
Comparison of Current Regulatory Status for Gene-Based Vaccines in the U.S., Europe and Japan

Abstract
:1. Introduction
2. Experimental Section
2.1. Analysis on Clinical Trials of Gene-Based Prophylactic Vaccines
2.2. Comparison of the Current Regulatory Context for Gene Therapy and Vaccines
3. Results
3.1. Analysis of Clinical Trials of Gene-Based Prophylactic Vaccines
3.1.1. Number of Clinical Trials Registered per Year and Their Phases
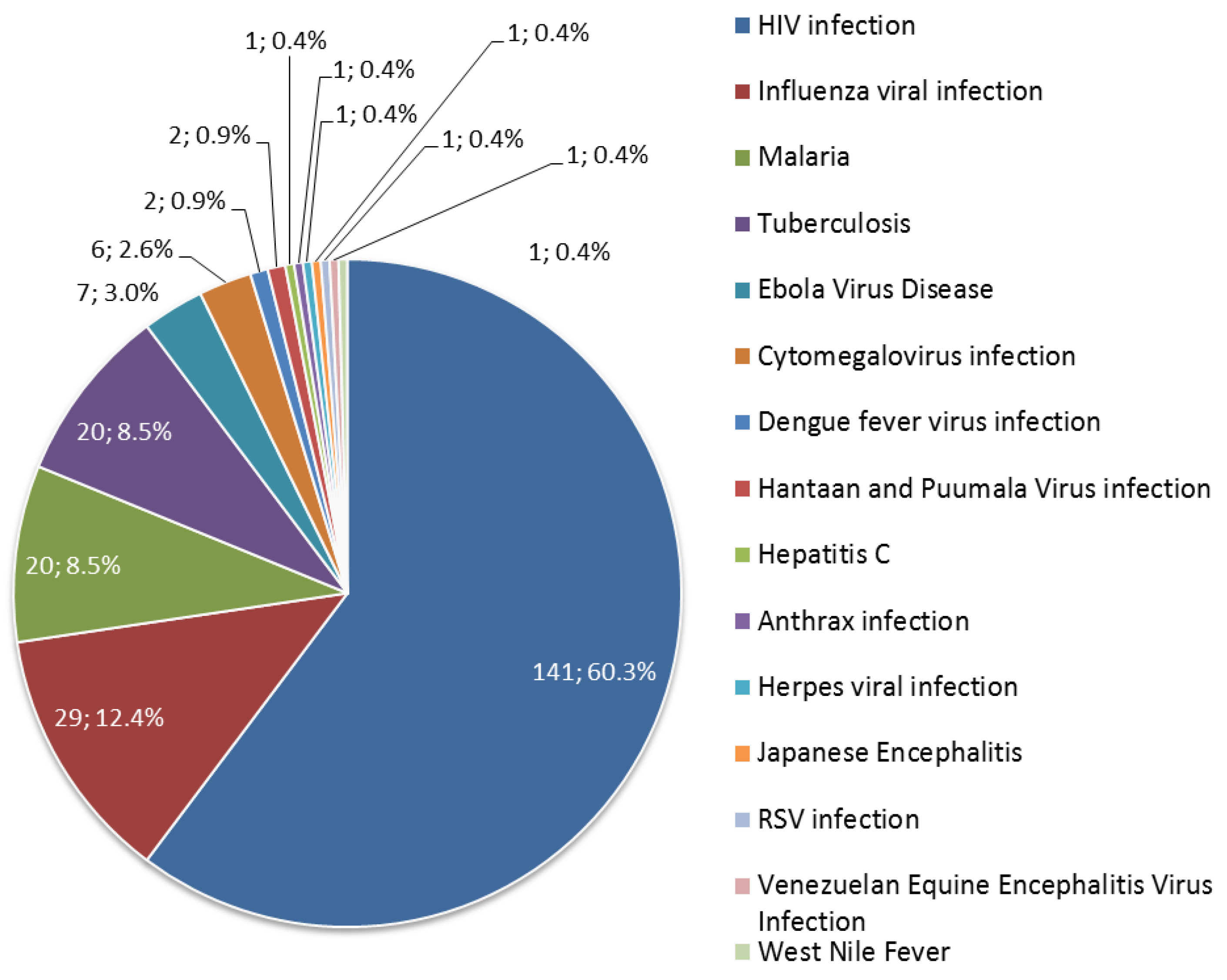
3.1.2. Diseases Targeted by Prophylactic Gene-Based Vaccines


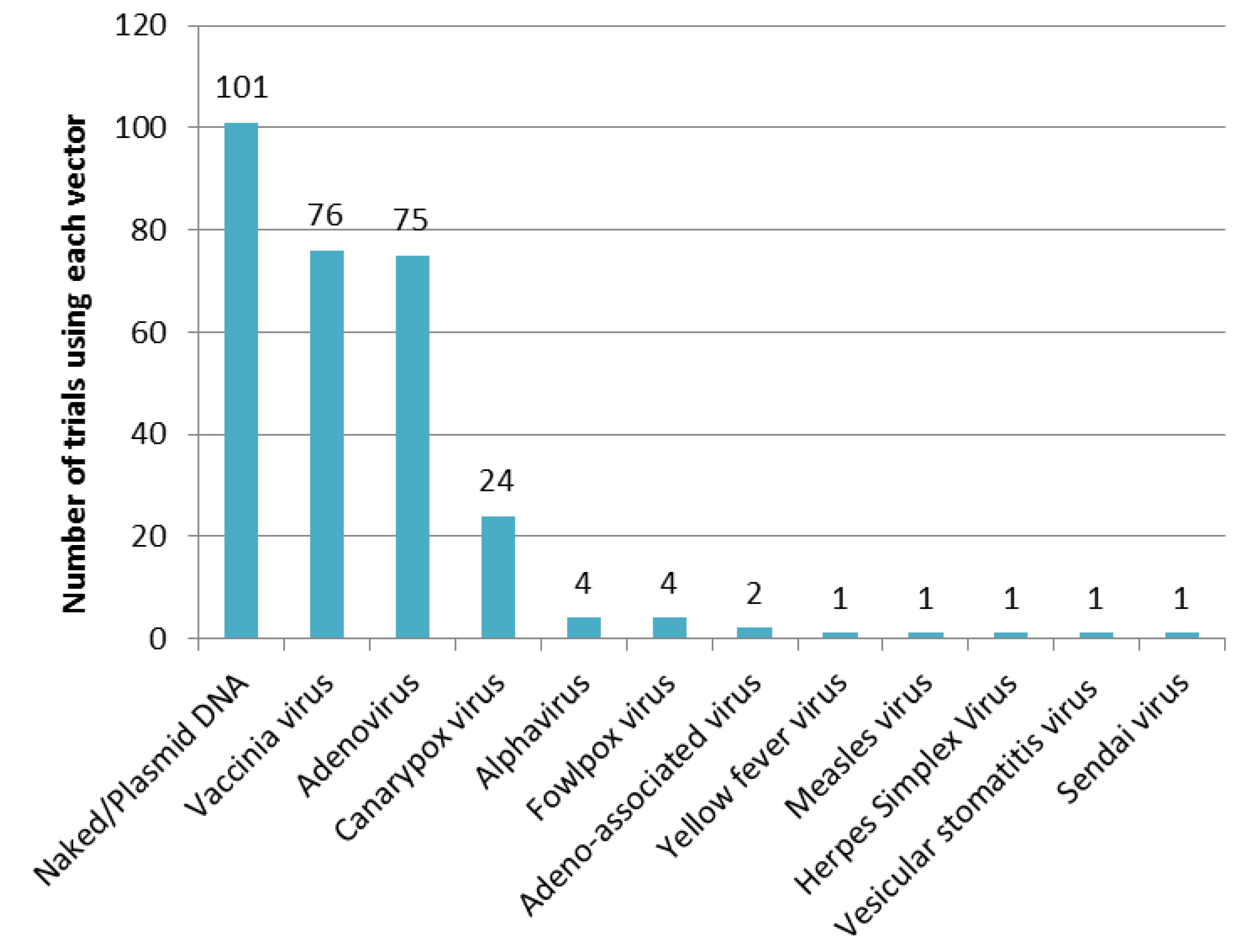
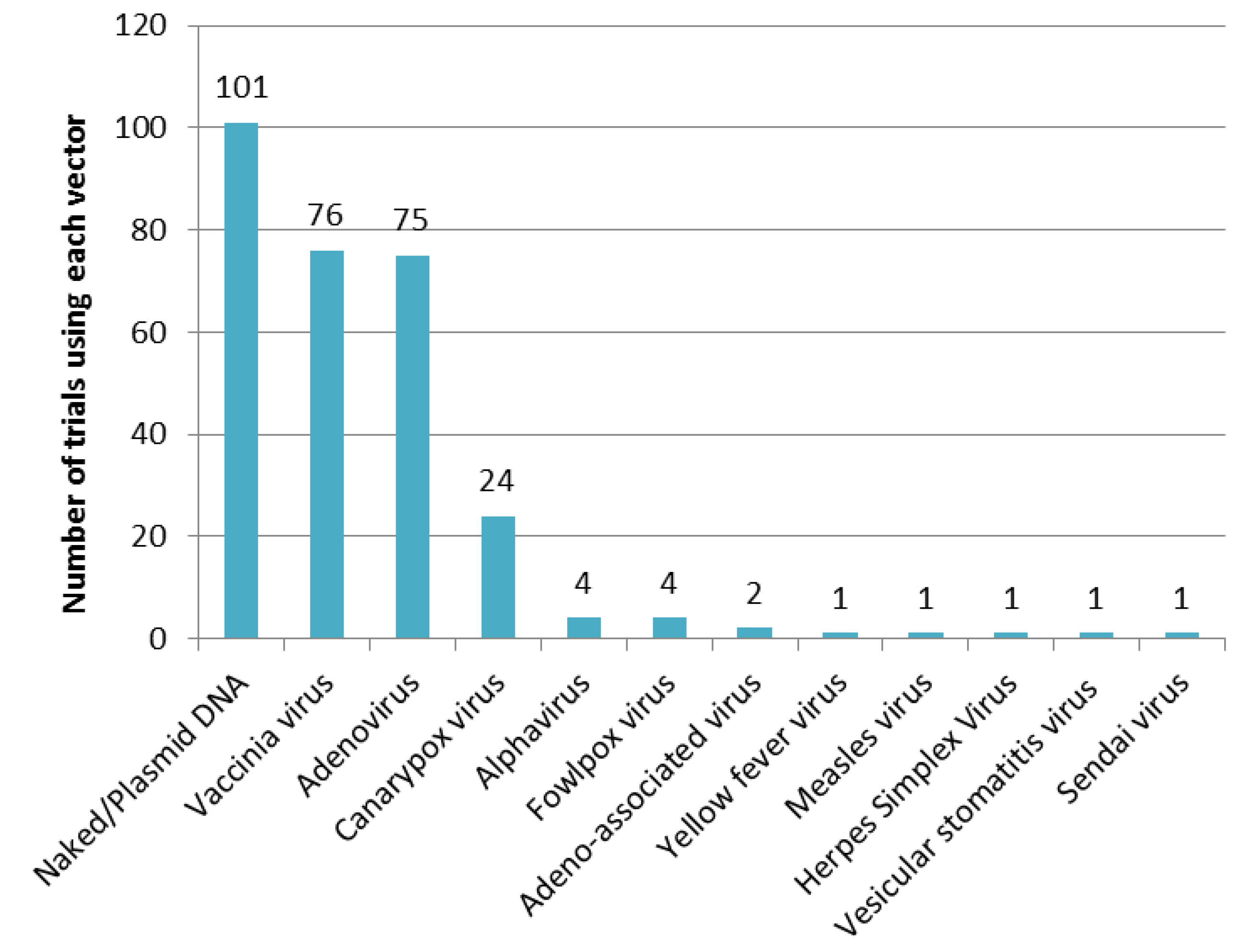
3.1.3. Vectors Used in Prophylactic Gene-Based Vaccines
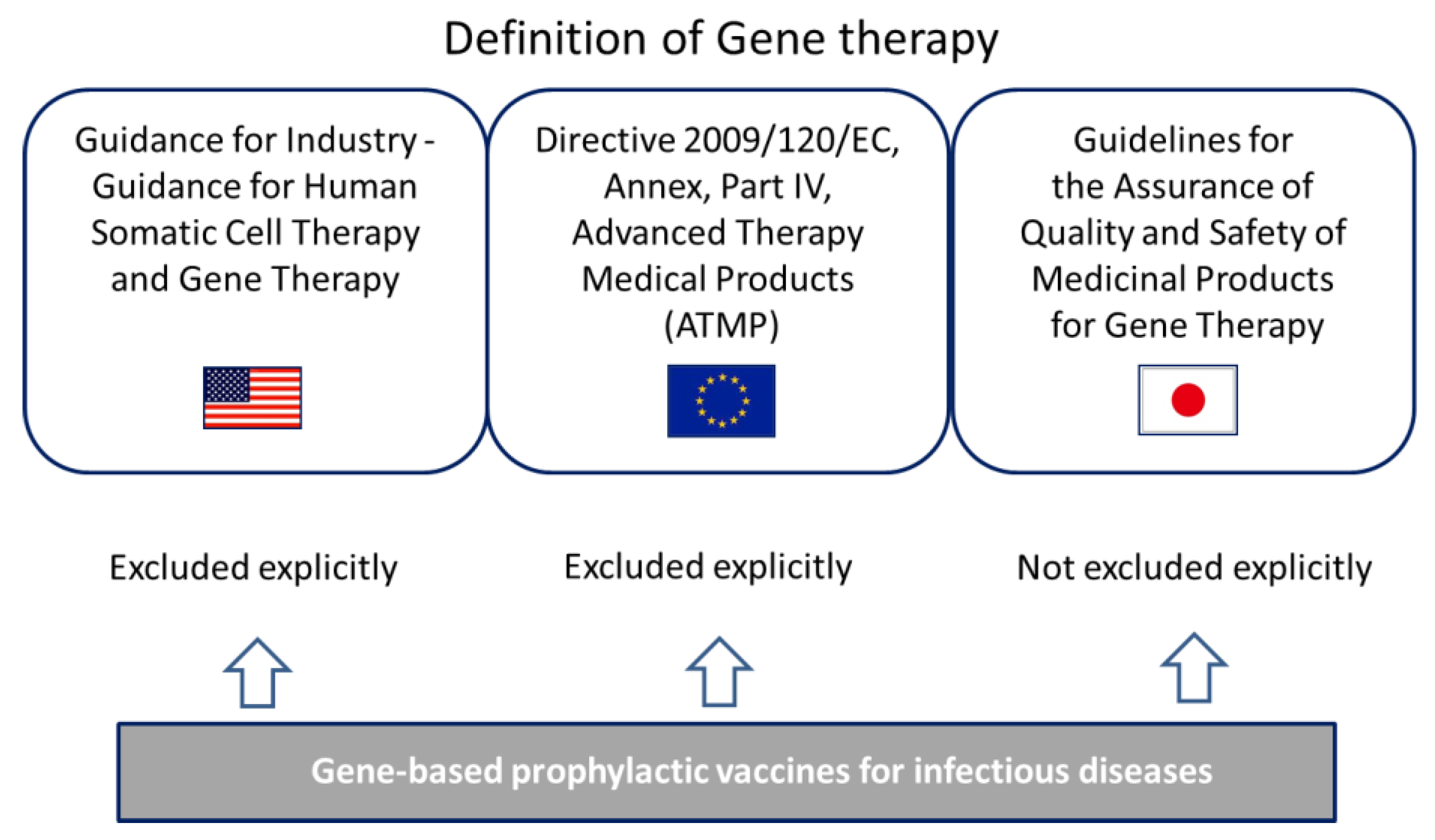
3.2. Definition of Gene Therapy in the U.S., Europe and Japan
3.2.1. Definition in the U.S.
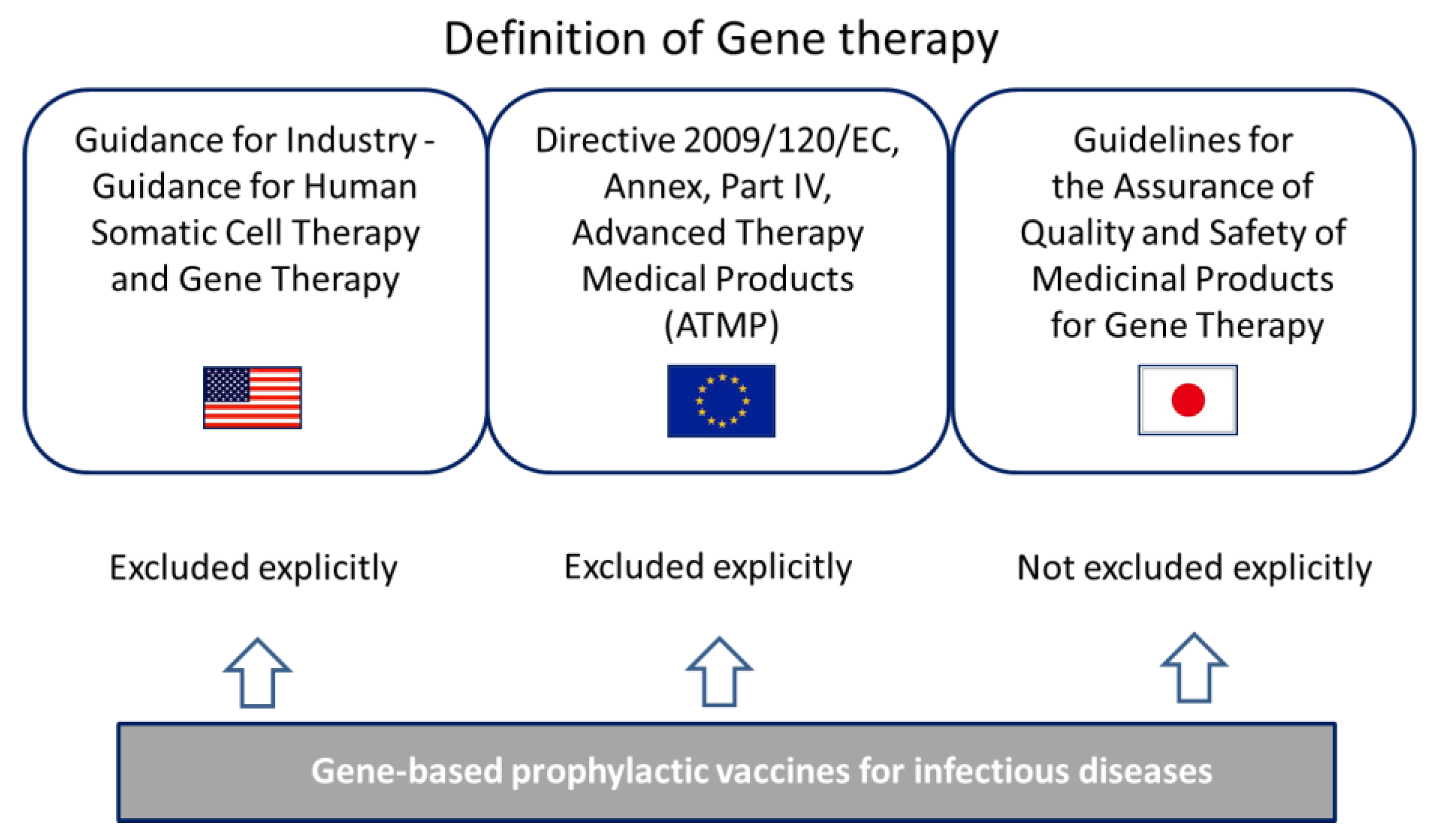
3.2.2. Definition in the Europe
3.2.3. Definition in Japan
3.3. Current Status of Guidelines Specific for the Development of Plasmid DNA Vaccines and Viral-Vectored Vaccines in the U.S., Europe and Japan
3.3.1. Vaccine-Related Guidance in the U.S.
- -
- studies of immunogenicity
- -
- studies of unintended adverse consequences come from immunomodulatory genes in DNA vaccine
- -
- prime-boost strategies
- -
- assessment whether vaccination causes autoimmune disease
- -
- local reactogenicity and systemic toxicity studies
- -
- biodistribution, persistence, and integration analysis
3.3.2. Vaccine-Related Guideline in Europe
- -
- General considerations
- -
- Pharmacodynamic studies (protection and immunogenicity), Pre-existing immunity
- -
- Non-clinical safety studies (toxicity testing)
- -
- Single and repeated dose toxicity
- -
- Distribution studies
- -
- Reproduction and developmental toxicity studies
- -
- Local tolerance
3.3.3. Vaccine-Related Guideline in Japan
- -
- Possibility of the proliferative viral expression
- -
- Cellular cytotoxicity
- -
- Genetic integration with chromosomes
- -
- Safety caused by abnormal expression of expression product
- -
- Carcinogenicity
- -
- Immunogenicity
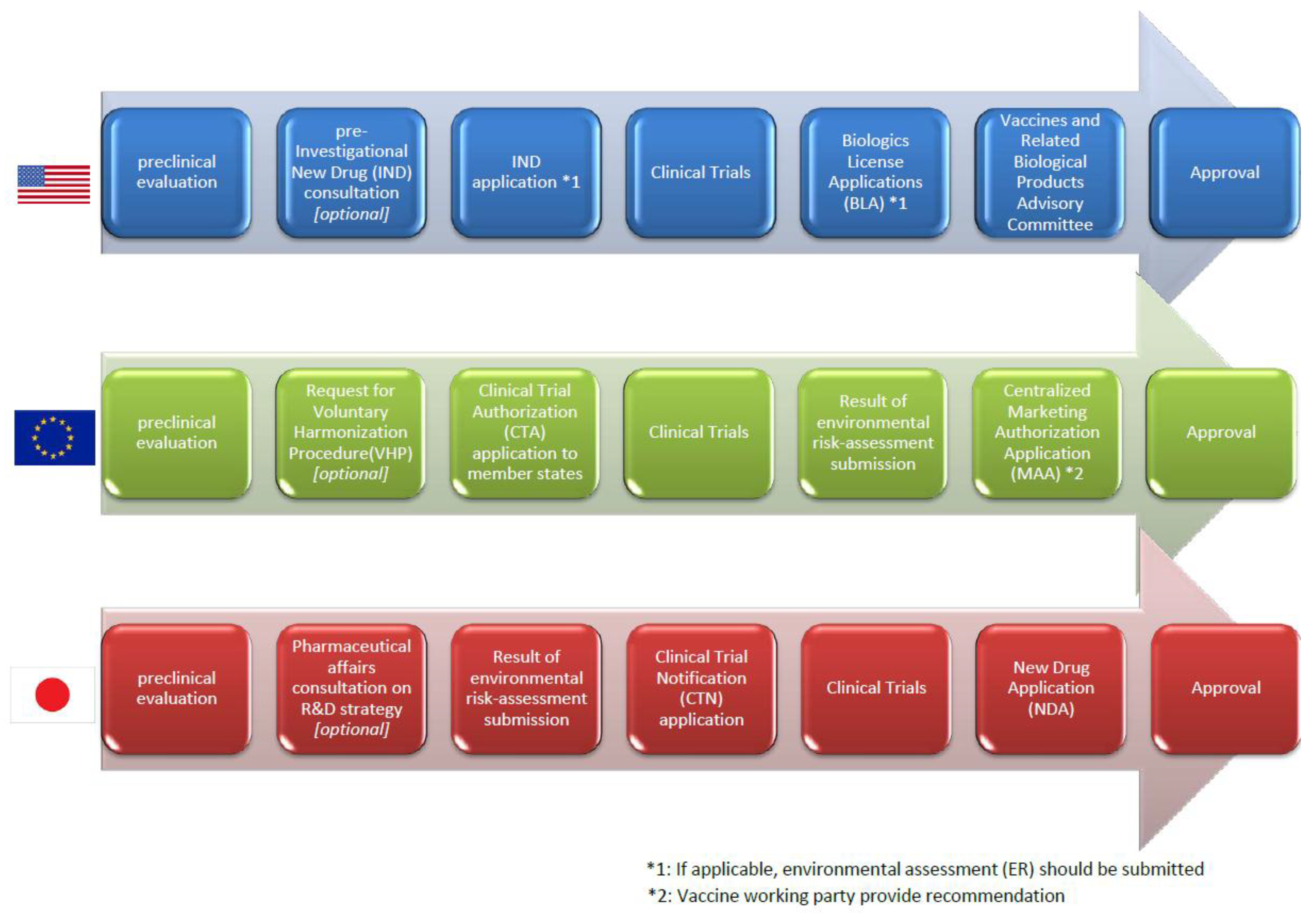
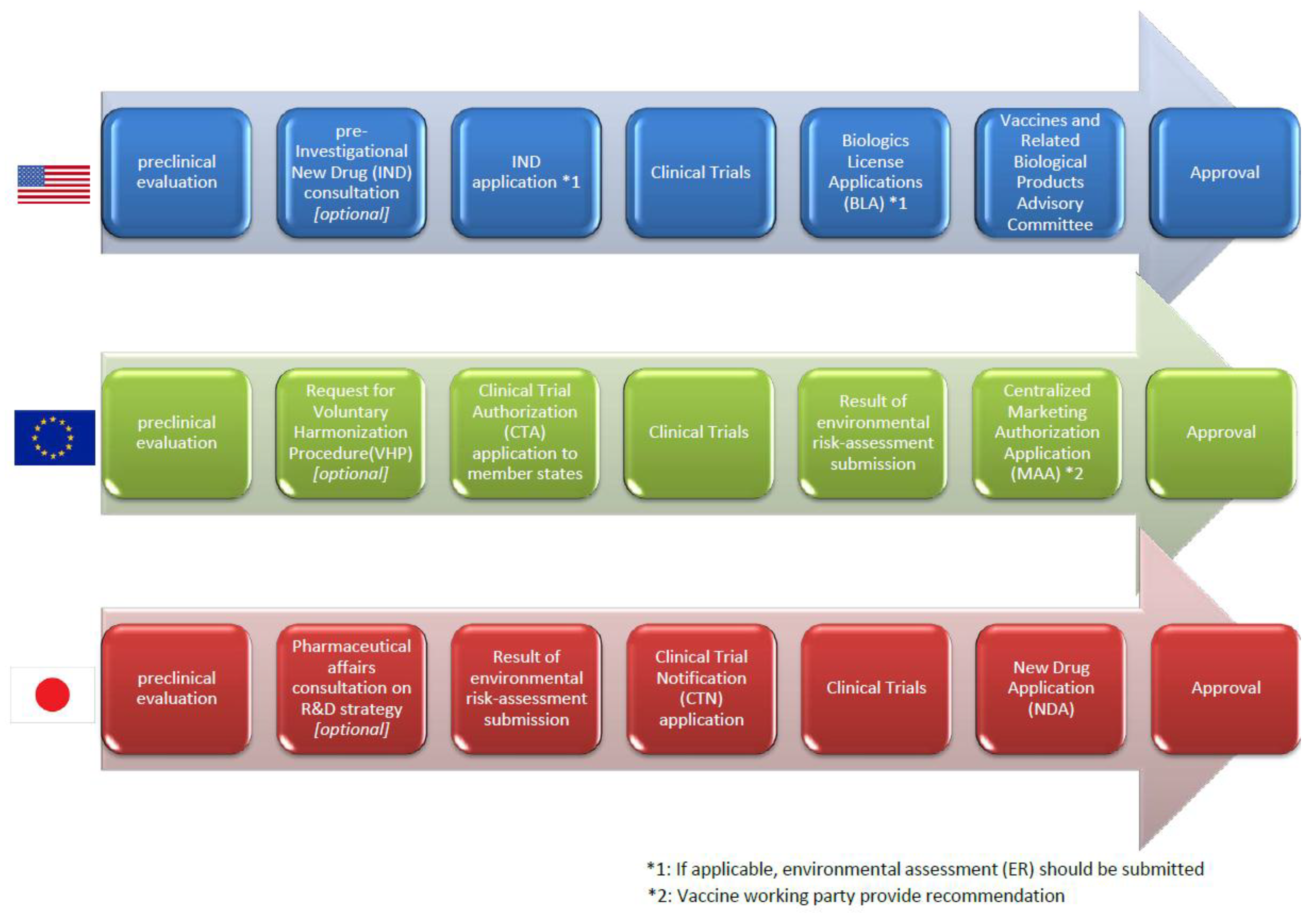
3.4. Regulatory Pathway from Clinical Trials to the Marketing Authorization in Each Region

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulatory agency | Conventional Vaccines * | Plasmid DNA Vaccines-Specific Guidelines | Viral-Vectored Vaccine Specific Guidelines |
|---|---|---|---|
| FDA (U.S.) | None | Considerations for plasmid DNA vaccines for infectious disease indications (2007) | Characterization and qualification of cell substrates and other biological materials used in the production of viral vaccines for infectious disease indications (2010) |
| Contents: Quality and Preclinical considerations | |||
| Contents: Quality c onsiderations | |||
| EMA (EU) | Note for guidance on preclinical pharmacological and toxicological testing of vaccines (1995) | None | Guideline on quality, non-clinical and clinical aspects of live recombinant viral vectored vaccines (2010) |
| Applicable for viral-vectored vaccine but DNA vaccine is out of scope | Contents: Quality, Preclinical and Clinical considerations | ||
| MHLW (Japan) | Guideline for non-clinical studies of vaccines for preventing infectious diseases (2010) | None | None |
| Plasmid DNA vaccine and viral-vectored vaccine are out of scope | |||
| WHO | WHO guideline on nonclinical evaluation of vaccines (2003) | Guidelines for assuring the quality and preclinical safety evaluation of DNA vaccines. WHO technical report series No 941, (2007) | None |
| Applicable for plasmid DNA vaccine and viral-vectored vaccine | Contents: Quality and preclinical considerations |
3.5. Assessment of the Environmental Impact of Medicinal Products
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
References and Notes
- Delany, I.; Rappuoli, R.; Gregorio, E.D. Vaccines for the 21st century. EMBO Mol. Med. 2014, 6, 708–720. [Google Scholar] [PubMed]
- Whalen, R.G. DNA Vaccines for emerging infectious diseases: What If? Emerg. Infect. Dis. 1996, 2, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Wada, Y.; Shimada, M. Recent developments in preclinical DNA vaccination. Vaccines 2014, 2, 89–106. [Google Scholar] [CrossRef]
- Wahren, B.; Liu, M.A. DNA vaccines: Recent developments and the future. Vaccines 2014, 2, 785–796. [Google Scholar] [CrossRef]
- Rollier, C.S.; Reyes-Sandoval, A.; Cottingham, M.G.; Ewer, Katie; Hill, A.V. Viral vectors as vaccine platforms: Deployment in sight. Curr. Opin. Immunol. 2011, 23, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Okuda, K.; Shimada, M. Developments in viral vector-based vaccines. Vaccines 2014, 2, 624–641. [Google Scholar] [CrossRef]
- WHO, Safety profile of Japanese encephalitis (JE) chimeric vaccine. Available online: http://www.who.int/vaccine_safety/committee/topics/japanese_encephalitis/Dec_2013/en/ (accessed on 29 November 2014).
- Gin, S.L.; Alexander, I.E.; Edelstein, M.L.; Abedi, M.R.; Wixon, J. Gene therapy clinical trials worldwide to 2012–An update. J. Gene Med. 2013, 15, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Gruber, M.; Matsumoto, M. Overview of global regulatory toxicology requirements for vaccines and adjuvants. J. Pharm. Tox. Methods 2012, 65, 49–57. [Google Scholar] [CrossRef]
- ClinicalTrial.gov. A Study to Evaluate a Therapeutic Vaccine, ASP0113, in Cytomegalovirus (CMV)-Seropositive Recipients Undergoing Allogeneic, Hematopoietic Cell Transplant (HCT) (HELIOS). Available online: http://clinicaltrials.gov/show/NCT01877655 (accessed on 29 November 2014).
- FDA: Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy. Available online: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/ucm081670.pdf (accessed on 29 November 2014).
- EU, Regulations, Directives and other acts. Available online: http://europa.eu/eu-law/decision-making/legal-acts/index_en.htm (accessed on 29 November 2014).
- EMA, Scientific guidelines. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000043.jsp (accessed on 29 November 2014).
- DIRECTIVE 2009/120/EC. Available online: http://ec.europa.eu/health/files/eudralex/vol-1/dir_2009_120/dir_2009_120_en.pdf (accessed on 29 November 2014).
- Directive 2001/83/EC. Available online: http://ec.europa.eu/health/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf (accessed on 10 February 2015).
- MHLW: Guidelines regarding the assurance of quality and safety of drugs for gene therapy (in Japanese). Available online: http://www.pmda.go.jp/operations/shonin/info/idenshi-chiryou/pdf/H250701_0000004.pdf (accessed on 29 November 2014).
- List of clinical research in Japan (in Japanese). Available online: http://www.nihs.go.jp/cgtp/cgtp/sec1/gt_prtcl/20120625_giji.pdf (accessed on 29 November 2014).
- WHO Guidelines on nonclinical evaluation of vaccines. Available online: http://www.who.int/biologicals/publications/nonclinical_evaluation_vaccines_nov_2003.pdf (accessed on 29 November 2014).
- FDA: Guidance for Industry: Considerations for Plasmid DNA Vaccines for Infectious Disease Indications. Available online: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/ucm091968.pdf (accessed on 29 November 2014).
- FDA: Points to Consider on Plasmid DNA Vaccines for Preventive Infectious Disease Indications. Available online: https://www.pharmamedtechbi.com/~/media/Images/Publications/Archive/The%20Pink%20Sheet%20Daily/2004/10/13/14041013011/041018_plasmid_dna_guidance.pdf (accessed on 29 November 2014).
- Klinman, D.M.; Klaschik, S.; Tross, D.; Shirota, H.; Steinhagen, F. FDA guidance on prophylactic DNA vaccines: Analysis and recommendations. Vaccine 2014, 28, 2801–2805. [Google Scholar] [CrossRef]
- Smith, H.A.; Klinman, D.M. The regulation of DNA vaccines. Curr. Opin. Biotechnol. 2001, 12, 299–303. [Google Scholar] [CrossRef] [PubMed]
- FDA: Guidance for Industry: Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease. Available online: http://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/vaccines/ucm202439.pdf (accessed on 29 November 2014).
- EMA: Note for guidance on preclinical pharmacological and toxicological testing of vaccines (CPMP/SWP/465/95). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500004004.pdf (accessed on 29 November 2014).
- EMA: Note for guidance on the quality, preclinical and clinical aspects of gene transfer medical products (CPMP/BWP/3088/99). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500003987.pdf (accessed on 29 November 2014).
- EMEA: Guideline of the non-clinical studies required before first clinical use of gene therapy medical products (EMEA/CHMP/GTWP/125459//2006). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003942.pdf (accessed on 29 November 2014).
- EMA: Guideline on Quality, Non-clinical and Clinical Aspects of Live Recombinant Viral Vectored Vaccines (EMA/CHMP/VWP/141697/2009). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/08/WC500095721.pdf (accessed on 29 November 2014).
- EMA: Concept paper on guidance for DNA vaccines (EMEA/CHMP/308136//2007). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/03/WC500124898.pdf (accessed on 2 March 2015).
- MHLW: Guideline for Non-clinical Studies of Preventive Vaccines for Infectious Diseases (in Japanese). Available online: http://www.pmda.go.jp/operations/notice/2010/file/20100527-04.pdf (accessed on 10 February 2015).
- MHLW: Guideline for Clinical Studies of Preventive Vaccines for Infectious Diseases (in Japanese). Available online: https://www.nibio.go.jp/news/data/100601_1.pdf (accessed on 10 February 2015).
- ICH: Guidance for Industry, S6 Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm074957.pdf (accessed on 29 November 2014).
- FDA: IND Process and Review Procedures (Including Clinical Holds), MAPP 6030.1. Available online: http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/StaffPoliciesandProcedures/ucm082022.pdf (accessed on 10 February 2015).
- FDA: Pre-IND Consultation Program. Available online: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/InvestigationalNewDrugINDApplication/Overview/default.htm (accessed on 10 February 2015).
- FDA: Vaccine Product Approval Process. Available online: http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/BiologicsLicenseApplicationsBLAProcess/ucm133096.htm (accessed on 10 February 2015).
- HMAs Clinical Trials Facilitation Group, Guidance document for sponsors for a Voluntary Har-monisation Procedure (VHP) for the assessment of mul-tinational Clinical Trial Applications (CTFG//VHP/2013/Rev1). Available online: http://www.hma.eu/fileadmin/dateien/Human_Medicines/01-About_HMA/Working_Groups/CTFG/2013_06_CTFG_VHP.pdf (accessed on 10 February 2015).
- Schneider, C.K.; Schäffner-Dallmann, G. Typical pitfalls in applications for marketing authorization of biotechnological products in Europe. Nat. Rev. Drug Discov. 2008, 7, 893–899. [Google Scholar] [CrossRef] [PubMed]
- PMDA: Information in English of Japanese Regulatory Affairs. Available online: http://www.jpma.or.jp/english/parj/pdf/2014.pdf (accessed on 10 February 2015).
- DIRECTIVE 2001/18/EC. Available online: http://www.biosafety.be/PDF/2001_18.pdf (accessed on 10 February 2015).
- Regulation (EC) 726/2004. Available online: http://www.biosafety.be/EMEA/726_2004_EN.pdf (accessed on 10 February 2015).
- EMEA: Environmental risk assessments for medicinal products containing, or consisting of, genetically modified organisms (GMOs) (EMEA/CHMP/BWP/135148/2004). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003806.pdf (accessed on 10 February 2015).
- FDA: Draft Guidance for Industry: Determining the Need for and Content of Environmental Assessments for Gene Therapies, Vectored Vaccines, and Related Recombinant Viral or Microbial Products. Available online: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM402063.pdf (accessed on 10 February 2015).
- Act on the Conservation and Sustainable Use of Biological Diversity through Regulations on the Use of Living Modified Organisms. Available online: http://www.bch.biodic.go.jp/download/en_law/en_law_ver_2.doc (accessed on 10 February 2015).
- The Guidance of Implementation of Assessment of Adverse Effect on Biological Diversity of Type 1 Use of Living Modified Organisms. Available online: http://www.bch.biodic.go.jp/download/en_law/en_assessment_guidence.doc (accessed on 10 February 2015).
- ICH: Guidance on nonclinical safety studies for the conduct of human clinical trial and marketing authorization for pharmaceuticals M3 (R2). Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf (accessed on 29 November 2014).
- McElrath, M.J.; Rosa, S.C.D.; Moodie, Z.; Dubey, S.; Kierstead, L.; Janes, H.; Defawe, O.D.; Carter, D.K.; Hural, J.; Akondy, R.; et al. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: A case–cohort analysis. Lancet 2008, 372, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, Y.; Aruga, A. Comparison of Current Regulatory Status for Gene-Based Vaccines in the U.S., Europe and Japan. Vaccines 2015, 3, 186-202. https://doi.org/10.3390/vaccines3010186
Nakayama Y, Aruga A. Comparison of Current Regulatory Status for Gene-Based Vaccines in the U.S., Europe and Japan. Vaccines. 2015; 3(1):186-202. https://doi.org/10.3390/vaccines3010186
Chicago/Turabian StyleNakayama, Yoshikazu, and Atsushi Aruga. 2015. "Comparison of Current Regulatory Status for Gene-Based Vaccines in the U.S., Europe and Japan" Vaccines 3, no. 1: 186-202. https://doi.org/10.3390/vaccines3010186