
Nanoparticle Drug Delivery Systems Designed to Improve Cancer Vaccines and Immunotherapy

Abstract
:1. Introduction
2. Principles of Adaptive Immunity against Cancer
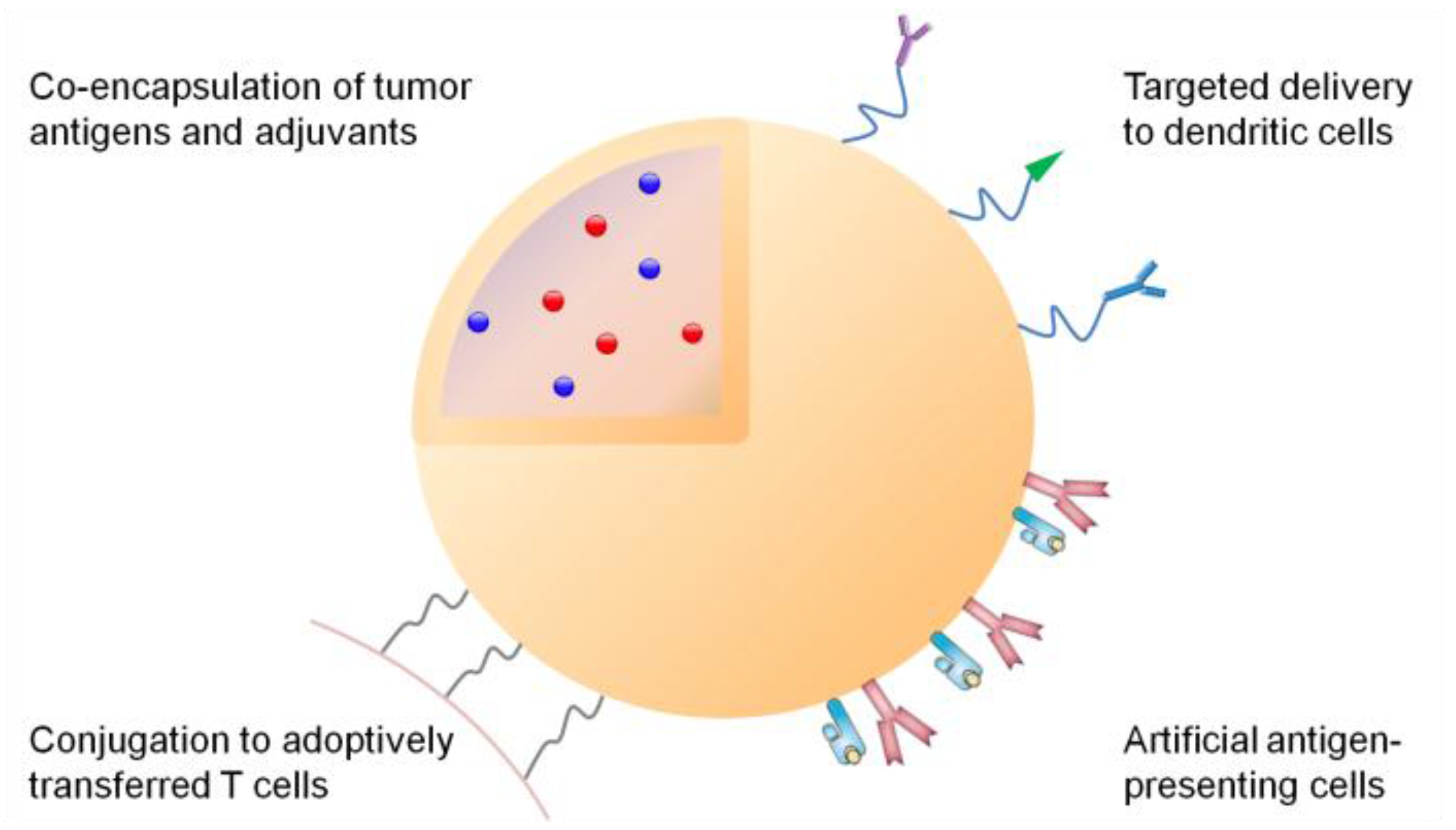
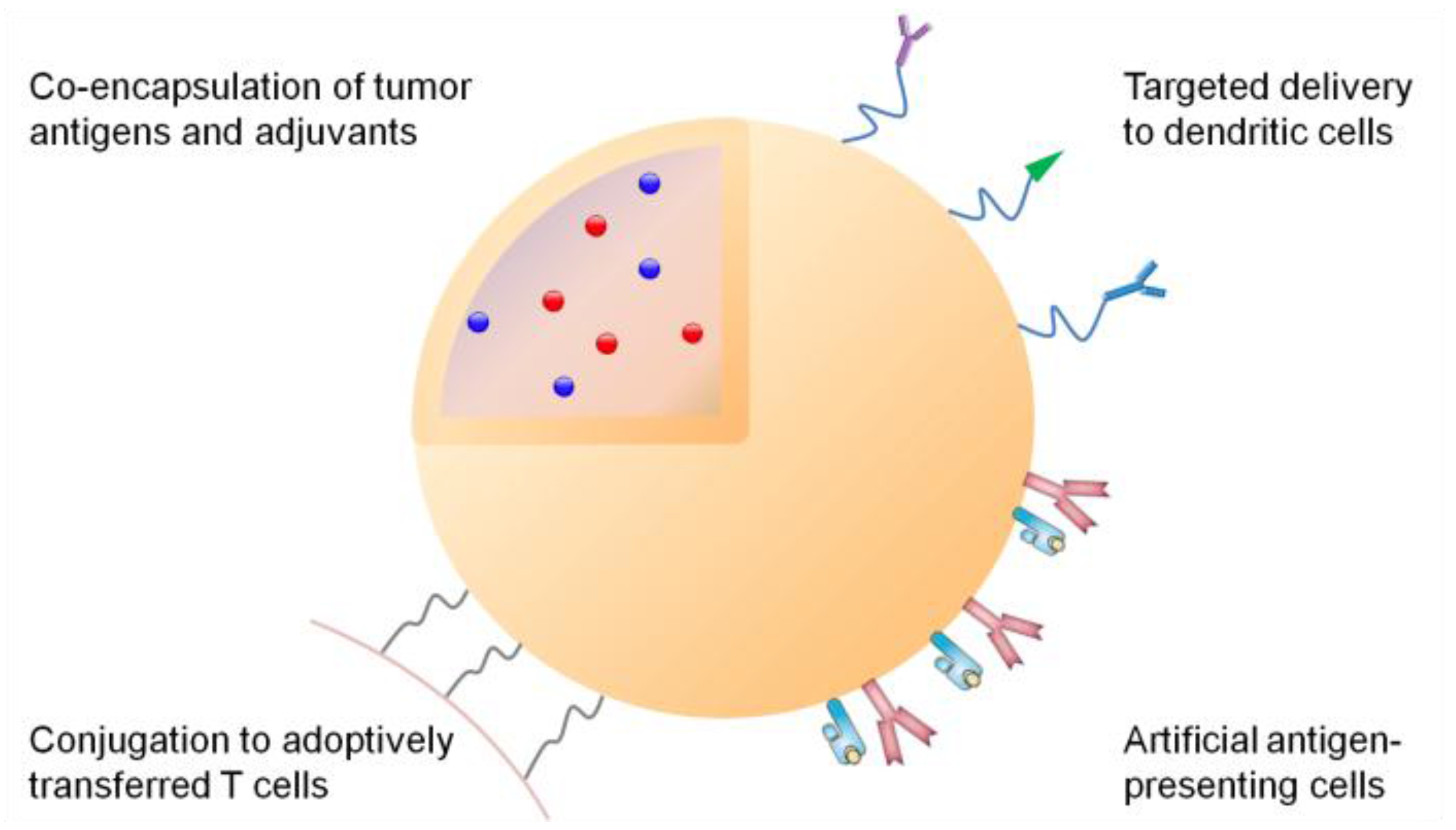
3. Synthetic Systems for Delivery of Tumor Antigens

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor antigens | Advantages | Challenges | |
|---|---|---|---|
| Subunit antigens | Polysaccharides | Defined chemical synthesis | Elicitation of humoral rather than cellular immune responses |
| Peptides | Ease of production | Poor delivery efficiency | |
| Stable vaccine formulations | Monovalent immune response | ||
| May not require antigen-processing by APCs | Subject to HLA-specificity | ||
| Proteins | Broad-epitope immune responses Wide HLA-specificity | Poor delivery efficiency | |
| Suboptimal for CD8+ T cell responses | |||
| Weak immunogenicity of self-antigens | |||
| DNA and mRNA | Ease of production | Poor delivery efficiency | |
| In situ expression of full-length antigens | Poor in vivo stability | ||
| Flexible to encode immune stimulators | Limited transfection efficiency | ||
| Whole-cell antigens | Tumor-cell lysate | Broad-epitope immune responses Potential for “personalized” therapy | Requires tissue biopsy |
| Manufacturing challenges | |||
| Loss of antigenicity during production | |||
| Presence of self-antigens | |||
| Immunogenically dying tumor cells | Broad-epitope immune responses | Requires additional therapeutic interventions Presence of self-antigens and immunosuppressive molecules, e.g., PD-L1 | |
| Full preservation of tumor antigens | |||
| Potential for “personalized” therapy | |||
3.1. Efficient Draining to Lymphoid Tissues
3.2. Targeted Delivery to Dendritic Cells
3.3. Promotion of Cross-Presentation
3.4. Co-Delivery of Adjuvants

3.5. Delivery of DNA and mRNA Tumor Antigens
3.6. Delivery of Whole-Cell Cancer Vaccines
4. Synthetic Delivery Systems for DC-Based Cancer Vaccines
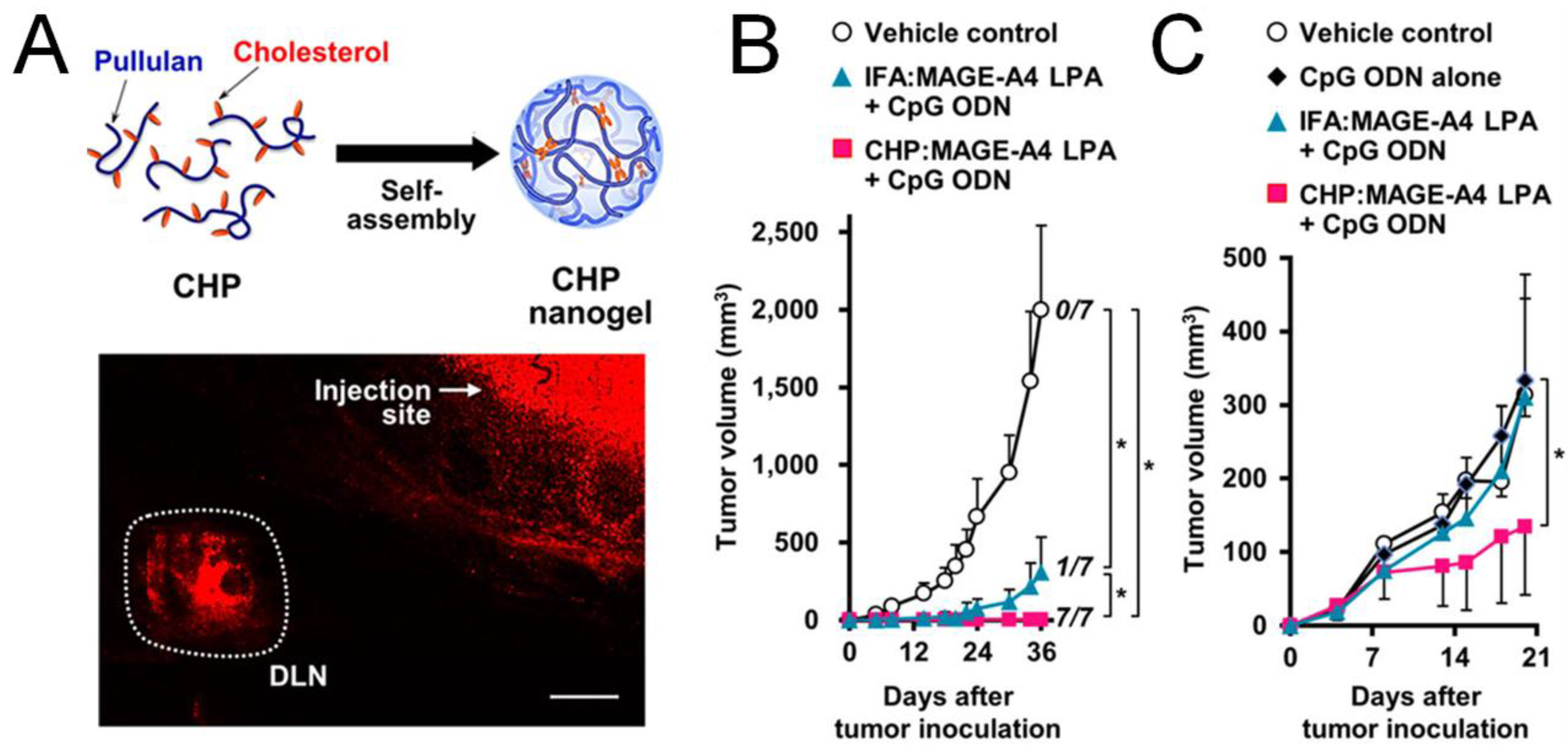
5. Synthetic Delivery Approaches to Induce Immunogenic Cell Death (ICD)

6. Synthetic Delivery Systems Targeted to Immune Checkpoints
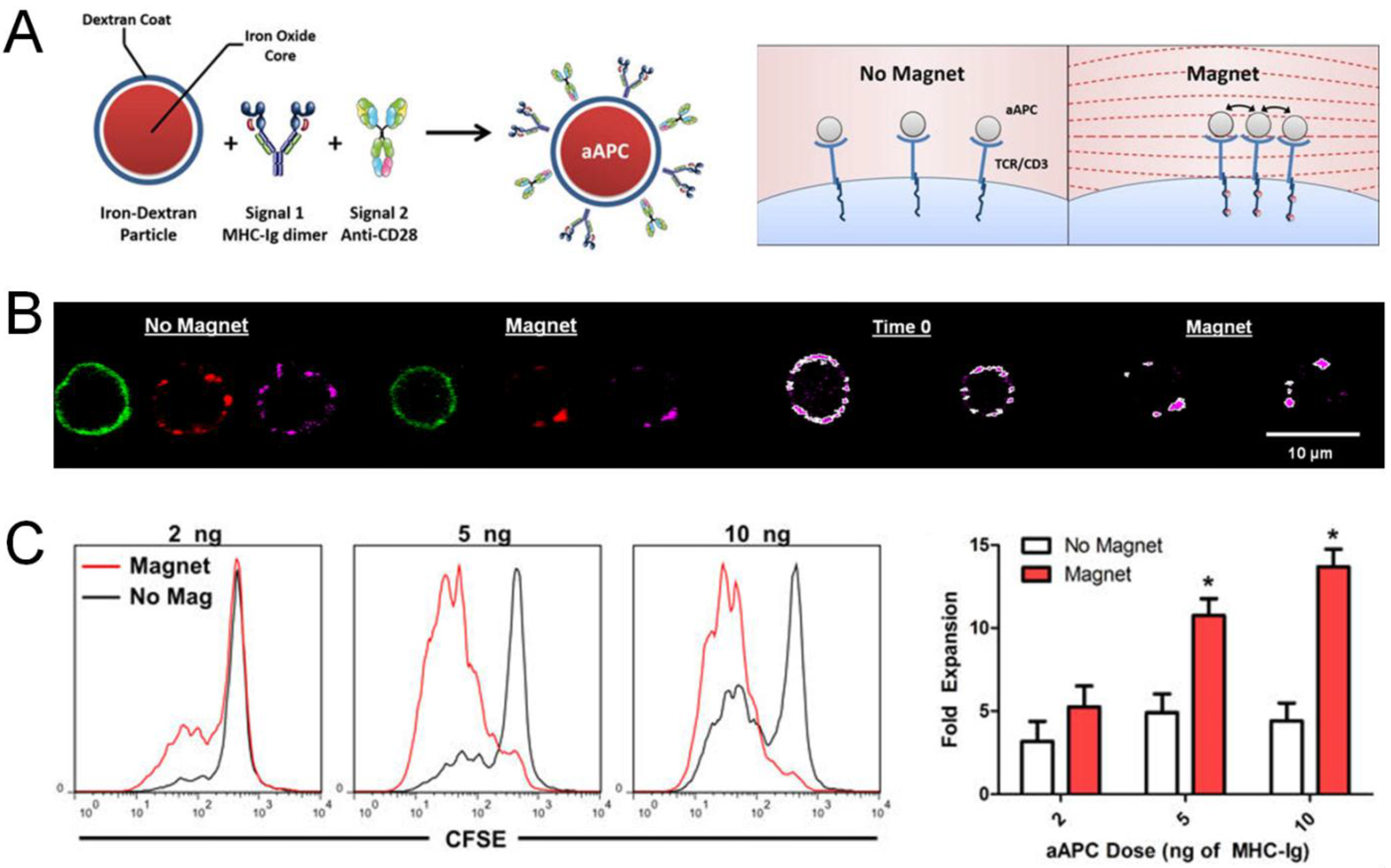
7. Synthetic Delivery Systems for Adoptively Transferred T Cells
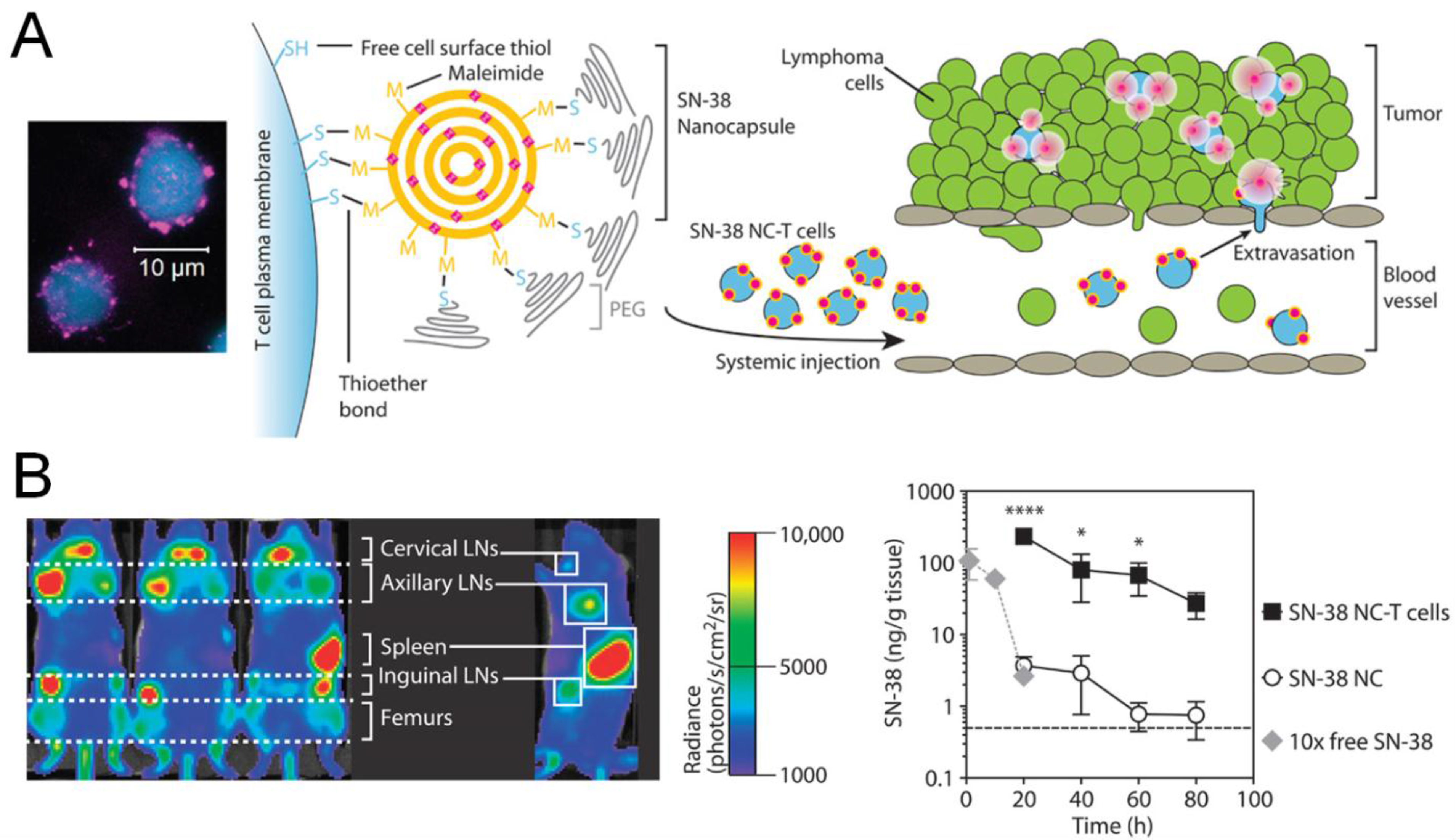
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| aAPC | Artificial antigen-presenting cell |
| ACT | Adoptive cell therapy |
| APCs | Antigen-presenting cells |
| CpG | Unmethylated oligonucleotide containing CpG motif |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CTL | Cytotoxic T lymphocyte |
| DCs | Dendritic cells |
| EBV | Epstein-Barr virus |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| HLA | Human leukocyte antigen |
| HPV | Human papilloma virus |
| ICD | Immunogenic cell death |
| LPS | Lipopolysaccharide |
| MDSCs | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| MPLA | Monophosphoryl lipid A |
| OVA | Ovalbumin |
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death ligand-1 |
| PLGA | Poly(lactic-co-glycolic acid) |
| siRNA | Small-interfering RNA |
| TCRs | T-cell receptors |
| TNF | Tumor necrosis factor |
| TH cells | Helper T cells |
| TLRs | Toll-like receptors |
| TNF | Tumor necrosis factor |
| Tregs | Regulatory T cells |
| Trp-2 | Tyrosine-related protein 2 |
References
- Starnes, C.O. Coley’s toxins in perspective. Nature 1992, 357, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Downey, S.G.; Klapper, J.A.; Smith, F.O.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Kammula, U.S.; Hughes, M.S.; Allen, T.E.; Levy, C.L.; et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clin. Cancer Res. 2007, 13, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.S.; Mohammad, F. Magnetic nanomaterials for hyperthermia-based therapy and controlled drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Du, W.; He, B.; Fu, F.; Yuan, L.; Wu, H.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; et al. The reduction of tumor interstitial fluid pressure by liposomal imatinib and its effect on combination therapy with liposomal doxorubicin. Biomaterials 2013, 34, 2277–2288. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Miura, S.; Na, K.; Bae, Y.H. pH-responsive and charge shielded cationic micelle of poly(L-histidine)-block-short branched PEI for acidic cancer treatment. J. Control. Release 2013, 172, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Van der Meel, R.; Vehmeijer, L.J.; Kok, R.J.; Storm, G.; van Gaal, E.V. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: Current status. Adv. Drug Deliv. Rev. 2013, 65, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, S.; Shi, Y.; Chuan, X.; Li, J.; Zhong, T.; Zhang, H.; Dai, W.; He, B.; Zhang, Q. The development of site-specific drug delivery nanocarriers based on receptor mediation. J. Control. Release 2014, 193, 139–153. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Schulz, A.; Wan, X.; Seitz, J.; Bludau, H.; Alakhova, D.Y.; Darr, D.B.; Perou, C.M.; Jordan, R.; Ojima, I.; et al. Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: Preparation, in vitro and in vivo evaluation. J. Control. Release 2015, 208, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Huang, B.; Irvine, D.J. Engineering nano- and microparticles to tune immunity. Adv. Mater. 2012, 24, 3724–3746. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Swartz, M.A.; Szeto, G.L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013, 12, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Immunoengineering: How nanotechnology can enhance cancer immunotherapy. Cell 2015, 161, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Videira, M.; Gaspar, R.; Preat, V.; Florindo, H.F. Immune system targeting by biodegradable nanoparticles for cancer vaccines. J. Control. Release 2013, 168, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Dewitte, H.; Verbeke, R.; Breckpot, K.; de Smedt, S.C.; Lentacker, I. Nanoparticle design to induce tumor immunity and challenge the suppressive tumor microenvironment. Nano Today 2014, 9, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Nune, S.K.; Gunda, P.; Majeti, B.K.; Thallapally, P.K.; Forrest, M.L. Advances in lymphatic imaging and drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Hirosue, S.; Hubbell, J.A. Engineering approaches to immunotherapy. Sci. Trans. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Hanson, M.C.; Rakhra, K.; Tokatlian, T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Leleux, J.; Roy, K. Micro and nanoparticle-based delivery systems for vaccine immunotherapy: An immunological and materials perspective. Adv. Healthc. Mater. 2013, 2, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Sahdev, P.; Ochyl, L.J.; Moon, J.J. Biomaterials for nanoparticle vaccine delivery systems. Pharm. Res. 2014, 31, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Green, J.J. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine 2013, 8, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Singha, S.; Clemente-Casares, X.; Tsai, S.; Yang, Y.; Santamaria, P. Nanoparticle-based immunotherapy for cancer. ACS Nano 2015, 9, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Irvine, D.J. Enhancing cell therapies from the outside in: Cell surface engineering using synthetic nanomaterials. Nano Today 2011, 6, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.; de los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef]
- Cebrian, I.; Visentin, G.; Blanchard, N.; Jouve, M.; Bobard, A.; Moita, C.; Enninga, J.; Moita, L.F.; Amigorena, S.; Savina, A. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 2011, 147, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Samie, M.; Cresswell, P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat. Immunol. 2015, 16, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L. The ins and outs of cross-presentation. Nat. Immunol. 2003, 4, 941–943. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Perez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CDL interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Huster, K.M.; Busch, V.; Schiemann, M.; Linkemann, K.; Kerksiek, K.M.; Wagner, H.; Busch, D.H. Selective expression of IL-7 receptor on memory T cells identifies early CDL-dependent generation of distinct CD8+ memory T cell subsets. Proc. Natl. Acad. Sci. USA. 2004, 101, 5610–5615. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Maeurer, M.J.; Ferrone, S. Antigen-processing machinery breakdown and tumor growth. Immunol. Today 2000, 21, 455–464. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Bloy, N.; Buque, A.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; et al. Trial Watch: Peptide-based anticancer vaccines. Oncoimmunology 2015, 4, e974411. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
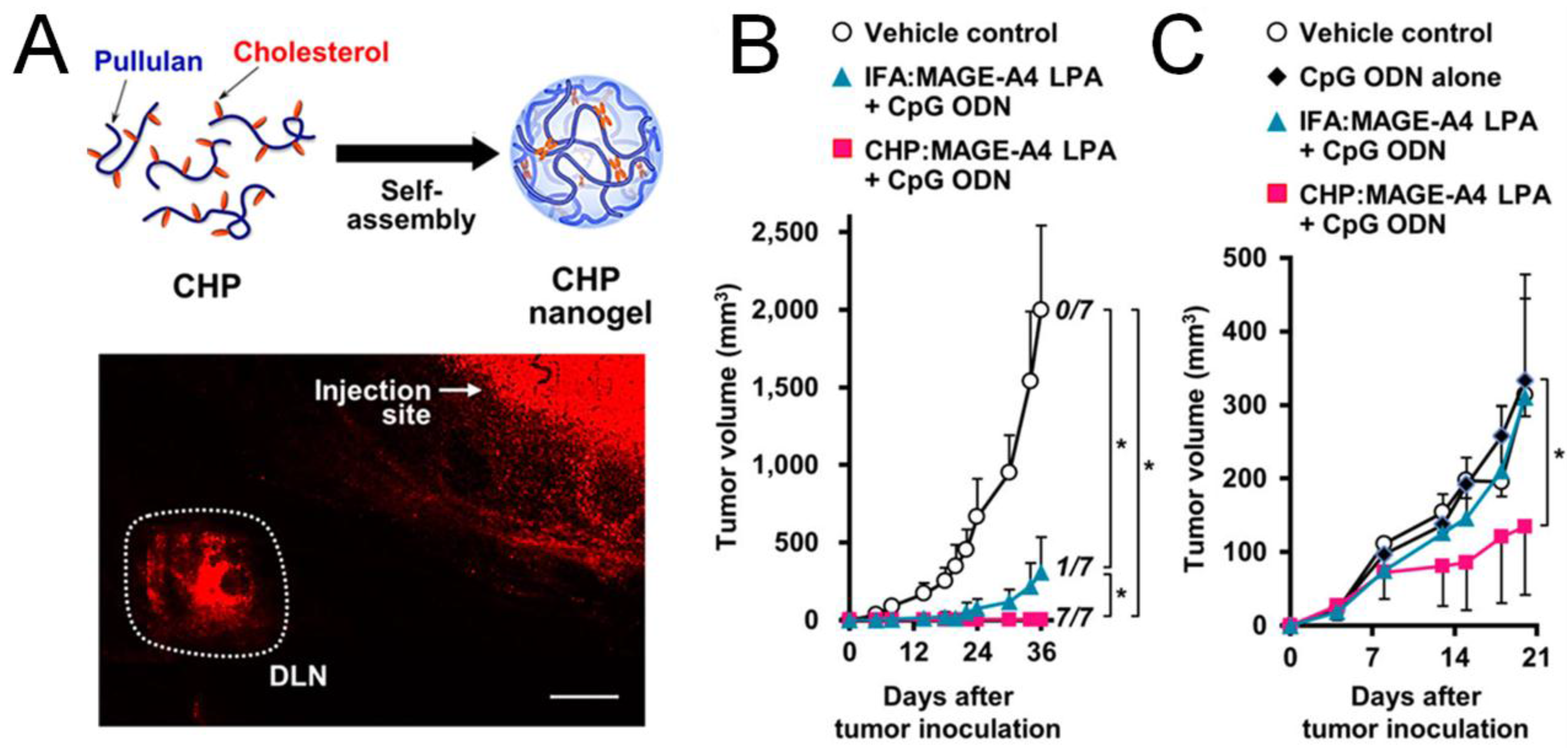
- Muraoka, D.; Harada, N.; Hayashi, T.; Tahara, Y.; Momose, F.; Sawada, S.; Mukai, S.A.; Akiyoshi, K.; Shiku, H. Nanogel-based immunologically stealth vaccine targets macrophages in the medulla of lymph node and induces potent antitumor immunity. ACS Nano 2014, 8, 9209–9218. [Google Scholar] [CrossRef] [PubMed]
- Jeanbart, L.; Ballester, M.; de Titta, A.; Corthesy, P.; Romero, P.; Hubbell, J.A.; Swartz, M.A. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res. 2014, 2, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, S.; Kleinovink, J.W.; Fransen, M.F.; Mezzanotte, L.; Gold, H.; Wisse, P.; Overkleeft, H.; Amidi, M.; Jiskoot, W.; Lowik, C.W.; et al. Near-infrared labeled, ovalbumin loaded polymeric nanoparticles based on a hydrophilic polyester as model vaccine: In vivo tracking and evaluation of antigen-specific CD8(+) T cell immune response. Biomaterials 2015, 37, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Zeelenberg, I.S.; Srinivas, M.; Bonetto, F.; Weigelin, B.; Eich, C.; de Vries, I.J.; Figdor, C.G. Tracking targeted bimodal nanovaccines: Immune responses and routing in cells, tissue, and whole organism. Mol. Pharm. 2014, 11, 4299–4313. [Google Scholar] [CrossRef] [PubMed]
- Mariani, G.; Moresco, L.; Viale, G.; Villa, G.; Bagnasco, M.; Canavese, G.; Buscombe, J.; Strauss, H.W.; Paganelli, G. Radioguided sentinel lymph node biopsy in breast cancer surgery. J. Nucl. Med. 2001, 42, 1198–1215. [Google Scholar] [PubMed]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Trans. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Unger, W.J.; Storm, G.; van Kooyk, Y.; Mastrobattista, E. Targeting tumor antigens to dendritic cells using particulate carriers. J. Control. Release 2012, 161, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.L.; Lustgarten, J. Targeting the tumor microenvironment with anti-neu/anti-CD40 conjugated nanoparticles for the induction of antitumor immune responses. Vaccine 2010, 28, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Rosalia, R.A.; Cruz, L.J.; van Duikeren, S.; Tromp, A.T.; Silva, A.L.; Jiskoot, W.; de Gruijl, T.; Lowik, C.; Oostendorp, J.; van der Burg, S.H.; et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials 2015, 40, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Rosalia, R.A.; Kleinovink, J.W.; Rueda, F.; Lowik, C.W.; Ossendorp, F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: A comparative study. J. Control. Release 2014, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, K.; Dhodapkar, K.M.; Dhodapkar, M.V. Targeting human dendritic cells in situ to improve vaccines. Immunol. Lett. 2014, 162, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; von Zastrow, M. Signal transduction and endocytosis: Close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 2002, 3, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E.; Tajima, N.; Yoshizaki, Y.; Harada, A.; Hayashi, H.; Kono, K. Dextran derivative-based pH-sensitive liposomes for cancer immunotherapy. Biomaterials 2014, 35, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Wilson, J.T.; Patilea, G.I.; Kern, H.B.; Convertine, A.J.; Stayton, P.S. Neutral polymer micelle carriers with pH-responsive, endosome-releasing activity modulate antigen trafficking to enhance CD8(+) T cell responses. J. Control. Release 2014, 191, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.A.; Stano, A.; Gillard, M.; Maio-Liu, A.C.; Swartz, M.A.; Hubbell, J.A. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials 2012, 33, 6211–6219. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ono, K.; Suzuki, Y.; Moriguchi, R.; Kogure, K.; Harashima, H. Octaarginine-modified liposomes enhance cross-presentation by promoting the C-terminal trimming of antigen peptide. Mol. Pharm. 2014, 11, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lee, I.H.; Kang, S.; Kim, D.; Choi, M.; Saw, P.E.; Shin, E.C.; Jon, S. Gold nanoparticles displaying tumor-associated self-antigens as a potential vaccine for cancer immunotherapy. Adv. Healthc. Mater. 2014, 3, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Mandraju, R.; Murray, S.; Forman, J.; Pasare, C. Differential ability of surface and endosomal TLRs to induce CD8 T cell responses in vivo. J. Immunol. 2014, 192, 4303–4315. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Z.; Kurche, J.S.; Burchill, M.A.; Kedl, R.M. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 2011, 118, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
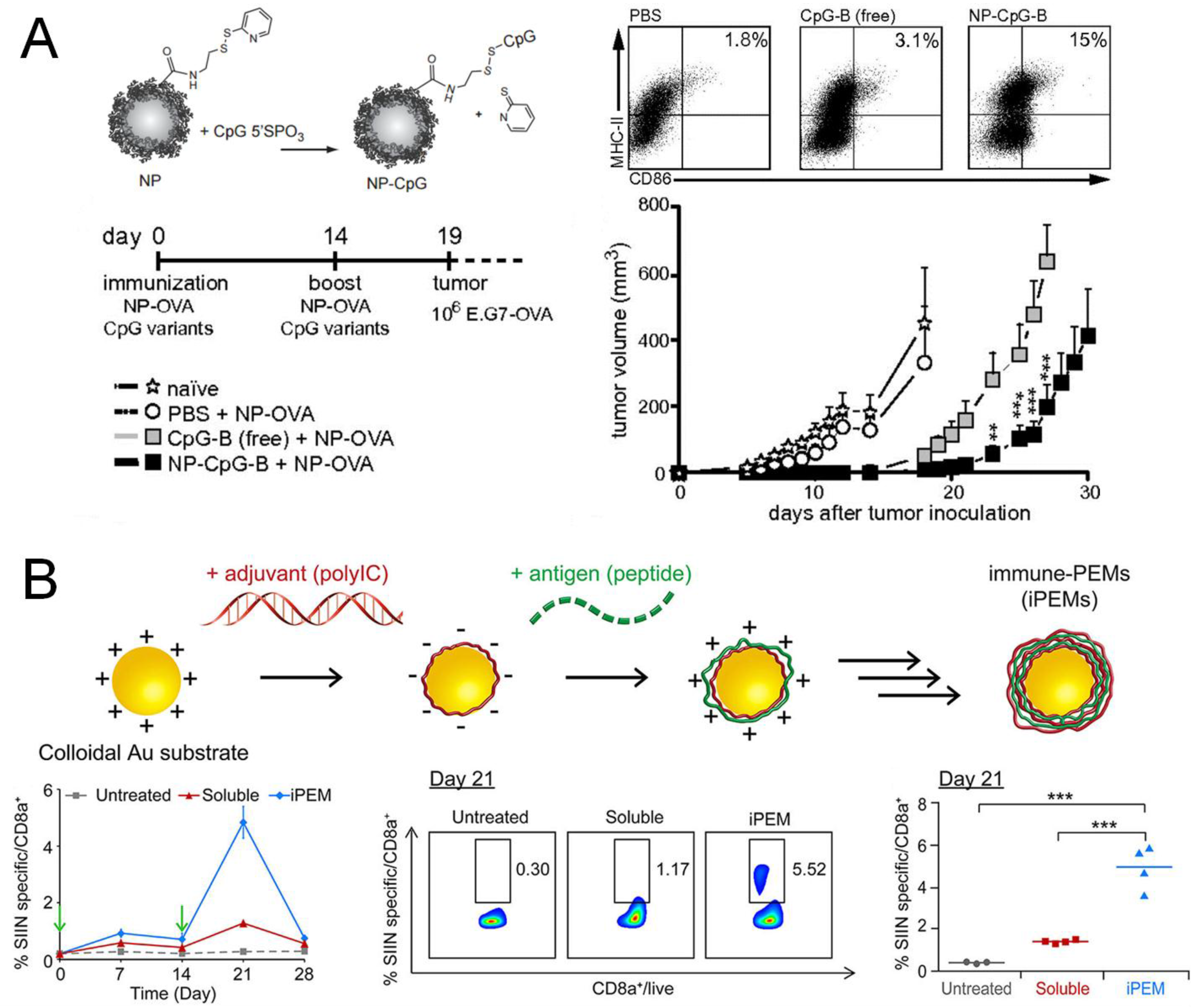
- De Titta, A.; Ballester, M.; Julier, Z.; Nembrini, C.; Jeanbart, L.; van der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc. Natl. Acad Sci. USA 2013, 110, 19902–19907. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.T.; Keller, S.; Manganiello, M.J.; Cheng, C.; Lee, C.C.; Opara, C.; Convertine, A.; Stayton, P.S. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 2013, 7, 3912–3925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chiu, Y.C.; Tostanoski, L.H.; Jewell, C.M. Polyelectrolyte multilayers assembled entirely from immune signals on gold nanoparticle templates promote antigen-specific T cell response. ACS Nano 2015, 9, 6465–6477. [Google Scholar] [CrossRef] [PubMed]
- Molino, N.M.; Anderson, A.K.; Nelson, E.L.; Wang, S.W. Biomimetic protein nanoparticles facilitate enhanced dendritic cell activation and cross-presentation. ACS Nano 2013, 7, 9743–9752. [Google Scholar] [CrossRef] [PubMed]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Zupancic, E.; Vandermeulen, G.; Oliveira, V.G.; Salgado, A.; Videira, M.; Gaspar, M.; Graca, L.; Preat, V.; Florindo, H.F. In vivo delivery of peptides and Toll-like receptor ligands by mannose-functionalized polymeric nanoparticles induces prophylactic and therapeutic anti-tumor immune responses in a melanoma model. J. Control Release 2015, 198, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Sivananthan, S.J.; Duthie, M.S.; Vergara, J.; Guderian, J.A.; Moon, E.; Coblentz, D.; Reed, S.G.; Carter, D. A nanoliposome delivery system to synergistically trigger TLR4 AND TLR7. J. Nanobiotechnol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, P.; Qin, H.; Leleux, J.A.; Gwak, D.; Sakamaki, I.; Kwak, L.W.; Roy, K. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials 2014, 35, 5491–5504. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Ottensmeier, C.H.; Stevenson, F.K. DNA vaccines: Precision tools for activating effective immunity against cancer. Nat. Rev. Cancer 2008, 8, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Senovilla, L.; Vacchelli, E.; Garcia, P.; Eggermont, A.; Fridman, W.H.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: DNA vaccines for cancer therapy. Oncoimmunology 2013, 2, e23803. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Hogberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Shu, Y.; Haque, F.; Abdelmawla, S.; Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011, 6, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xu, B.H.; Xu, J.J.; Shou, D.; Liu, E.G.; Gao, J.Q.; Liang, W.Q.; Huang, Y.Z. Microneedle-assisted dendritic cell-targeted nanoparticles for transcutaneous DNA immunization. Polym. Chem. 2015, 6, 373–379. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, D.; Liu, S.; Gong, J.; Wang, D.; Xiong, M.; Chen, X.; Xiang, R.; Tan, X. Alginic acid-coated chitosan nanoparticles loaded with legumain DNA vaccine: Effect against breast cancer in mice. PLoS ONE 2013, 8, e60190. [Google Scholar] [CrossRef] [PubMed]
- Phua, K.K.; Staats, H.F.; Leong, K.W.; Nair, S.K. Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffres, P.A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccination against BF10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 2011, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhuang, Y.; Zhang, Y.; Luo, Z.; Gao, N.; Li, P.; Pan, H.; Cai, L.; Ma, Y. Toll-like receptor 3 agonist complexed with cationic liposome augments vaccine-elicited antitumor immunity by enhancing TLR3-IRF3 signaling and type I interferons in dendritic cells. Vaccine 2012, 30, 4790–4799. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.P.; Wongrakpanich, A.; Francis, M.B.; Salem, A.K.; Norian, L.A. A therapeutic microparticle-based tumor lysate vaccine reduces spontaneous metastases in murine breast cancer. AAPS J. 2014, 16, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.A.; Huebsch, N.; Cao, L.; Dranoff, G.; Mooney, D.J. Infection-mimicking materials to program dendritic cells in situ. Nat. Mater. 2009, 8, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Hu, C.M.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Ishii, Y.; Matsuo, K.; Yoshinaga, T.; Akashi, M.; Mukai, Y.; Yoshioka, Y.; Okada, N.; Nakagawa, S. The utility of poly(gamma-glutamic acid) nanoparticles as antigen delivery carriers in dendritic cell-based cancer immunotherapy. Biol. Pharm. Bull. 2010, 33, 2003–2007. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Mai, J.; Xu, R.; Perez, J.E.; Guevara, M.L.; Shen, Q.; Mu, C.; Tung, H.Y.; Corry, D.B.; Evans, S.E.; et al. Porous silicon microparticle potentiates anti-tumor immunity by enhancing cross-presentation and inducing type I interferon response. Cell Rep. 2015, 11, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Dewitte, H.; Van Lint, S.; Heirman, C.; Thielemans, K.; de Smedt, S.C.; Breckpot, K.; Lentacker, I. The potential of antigen and TriMix sonoporation using mRNA-loaded microbubbles for ultrasound-triggered cancer immunotherapy. J. Control. Release 2014, 194, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Steenblock, E.R.; Fahmy, T.M. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol. Ther. 2008, 16, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Prakken, B.; Wauben, M.; Genini, D.; Samodal, R.; Barnett, J.; Mendivil, A.; Leoni, L.; Albani, S. Artificial antigen-presenting cells as a tool to exploit the immune “synapse”. Nat. Med. 2000, 6, 1406–1410. [Google Scholar] [PubMed]
- Perica, K.; Tu, A.; Richter, A.; Bieler, J.G.; Edidin, M.; Schneck, J.P. Magnetic field-induced T cell receptor clustering by nanoparticles enhances T cell activation and stimulates antitumor activity. ACS Nano 2014, 8, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Fadel, T.R.; Sharp, F.A.; Vudattu, N.; Ragheb, R.; Garyu, J.; Kim, D.; Hong, E.; Li, N.; Haller, G.L.; Pfefferle, L.D.; et al. A carbon nanotube-polymer composite for T-cell therapy. Nat. Nanotechnol. 2014, 9, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A.; Sunshine, J.C.; Perica, K.; Kosmides, A.K.; Aje, K.; Schneck, J.P.; Green, J.J. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small 2015, 11, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Perica, K.; Schneck, J.P.; Green, J.J. Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials 2014, 35, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spisek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Vitale, I.; Tartour, E.; Eggermont, A.; Sautes-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Anticancer radioimmunotherapy. Oncoimmunology 2013, e25595. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Galluzzi, L.; Fridman, W.H.; Galon, J.; Sautes-Fridman, C.; Tartour, E.; Kroemer, G. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2012, 1, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Joshi, V.B.; Wongrakpanich, A.; Lemke, C.D.; Gross, B.P.; Salem, A.K.; Weiner, G.J. Biodegradable microparticles loaded with doxorubicin and CpG ODN for in situ immunization against cancer. AAPS J. 2015, 17, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Joshi, V.B.; Lemke, C.D.; Wongrakpanich, A.; Olivier, A.K.; Blackwell, S.E.; Salem, A.K.; Weiner, G.J. Three steps to breaking immune tolerance to lymphoma: A microparticle approach. Cancer Immunol. Res. 2015, 3, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Liu, P.; Chen, B.; Mao, Y.; Engelmann, H.; Shin, Y.; Jaffar, J.; Hellstrom, I.; Liu, J.; Hellstrom, K.E. Local release of highly loaded antibodies from functionalized nanoporous support for cancer immunotherapy. J. Am. Chem. Soc. 2010, 132, 6906–6907. [Google Scholar] [CrossRef] [PubMed]
- Roeven, M.W.; Hobo, W.; van der Voort, R.; Fredrix, H.; Norde, W.J.; Teijgeler, K.; Ruiters, M.H.; Schaap, N.; Dolstra, H. Efficient nontoxic delivery of PD-L1 and PD-L2 siRNA into dendritic cell vaccines using the cationic lipid SAINT-18. J. Immunother. 2015, 38, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Teo, P.Y.; Yang, C.; Whilding, L.M.; Parente-Pereira, A.C.; Maher, J.; George, A.J.; Hedrick, J.L.; Yang, Y.Y.; Ghaem-Maghami, S. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: Strategies to enhance T cell killing. Adv. Healthc. Mater. 2015, 4, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.E.; Kovacsovics-Bankowski, M.; Weinberg, A.D. The TNFRs OX40, 4–1BB, and CD40 as targets for cancer immunotherapy. Curr. Opin. Immunol. 2013, 25, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Gong, W.; Demirci, G.; Liu, W.; Spoerl, S.; Chu, X.; Bishop, D.K.; Turka, L.A.; Li, X.C. New insights on OX40 in the control of T cell immunity and immune tolerance in vivo. J. Immunol. 2012, 188, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Park, Y.; Song, A.; Cheroutre, H.; Kwon, B.S.; Croft, M. Functional dichotomy between OX40 and 4–1BB in modulating effector CD8 T cell responses. J. Immunol. 2006, 177, 4464–4472. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, A.T.; Mufti, G.J.; Guinn, B.A. Role of 4-1BB:4-1BB ligand in cancer immunotherapy. Cancer Gene Ther. 2004, 11, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ouyang, H.; Zhou, S.; Li, J.; Ye, Y. PLGA-nanoparticle mediated delivery of anti-OX40 monoclonal antibody enhances anti-tumor cytotoxic T cell responses. Cellular Immunol. 2014, 287, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Kwong, B.; Gai, S.A.; Elkhader, J.; Wittrup, K.D.; Irvine, D.J. Localized immunotherapy via liposome-anchored Anti-CD137+ IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 2013, 73, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Res. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Moon, J.J.; Um, S.H.; Bershteyn, A.; Irvine, D.J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 2010, 16, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Stephan, S.B.; Bak, P.; Chen, J.; Irvine, D.J. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials 2012, 33, 5776–5787. [Google Scholar] [CrossRef] [PubMed]
- Stephan, S.B.; Taber, A.M.; Jileaeva, I.; Pegues, E.P.; Sentman, C.L.; Stephan, M.T. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 2015, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Abraham, W.D.; Zheng, Y.; Bustamante Lopez, S.C.; Luo, S.S.; Irvine, D.J. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Trans. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Lower, M.; Diekmann, J.; Boegel, S.; Schrors, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Moon, J.J. Nanoparticle Drug Delivery Systems Designed to Improve Cancer Vaccines and Immunotherapy. Vaccines 2015, 3, 662-685. https://doi.org/10.3390/vaccines3030662
Fan Y, Moon JJ. Nanoparticle Drug Delivery Systems Designed to Improve Cancer Vaccines and Immunotherapy. Vaccines. 2015; 3(3):662-685. https://doi.org/10.3390/vaccines3030662
Chicago/Turabian StyleFan, Yuchen, and James J. Moon. 2015. "Nanoparticle Drug Delivery Systems Designed to Improve Cancer Vaccines and Immunotherapy" Vaccines 3, no. 3: 662-685. https://doi.org/10.3390/vaccines3030662