Immune System Regulation in the Induction of Broadly Neutralizing HIV-1 Antibodies

{kind=link}
Abstract
:1. Introduction
2. HIV-1 Envelope Antigenicity and Immunogenicity
3. Designing New Vaccine Strategies
3.1. HIV-1 Broadly Neutralizing Antibodies
3.2. Wolves in Sheep’s Clothing
3.3. Physiological B-Cell Tolerance
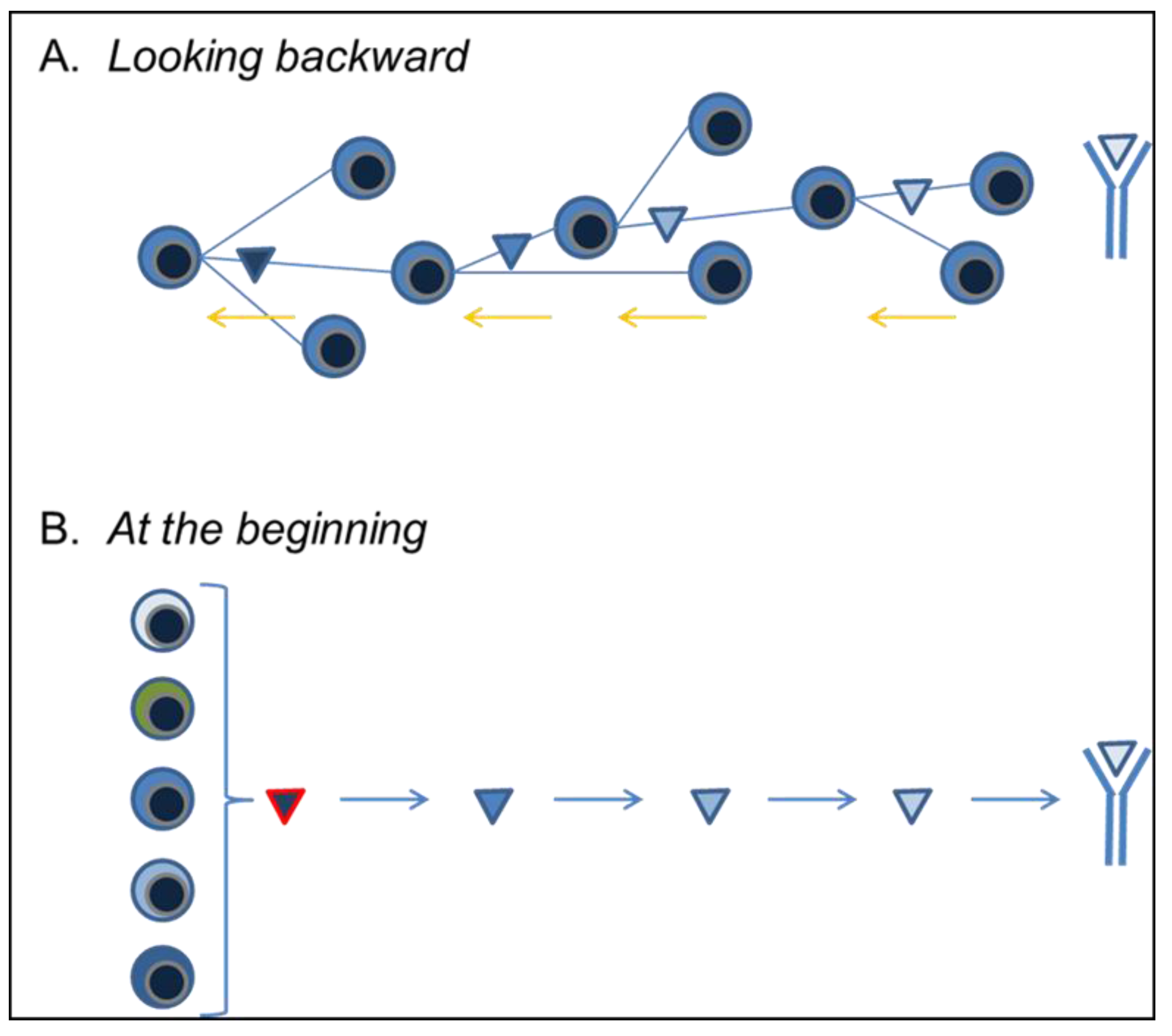
3.4. Looking Backward
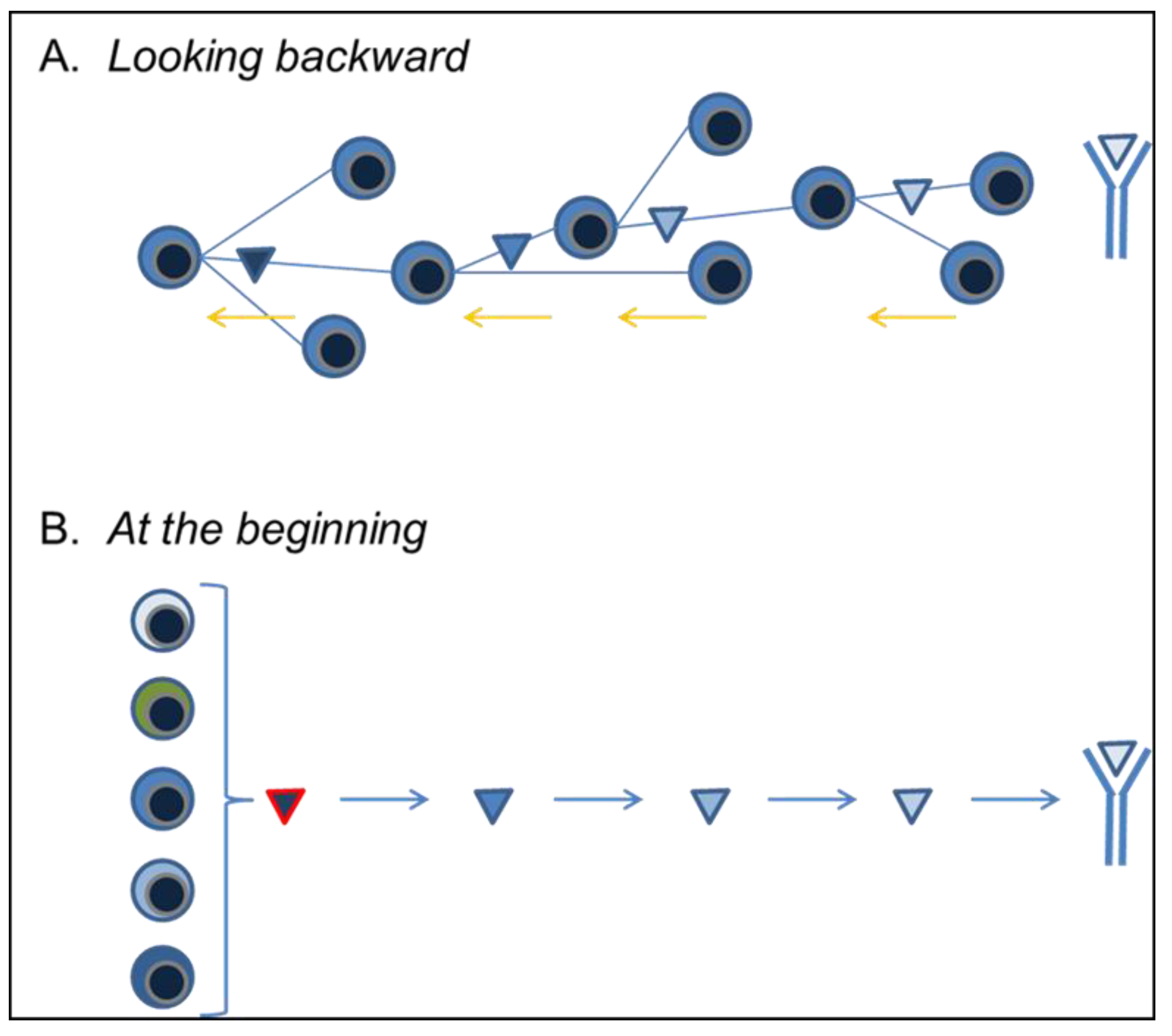
3.5. At the Beginning
4. Moving Forward
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mascola, J.R.; Haynes, B.F. HIV-1 neutralizing antibodies: Understanding nature’s pathways. Immunol. Rev. 2013, 254, 225–244. [Google Scholar] [CrossRef]
- Haynes, B.F.; Kelsoe, G.; Harrison, S.C.; Kepler, T.B. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat. Biotechnol. 2012, 30, 423–433. [Google Scholar] [CrossRef]
- Yang, G.; Holl, T.M.; Liu, Y.; Li, Y.; Lu, X.; Nicely, N.I.; Kepler, T.B.; Alam, S.M.; Liao, H.X.; Cain, D.W.; et al. Identification of autoantigens recognized by the 2F5 and 4E10 broadly neutralizing HIV-1 antibodies. J. Exp. Med. 2013, 210, 241–256. [Google Scholar] [CrossRef]
- Gorny, M.K.; Stamatatos, L.; Volsky, B.; Revesz, K.; Williams, C.; Wang, X.H.; Cohen, S.; Staudinger, R.; Zolla-Pazner, S. Identification of a new quaternary neutralizing epitope on human immunodeficiency virus type 1 virus particles. J. Virol. 2005, 79, 5232–5237. [Google Scholar] [CrossRef]
- Hicar, M.D.; Kalams, S.A.; Spearman, P.W.; Crowe, J.E., Jr. Emerging studies of human HIV-specific antibody repertoires. Vaccine 2010, 28, B18–B23. [Google Scholar] [CrossRef]
- Briney, B.S.; Willis, J.R.; Crowe, J.E., Jr. Human peripheral blood antibodies with long HCDR3s are established primarily at original recombination using a limited subset of germline genes. PLoS One 2012, 7, e36750. [Google Scholar] [CrossRef]
- Wyatt, R.; Kwong, P.D.; Desjardins, E.; Sweet, R.W.; Robinson, J.; Hendrickson, W.A.; Sodroski, J.G. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 1998, 393, 705–711. [Google Scholar] [CrossRef]
- Kwong, P.D.; Doyle, M.L.; Casper, D.J.; Cicala, C.; Leavitt, S.A.; Majeed, S.; Steenbeke, T.D.; Venturi, M.; Chaiken, I.; Fung, M.; et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 2002, 420, 678–682. [Google Scholar] [CrossRef]
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312. [Google Scholar] [CrossRef]
- Van Duin, D.; Medzhitov, R.; Shaw, A.C. Triggering TLR signaling in vaccination. Trends Immunol. 2006, 27, 49–55. [Google Scholar] [CrossRef]
- Palm, N.W.; Medzhitov, R. Immunostimulatory activity of haptenated proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 4782–4787. [Google Scholar] [CrossRef]
- Palm, N.W.; Medzhitov, R. Pattern recognition receptors and control of adaptive immunity. Immunol. Rev. 2009, 227, 221–233. [Google Scholar] [CrossRef]
- Ofek, G.; Guenaga, F.J.; Schief, W.R.; Skinner, J.; Baker, D.; Wyatt, R.; Kwong, P.D. Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA 2010, 107, 17880–17887. [Google Scholar] [CrossRef]
- Azoitei, M.L.; Correia, B.E.; Ban, Y.E.; Carrico, C.; Kalyuzhniy, O.; Chen, L.; Schroeter, A.; Huang, P.S.; McLellan, J.S.; Kwong, P.D.; et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science 2011, 334, 373–376. [Google Scholar] [CrossRef]
- Guenaga, J.; Dosenovic, P.; Ofek, G.; Baker, D.; Schief, W.R.; Kwong, P.D.; Karlsson Hedestam, G.B.; Wyatt, R.T. Heterologous epitope-scaffold prime:boosting immuno-focuses B cell responses to the HIV-1 gp41 2F5 neutralization determinant. PLoS One 2011, 6, e16074. [Google Scholar]
- Zolla-Pazner, S.; Kong, X.P.; Jiang, X.; Cardozo, T.; Nadas, A.; Cohen, S.; Totrov, M.; Seaman, M.S.; Wang, S.; Lu, S. Cross-clade HIV-1 neutralizing antibodies induced with V3-scaffold protein immunogens following priming with gp120 DNA. J. Virol. 2011, 85, 9887–9898. [Google Scholar] [CrossRef]
- Jardine, J.; Julien, J.P.; Menis, S.; Ota, T.; Kalyuzhniy, O.; McGuire, A.; Sok, D.; Huang, P.S.; MacPherson, S.; Jones, M.; et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 2013, 340, 711–716. [Google Scholar] [CrossRef]
- Schiffner, T.; Kong, L.; Duncan, C.J.; Back, J.W.; Benschop, J.J.; Shen, X.; Huang, P.S.; Stewart-Jones, G.B.; Destefano, J.; Seaman, M.S.; et al. Immune focusing and enhanced neutralization induced by HIV-1 gp140 chemical cross-linking. J. Virol. 2013, 87, 10163–10172. [Google Scholar] [CrossRef]
- McGuire, A.T.; Hoot, S.; Dreyer, A.M.; Lippy, A.; Stuart, A.; Cohen, K.W.; Jardine, J.; Menis, S.; Scheid, J.F.; West, A.P.; et al. Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J. Exp. Med. 2013, 210, 655–663. [Google Scholar] [CrossRef]
- Dennison, S.M.; Sutherland, L.L.; Jaeger, F.; Anasti, K.; Parks, R.; Stewart, S.M.; Bowman, C.; Xia, S.; Zhang, R.; Shen, X.; et al. Induction of antibodies in rhesus macaques that recognize a fusion-intermediate conformation of HIV-1 gp41. PLoS One 2011, 6, e27824. [Google Scholar] [CrossRef]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef]
- Liu, M.; Yang, G.; Cain, D.; Wiehe, K.; Nicely, N.; Gao, J.; Haynes, B.F.; Conners, M.J.; Mascola, J.R.; Bjorkman, P.; et al. Duke University School of Medicine: Durham, NC, USA, 2013; Unpublished work.
- Verkoczy, L.; Diaz, M.; Holl, T.M.; Ouyang, Y.B.; Bouton-Verville, H.; Alam, S.M.; Liao, H.X.; Kelsoe, G.; Haynes, B.F. Autoreactivity in an HIV-1 broadly reactive neutralizing antibody variable region heavy chain induces immunologic tolerance. Proc. Natl. Acad. Sci. USA 2010, 107, 181–186. [Google Scholar] [CrossRef]
- Verkoczy, L.; Chen, Y.; Bouton-Verville, H.; Zhang, J.; Diaz, M.; Hutchinson, J.; Ouyang, Y.B.; Alam, S.M.; Holl, T.M.; Hwang, K.K.; et al. Rescue of HIV-1 broad neutralizing antibody-expressing B cells in 2F5 VH × VL knockin mice reveals multiple tolerance controls. J. Immunol. 2011, 187, 3785–3797. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Hwang, K.K.; Bouton-Verville, H.; Xia, S.M.; Newman, A.; Ouyang, Y.B.; Haynes, B.F.; Verkoczy, L. Common tolerance mechanisms, but distinct cross-reactivities associated with gp41 and lipids, limit production of HIV-1 broad neutralizing antibodies 2F5 and 4E10. J. Immunol. 2013, 191, 1260–1275. [Google Scholar] [CrossRef]
- Alam, S.M.; Morelli, M.; Dennison, S.M.; Liao, H.X.; Zhang, R.; Xia, S.M.; Rits-Volloch, S.; Sun, L.; Harrison, S.C.; Haynes, B.F.; et al. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2009, 106, 20234–20239. [Google Scholar] [CrossRef]
- Haynes, B.F.; Fleming, J.; St Clair, E.W.; Katinger, H.; Stiegler, G.; Kunert, R.; Robinson, J.; Scearce, R.M.; Plonk, K.; Staats, H.F.; et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 2005, 308, 1906–1908. [Google Scholar] [CrossRef]
- Verkoczy, L.; Chen, Y.; Zhang, J.; Bouton-Verville, H.; Newman, A.; Lockwood, B.; Scearce, R.M.; Montefiori, D.C.; Dennison, S.M.; Xia, S.M.; et al. Induction of HIV-1 broad neutralizing antibodies in 2F5 knock-in mice: Selection against membrane proximal external region-associated autoreactivity limits T-dependent responses. J. Immunol. 2013, 191, 2538–2550. [Google Scholar] [CrossRef]
- Doyle-Cooper, C.; Hudson, K.E.; Cooper, A.B.; Ota, T.; Skog, P.; Dawson, P.E.; Zwick, M.B.; Schief, W.R.; Burton, D.R.; Nemazee, D. Immune tolerance negatively regulates B cells in knock-in mice expressing broadly neutralizing HIV antibody 4E10. J. Immunol. 2013, 191, 3186–3191. [Google Scholar] [CrossRef]
- Haynes, B.F.; Moody, M.A.; Verkoczy, L.; Kelsoe, G.; Alam, S.M. Antibody polyspecificity and neutralization of HIV-1: A hypothesis. Hum. Antib. 2005, 14, 59–67. [Google Scholar]
- Klein, F.; Diskin, R.; Scheid, J.F.; Gaebler, C.; Mouquet, H.; Georgiev, I.S.; Pancera, M.; Zhou, T.; Incesu, R.B.; Fu, B.Z.; et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 2013, 153, 126–138. [Google Scholar] [CrossRef]
- Di Noia, J.M.; Neuberger, M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef]
- Dal Porto, J.M.; Haberman, A.M.; Kelsoe, G.; Shlomchik, M.J. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J. Exp. Med. 2002, 195, 1215–1221. [Google Scholar] [CrossRef]
- Shih, T.A.; Meffre, E.; Roederer, M.; Nussenzweig, M.C. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat. Immunol. 2002, 3, 570–575. [Google Scholar] [CrossRef]
- Victora, G.D.; Schwickert, T.A.; Fooksman, D.R.; Kamphorst, A.O.; Meyer-Hermann, M.; Dustin, M.L.; Nussenzweig, M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010, 143, 592–605. [Google Scholar] [CrossRef]
- Jacob, J.; Kelsoe, G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J. Exp. Med. 1992, 176, 679–687. [Google Scholar] [CrossRef]
- Jacob, J.; Przylepa, J.; Miller, C.; Kelsoe, G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. J. Exp. Med. 1993, 178, 1293–1307. [Google Scholar] [CrossRef]
- Wardemann, H.; Nussenzweig, M.C. B-cell self-tolerance in humans. Adv. Immunol. 2007, 95, 83–110. [Google Scholar] [CrossRef]
- Kain, R.; Exner, M.; Brandes, R.; Ziebermayr, R.; Cunningham, D.; Alderson, C.A.; Davidovits, A.; Raab, I.; Jahn, R.; Ashour, O.; et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat. Med. 2008, 14, 1088–1096. [Google Scholar] [CrossRef]
- Yu, R.K.; Ariga, T.; Usuki, S.; Kaida, K. Pathological roles of ganglioside mimicry in Guillain-Barre syndrome and related neuropathies. Adv. Exp. Med. Biol. 2011, 705, 349–365. [Google Scholar] [CrossRef]
- Bowes, T.; Wagner, E.R.; Boffey, J.; Nicholl, D.; Cochrane, L.; Benboubetra, M.; Conner, J.; Furukawa, K.; Furukawa, K.; Willison, H.J. Tolerance to self gangliosides is the major factor restricting the antibody response to lipopolysaccharide core oligosaccharides in Campylobacter jejuni strains associated with Guillain-Barre syndrome. Infect. Immun. 2002, 70, 5008–5018. [Google Scholar] [CrossRef]
- Verkoczy, L.; Bouton-Verville, H.; Diaz, M.; Haynes, B.F. Duke University School of Medicine: Durham, NC, USA, 2013; Unpublished work.
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant autoantibody production by early human B cell precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef]
- Lai, A.Y.; Kondo, M. Asymmetrical lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. J. Exp. Med. 2006, 203, 1867–1873. [Google Scholar] [CrossRef]
- Nemazee, D.A.; Burki, K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature 1989, 337, 562–566. [Google Scholar] [CrossRef]
- Erikson, J.; Radic, M.Z.; Camper, S.A.; Hardy, R.R.; Carmack, C.; Weigert, M. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature 1991, 349, 331–334. [Google Scholar] [CrossRef]
- Melchers, F. The pre-B-cell receptor: Selector of fitting immunoglobulin heavy chains for the B-cell repertoire. Nat. Rev. Immunol. 2005, 5, 578–584. [Google Scholar] [CrossRef]
- Melchers, F.; Rolink, A.R. B cell tolerance—How to make it and how to break it. Curr. Top. Microbiol. Immunol. 2006, 305, 1–23. [Google Scholar]
- Barthel, H.R.; Wallace, D.J. False-positive human immunodeficiency virus testing in patients with lupus erythematosus. Semin. Arthritis Rheum. 1993, 23, 1–7. [Google Scholar] [CrossRef]
- Mylonakis, E.; Paliou, M.; Greenbough, T.C.; Flaningan, T.P.; Letvin, N.L.; Rich, J.D. Report of a false-positive HIV test result and the potential use of additional tests in establishing HIV serostatus. Arch. Intern. Med. 2000, 160, 2386–2388. [Google Scholar] [CrossRef]
- Palacios, R.; Santos, J.; Valdivielso, P.; Marquez, M. Human immunodeficiency virus infection and systemic lupus erythematosus. An unusual case and a review of the literature. Lupus 2002, 11, 60–63. [Google Scholar] [CrossRef]
- Calza, L.; Manfredi, R.; Colangeli, V.; D’Antuono, A.; Passarini, B.; Chiodo, F. Systemic and discoid lupus erythematosus in HIV-infected patients treated with highly active antiretroviral therapy. Int. J. STD AIDS 2003, 14, 356–359. [Google Scholar] [CrossRef]
- Bonsignori, M.; Hwang, K.K.; Chen, X.; Tsao, C.Y.; Morris, L.; Gray, E.; Marshall, D.J.; Crump, J.A.; Kapiga, S.H.; Sam, N.E.; et al. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J. Virol. 2011, 85, 9998–10009. [Google Scholar] [CrossRef]
- Liao, H.X.; Lynch, R.; Zhou, T.; Gao, F.; Alam, S.M.; Boyd, S.D.; Fire, A.Z.; Roskin, K.M.; Schramm, C.A.; Zhang, Z.; et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 2013, 496, 469–476. [Google Scholar] [CrossRef]
- Gray, E.S.; Madiga, M.C.; Moore, P.L.; Mlisana, K.; Abdool Karim, S.S.; Binley, J.M.; Shaw, G.M.; Mascola, J.R.; Morris, L. Broad neutralization of human immunodeficiency virus type 1 mediated by plasma antibodies against the gp41 membrane proximal external region. J. Virol. 2009, 83, 11265–11274. [Google Scholar] [CrossRef]
- Gray, E.S.; Taylor, N.; Wycuff, D.; Moore, P.L.; Tomaras, G.D.; Wibmer, C.K.; Puren, A.; DeCamp, A.; Gilbert, P.B.; Wood, B.; et al. Antibody specificities associated with neutralization breadth in plasma from human immunodeficiency virus type 1 subtype C-infected blood donors. J. Virol. 2009, 83, 8925–8937. [Google Scholar] [CrossRef]
- Gray, E.S.; Madiga, M.C.; Hermanus, T.; Moore, P.L.; Wibmer, C.K.; Tumba, N.L.; Werner, L.; Mlisana, K.; Sibeko, S.; Williamson, C.; et al. The neutralization breadth of HIV-1 develops incrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection. J. Virol. 2011, 85, 4828–4840. [Google Scholar] [CrossRef]
- Mikell, I.; Sather, D.N.; Kalams, S.A.; Altfeld, M.; Alter, G.; Stamatatos, L. Characteristics of the earliest cross-neutralizing antibody response to HIV-1. PLoS Pathog. 2011, 7, e1001251. [Google Scholar] [CrossRef]
- Moore, P.L.; Gray, E.S.; Wibmer, C.K.; Bhiman, J.N.; Nonyane, M.; Sheward, D.J.; Hermanus, T.; Bajimaya, S.; Tumba, N.L.; Abrahams, M.R.; et al. Evolution of an HIV glycan-dependent broadly neutralizing antibody epitope through immune escape. Nat. Med. 2012, 18, 1688–1692. [Google Scholar] [CrossRef]
- Murphy, M.K.; Yue, L.; Pan, R.; Boliar, S.; Sethi, A.; Tian, J.; Pfafferot, K.; Karita, E.; Allen, S.A.; Cormier, E.; et al. Viral escape from neutralizing antibodies in early subtype A HIV-1 infection drives an increase in autologous neutralization breadth. PLoS Pathog. 2013, 9, e1003173. [Google Scholar] [CrossRef]
- Holl, T.M.; Haynes, B.F.; Kelsoe, G. Stromal cell independent B cell development in vitro: Generation and recovery of autoreactive clones. J. Immunol. Methods 2010, 354, 53–67. [Google Scholar] [CrossRef]
- Nojima, T.; Haniuda, K.; Moutai, T.; Matsudaira, M.; Mizokawa, S.; Shiratori, I.; Azuma, T.; Kitamura, D. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat. Commun. 2011, 2, e465. [Google Scholar] [CrossRef]
- Liao, H.X.; Bonsignori, M.; Alam, S.M.; McLellan, J.S.; Tomaras, G.D.; Moody, M.A.; Kozink, D.M.; Hwang, K.K.; Chen, X.; Tsao, C.Y.; et al. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity 2013, 38, 176–186. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.Y.; Li, Y.; Hogerkorp, C.M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef]
- McElrath, M.J.; Haynes, B.F. Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity 2010, 33, 542–554. [Google Scholar] [CrossRef]
- Burton, D.R.; Poignard, P.; Stanfield, R.L.; Wilson, I.A. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 2012, 337, 183–186. [Google Scholar] [CrossRef]
- Lingwood, D.; McTamney, P.M.; Yassine, H.M.; Whittle, J.R.; Guo, X.; Boyington, J.C.; Wei, C.J.; Nabel, G.J. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 2012, 489, 566–570. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kelsoe, G.; Verkoczy, L.; Haynes, B.F. Immune System Regulation in the Induction of Broadly Neutralizing HIV-1 Antibodies. Vaccines 2014, 2, 1-14. https://doi.org/10.3390/vaccines2010001
Kelsoe G, Verkoczy L, Haynes BF. Immune System Regulation in the Induction of Broadly Neutralizing HIV-1 Antibodies. Vaccines. 2014; 2(1):1-14. https://doi.org/10.3390/vaccines2010001
Chicago/Turabian StyleKelsoe, Garnett, Laurent Verkoczy, and Barton F. Haynes. 2014. "Immune System Regulation in the Induction of Broadly Neutralizing HIV-1 Antibodies" Vaccines 2, no. 1: 1-14. https://doi.org/10.3390/vaccines2010001