immunol. Immunother. 2011, 60, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Epple, L.M.; Dusto, N.L.; Lencioni, A.M.; Nega, M.; Herring, M.; Winston, B.; Madsen, H.; Bemis, L.T.; Anchordoquy, T.J. Circulating exosomes as new biomarkers for brain disease and injury. In Proceedings of SPIE Vol. 8723 Sensing Technologies for Global Health, Military Medicine, and Environmental Monitoring III, Baltimore, MD, USA, 29 Apr–1 May 2013; Southern, S.O., Ed.;
- Clayton, A.; Tabi, Z. Exosomes and the mica-NKG2D system in cancer. Blood Cells Mol. Dis. 2005, 34, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma t and b cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef] [PubMed]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [PubMed]
- Elsner, L.; Muppala, V.; Gehrmann, M.; Lozano, J.; Malzahn, D.; Bickeboller, H.; Brunner, E.; Zientkowska, M.; Herrmann, T.; Walter, L.; et al. The heat shock protein hsp70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J. Immunol. 2007, 179, 5523–5533. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated hsp72 from tumor-derived exosomes mediates stat3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, A.; Zhou, W.; Liu, L.; Fong, M.Y.; Champer, J.; van Haute, D.; Chin, A.R.; Ren, X.; Gugiu, B.G.; Meng, Z.; et al. Macrophage immunomodulation by breast cancer-derived exosomes requires toll-like receptor 2-mediated activation of NF-kappab. Sci. Rep. 2014. [Google Scholar] [CrossRef]
- De Vrij, J.; Maas, S.L.; Kwappenberg, K.M.; Schnoor, R.; Kleijn, A.; Dekker, L.; Luider, T.M.; de Witte, L.D.; Litjens, M.; van Strien, M.E.; et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int. J. Cancer 2015, 137, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Marton, A.; Vizler, C.; Kusz, E.; Temesfoi, V.; Szathmary, Z.; Nagy, K.; Szegletes, Z.; Varo, G.; Siklos, L.; Katona, R.L.; et al. Melanoma cell-derived exosomes alter macrophage and dendritic cell functions in vitro. Immunol. Lett. 2012, 148, 34–38. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Qin, H.; Poon, T.C.; Sze, S.C.; Ding, X.; Co, N.N.; Ngai, S.M.; Chan, T.F.; Wong, N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and rnas. Carcinogenesis 2015, 36, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microrna biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.B.; Li, Z.L.; Luo, D.H.; Huang, B.J.; Chen, Y.S.; Zhang, X.S.; Cui, J.; Zeng, Y.X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal micrornas in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.D.; Wu, Y.; Shen, H.Y.; Lv, M.M.; Chen, W.X.; Zhang, X.H.; Zhong, S.L.; Tang, J.H.; Zhao, J.H. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015, 106, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Scheerlinck, J.P.; Greenwood, D.L. Virus-sized vaccine delivery systems. Drug Discov. Today 2008, 13, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, L.; Federico, M. A strategy of antigen incorporation into exosomes: Comparing cross-presentation levels of antigens delivered by engineered exosomes and by lentiviral virus-like particles. Vaccine 2012, 30, 7229–7237. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.J.; Redzic, J.S.; Graner, M.W.; Anchordoquy, T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta 2014, 1838, 2954–2965. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface functionalization of exosomes using click chemistry. Bioconjugate Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Hung, M.E.; Breakefield, X.O.; Leonard, J.N. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439–464. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, U.; Naslund, T.I.; Hiltbrunner, S.; Larssen, P.; Gabrielsson, S. Harnessing the exosome-induced immune response for cancer immunotherapy. Semin. Cancer Biol. 2014, 28, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Mitragotri, S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Bianco, N.; Menon, R.; Lechman, E.R.; Shufesky, W.J.; Morelli, A.E.; Robbins, P.D. Exosomes derived from genetically modified dc expressing fasl are anti-inflammatory and immunosuppressive. Mol. Ther. 2006, 13, 289–300. [Google Scholar] [PubMed]
- Segura, E.; Guerin, C.; Hogg, N.; Amigorena, S.; Thery, C. CD8+ dendritic cells use lfa-1 to capture MHC-peptide complexes from exosomes in vivo. J. Immunol. 2007, 179, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Iero, M.; Valenti, R.; Huber, V.; Filipazzi, P.; Parmiani, G.; Fais, S.; Rivoltini, L. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008, 15, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Xie, X.; Lei, Y.; An, G.; He, L.; Chen, R. Consideration of dual characters of exosomes in the tumour immune response. Cell Biol. Int. 2014, 38, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Huang, B.; Hanash, S.M.; Onuchic, J.N.; Ben-Jacob, E. Modeling putative therapeutic implications of exosome exchange between tumor and immune cells. Proc. Natl. Acad. Sci. USA 2014, 111, E4165–E4174. [Google Scholar] [CrossRef] [PubMed]
- Marleau, A.M.; Chen, C.S.; Joyce, J.A.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R.; Raez, L.E. Allogeneic tumor-cell-based vaccines secreting endoplasmic reticulum chaperone gp96. Expert Opin. Biol. Ther. 2007, 7, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kondo, S.; Tada, K.; Kitano, S. Clinical development of immune checkpoint inhibitors. BioMed Res. Int. 2015. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunigelis, K.E.; Graner, M.W. The Dichotomy of Tumor Exosomes (TEX) in Cancer Immunity: Is It All in the ConTEXt? Vaccines 2015, 3, 1019-1051. https://doi.org/10.3390/vaccines3041019
Kunigelis KE, Graner MW. The Dichotomy of Tumor Exosomes (TEX) in Cancer Immunity: Is It All in the ConTEXt? Vaccines. 2015; 3(4):1019-1051. https://doi.org/10.3390/vaccines3041019
Chicago/Turabian StyleKunigelis, Katherine E., and Michael W. Graner. 2015. "The Dichotomy of Tumor Exosomes (TEX) in Cancer Immunity: Is It All in the ConTEXt?" Vaccines 3, no. 4: 1019-1051. https://doi.org/10.3390/vaccines3041019